Гипофосфатемический рахит
Добавил пользователь Евгений Кузнецов Обновлено: 07.01.2026
Гипофосфатемические рахиты (hypophosphatemic rickets, HR) – генетически гетерогенная группа заболеваний с различными типами наследования, причиной развития которых могут быть мутации в ряде генов. Выделяют аутосомно-доминантную форму: ADHR (193100), обусловленную мутациями в гене FGF23 (12p13.32; 605380) ; аутосомно-рецессивные формы: ARHR1 (241520), обусловленную мутациями в гене DMP1, (4q21; 600980), и ARHR2 (613312), обусловленную мутациями в гене ENPP1 (6q22-q23; 173335); Х-сцепленную доминантную форму (307800), обусловленную мутациями в гене PHEX (Xp22.11; 300550); Х-сцепленную рецессивную форму (300554), обусловленную мутациями в гене CLCN5 (Xp11.23; 300008).
Семейный гипофосфатемический рахит, или фосфат-диабет, или витамин-D-резистентный рахит (HYPOPHOSPHATEMIC VITAMIN D-RESISTANT RICKETS; 307800), впервые описал Albrigt и соавт. в 1937г. Заболевание имеет Х-сцепленный доминантный тип наследования. Характеризуется полной пенетрантностью для гипофосфатемии и неполной — для костных изменений. Женщины передают патологический признак дочерям и сыновьям c вероятностью 50%, мужчины — только дочерям с вероятностью 100%. У мальчиков болезнь протекает тяжелее, чем у девочек.
Х-сцепленный гипофосфатемический рахит – наиболее распространенная форма гипофосфатемии, с частотой встречаемости около 1 на 20000-25000.
Молекулярно-генетическая причина заболевания – мутации в гене PHEX (Phosphat regulating hormone with homologies to endopeptidases on the X-chromosome). Ген состоит из 18 экзонов и кодирует фосфат-регулирующую эндопептидазу (гомологичную нейтральным эндопептидазам, которые регулируют активность других протеинов), контролирующую мембранный транспорт фосфата в почечных канальцах, тонкой кишке и, возможно, в других органах. Каким образом мутации в гене PHEX через гипотетический фосфатурический гормон приводят к почечной потере фосфата и нарушениям обмена витамина D, еще остается неясным и мнения по этому вопросу весьма противоречивы. Гипотетическая модель: предполагается, что эндопептидаза PHEX обуславливает активацию фосфатуритического гормона. Если мутация в PHEX-гене приводит к потере активности эндопептидазы, то вследствие этого снижается активность фосфатонина и, в итоге, возникает потеря фосфата через почки и отсутствие подавления инактивации 1,25-(OH)3-витамин-D.
Генетический дефект приводит к нарушениям реабсорбции фосфата в канальцах почек и его всасывания в тонкой кишке.
Заболевание начинается в конце первого или начале второго года жизни. У больных детей наблюдается мышечная гипотония, варусные деформации костей, особенно нижних конечностей, рахитические “браслеты”, “четки”, нарушение походки - “утиная” походка, отставание в росте. Иногда наблюдаются спонтанные переломы. Зубы прорезываются нормально, но быстро поражаются кариесом. Психическое развитие не страдает. На рентгенограмме костей выявляются значительные изменения в эпи- и метафизарных зон. Структура диафизов отличается от таковой при обычном рахите: наряду с зонами роста имеются зоны остеосклероза. Наблюдается утолщение трубчатых костей за счет одностороннего, чаще медиального слоя периоста. После закрытия эпифизарных зон роста проявления болезни ослабевают, но у нелеченных больных в зрелом возрасте встречаются тяжелые поражения костей.
После пубертатного периода заболевание может проходить даже без активной терапии. У взрослых, перенесших в детстве витамин-D-резистентный рахит, сохраняется гипофосфатемия, низкорослость, пострахитические деформации конечностей таза, часто обусловливающие кесарево сечение у женщин. Рецидивы возможны в период напряжения минерального обмена (беременность, лактация).
Аутосомно-доминантный гипофосфатемический рахит (HYPOPHOSPHATEMIC RICKETS, AUTOSOMAL DOMINANT; 193100) характеризуется изолированной почечной недостаточностью фосфатов, гипофосфатемией и нетипично нормальным уровнем 1,25-дигидроксивитамина D3 (кальцитриола). Пациенты часто страдают от боли в костях, рахита и абсцесса зубов. В отличие от X-сцепленного доминантного гипофосфатемического рахита, для АД гипофосфатемического рахита характерна неполная пенетрантность, варьирующий возраст начала заболевания (от детского до взрослого) и, в редких случаях, восстановление дефекта истощения фосфатов.
Молеклярно-генетическая причина АД гипофосфатемического рахита – мутации гена FGF23 (fibroblast growth factor 23). Ген картирован в локусе 12p13.32, состоит из 3 экзонов, кодирует белок из семейства факторов роста фиброблатов.
В Центре молекулярной генетики проводится прямая ДНК-диагностика гипофосфатемического рахита методом прямого автоматического секвенирования кодирующих последовательней и прилежащих интронных областей генов FGF23 и PHEX.
Гипофосфатемический рахит
- Главная /
- Редкие болезни /
- Энциклопедия заболеваний /
- Гипофосфатемический рахит (ГФР)
Гипофосфатемический рахит (ГФР)
(Витамин-Д-резистентный рахит; Х-сцепленный доминантный гипофосфатемический рахит; Х-сцепленный рецессивный гипофосфатемический рахит или Х-сцепленный гиперкальциурический нефролитиаз; Аутосомно-доминантный гипофосфатемический рахит; Аутосомно-рецессивный гипофосфатемический рахит 1, 2; Гипофосфатемический рахит с гиперкальциурией; Гипофосфатемический рахит и гиперпаратиреоз; Гипофосфатемический рахит с нефролитиазом/остеопорозом)
Hypophosphatemic rickets; Vitamin D-Resistant Rickets (VDRR); X-Linked Hypophosphatemic rickets (XLHR); X-linked recessive hypophosphatemic rickets (XLR) or X-linked hypercalciuric nephrolithiasis; Autosomal dominant hypophosphatemic rickets (ADHR); Autosomal recessive hypophosphatemic rickets - 1 (ARHR1); Autosomal recessive hypophosphatemic rickets - 2 (ARHR2); Hypophosphatemic rickets with hypercalciuria (HHRH); Hypophosphatemic rickets and hyperparathyroidism; Hypophosphatemic nephrolithiasis/osteoporosis-1 (NPHLOP1).
Генетика: мутации различных генов: фосфатрегулирующего гена с гомологией к эндопептидазам на Х-хромосоме (PHEX); потенциал-зависимого канала хлоридов 5 (CLCN5); фактора роста фибробластов 23 (FGF23); дентин-матричного протеина 1 (DMP1); эктонуклеотид пирофосфатаза/фосфодиэстераза 1 (ENPP1); натрий-фосфорного котранспортера тип 2 (SLC34A3; Npt2c); клото (Klotho, KL); натрий-фосфорного транспортера тип 2 (SLC34A1, Npt2)
Гены картированы на Xp22.11, Хp.11.23-p11.22-хромосоме, 12p13.32, 4q22.1, 6q23.2, 9q34.3, 13q13.1, 5q35.3, соответственно.
Тип наследования: Х-сцепленный доминантный, Х-сцепленный рецессивный, аутосомно-доминантный, аутосомно-рецессивный, соответственно.
Эпидемиология: относятся к редким наследственным заболеваниям. Х-сцепленный доминантный гипофосфатемический рахит встречается с частотой 1:20 000 живых новорожденных. Частота остальных форм неизвестна.
Патогенез
За счет поломки генов нарушается реабсорбция фосфора (т.е. обратное всасывание фосфора из первичной мочи в кровь) в проксимальных почечных канальцах, что приводит к потере фосфора из организма. Недостаток фосфора является причиной нарушения минерализации костей скелета и зубов, что приводит к развитию рахита. При данном заболевании снижается активность 1альфа-гидроксилазы (CYP27b1) в почках, поэтому нарушается образование из неактивной формы витамина Д (т.е.холекальциферола) той формы, которая является биологически активной, т.е. кальцитриола (1,25(ОН)2D), в связи с чем назначение препаратов нативного витамина Д (холекальциферол, рыбий жир, аквадетрим, вигантол) является неэффективным.
При некоторых формах наряду с потерей фосфора нарушается регуляция синтеза паратгормона и 1,25(ОН)- витамина Д, что может приводить к развитию гиперпаратиреоза, гиперкальциурия с формированием нефрокальциноза.
Клинические проявления: как правило манифестация заболевания отмечается на 1-2 году жизни ребенка, при некоторых формах начало клинических проявлений отмечается в более позднем возрасте (5-6 лет). Корреляции между генотипом и фенотипом не отмечено. В одной семье тяжесть клинических проявлений между пострадавшими родственниками может быть различной.
Основными клиническими признаками ГФР являются:
- рахитические деформации грудной клетки, гипертрофия лобных бугров на первом году жизни;
- прогрессирующие деформации ног с момента начала ходьбы;
- «переваливающаяся» походка;
- мышечная слабость;
- задержка роста;
- боль в костях;
- позднее прорезывание зубов или частый кариес и абсцессы.
На первом году жизни у ребенка могут отмечаться характерные признаки рахита: гипотония, гипертрофия лобных бугров, рахитические «браслетки» на лучезапястных суставах, «четки» на ребрах, с началом самостоятельной опоры на ноги или самостоятельной ходьбы появляется варусная (или О-образная) деформация ног. Дети начинаются ходить широко расставив ноги, походка переваливающаяся (по типу «утиной»), при этом быстро устают. В последующем может сформироваться вальгусная (или Х-образная) деформация ног.
У некоторых пациентов в раннем детском возрасте может сформироваться краниостеноз. Прорезывание зубов у детей запаздывает, со 2-3 года жизни отмечается истончение эмали, частый кариес, часто - спонтанные абсцессы зубов по причине формирования прикорневых кист.
На 2-3 году отмечается замедление динамики роста, дети отстают от сверстников в физическом развитии, в последующем отставание в росте наростает, что может быть связано с прогрессирующими деформациями ног. Деформации ног ведут к диспропорциональному телосложению за счет укорочения нижнего сегмента.
На фоне назначения препаратов нативной формы витамина Д (холекальциферол, рыбий жир, вигантол, аквадетрим) даже в очень высоких дозах эффекта не наблюдается. Проведение коррегирующих остеотомий на костях ног в допубертатном периоде (до 13-14 лет), как правило, практически в 100% случаев характеризуется рецидивом деформаций или усугублением искривлений ног.
В взрослом возрасте у пациентов имеется значительно выраженный болевой синдром в области коленных суставов и их тугоподвижность (артоз), нефрокальциноз, кальцификация связок. При минимальных травмах возможны переломы костей ног, рук на фоне остеопороза. После 18 лет отмечаются частые абсцессы зубов, возможно до полного отсутствия собственных зубов. Кроме того по причине кальцификации связок в среднем у пациентов может отмечаться снижение слуха.
Лечение
В настоящее время в мире используются препараты фосфорного буфера в сочетании с активными формами витамина Д (альфакальцидолом или кальцитриолом). Данная схема терапии применяется у пациентов в период активного роста (до 18 лет). Следует помнить, что при данном заболевании прием препаратов фосфора должен быть распределен на 5-6 раз в сутки по причине быстрого выведения фосфора из организма. Дозы подбираются в зависимости от веса пациента и переносимости препарата. Эффективность лечения оценивается по отсутствию прогрессии деформаций ног, улучшению физической активности (уменьшению мышечной слабости), улучшению динамики роста. Нормализация уровня фосфора в крови не является целью лечения и, наоборот, достижение нормального показателя фосфора крови говорит о передозировке препаратами. На фоне проводимой терапии необходимо регулярное наблюдение лечащего доктора, проводить контроль уровня паратгормона крови, экскреции кальция в моче и УЗИ почек, с целью избежать побочных эффектов от терапии. Возможные побочные эффекты: ранние- жидкий стул, тошнота; поздние- торичный гиперпаратиреоз, гиперкальциурия, нефрокальциноз.
Пациентам, достигшим конечного роста (после 18 лет), рекомендуется назначать препараты фосфора по следующим показаниям: 1. Выраженная мышечная слабость и боли в костях,которые ограничивают активные движения. 2. При проведении оперативных вмешательств на костях (коррегирующие остеотомии) с целью улучшения регенерации костной ткани. 3. При выраженном остеопорозе.
Гипофосфатемический рахит: патогенез, диагностика и лечение
Гипофосфатемический рахит (ГФР) – группа заболеваний, характеризующихся развитием рахитических изменений костной ткани вследствие повышенного выведения фосфора из организма. Данная форма рахита является наиболее распространенной среди вариантов генетически детерминированных форм нарушений минерального обмена. ГФР является актуальной медико-социальной проблемой, требующей постоянного обновления знаний как эндокринологов, так и врачей другого профиля. Это обусловлено тем, что клиническая картина ГФР имеет значительную гетерогенность и может проявляться выраженными деформациями скелета, задержкой физического развития, тяжелой мышечной гипотонией, частыми переломами, абсцессами зубов, что в ряде случаев приводит к инвалидизации пациента и, соответственно, снижает качество жизни. Своевременная диагностика и адекватная терапия ГФР имеет крайне важное значение для предотвращения развития тяжелых осложнений.
В настоящее время известны более 10 генов-кандидатов, дефекты в которых приводят к развитию врожденных форм ГФР. Генетическая диагностика ГФР имеет большое значение для определения формы ГФР, а также для проведения генетического консультирования семей при планировании беременности.
Ключевые слова
Для цитирования:
For citation:
Введение
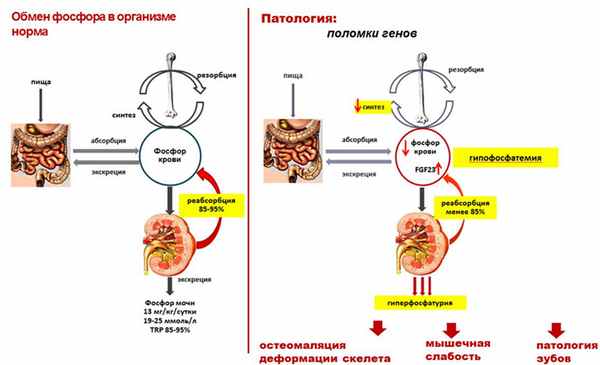
Витамин-Д-резистентный рахит (ВДРР), или гипофосфатемический рахит (ГФР), – группа заболеваний, характеризующихся развитием рахитических изменений костной ткани вследствие повышенного выведения фосфора из организма (рис. 1) [1].
Рис. 1. Схематичное изображение механизма развития гипофосфатемического рахита.
Особенностью данной группы заболеваний является гетерогенность клинической картины, причина которой в сложности регуляции обмена фосфора в организме.
Своевременная диагностика и адекватная терапия ГФР имеют крайне важное значение для предотвращения развития осложнений, а именно: тяжелых деформаций скелета, которые могут приводить к частым переломам и низкорослости, мышечной слабости, являющейся основным фактором ограничения активных движений, абсцессов зубов [1–3]. Данные проявления заболевания являются основными причинами инвалидизации пациентов с ГФР.
Значительная гетерогенность клинической картины ГФР требует привлечения широкого круга специалистов (терапевтов, педиатров, эндокринологов, ортопедов и стоматологов) к ведению таких больных, поэтому единое понимание проблемы является одним из основных факторов в достижении компенсации заболевания и, соответственно, улучшении качества жизни пациента.
Основные клинические признаки ГФР
Фосфор в организме человека играет важную роль, а именно: принимает участие в аккумуляции энергии в виде АТФ, является неотъемлемой частью молекул ДНК и РНК, необходим для построения кристаллов гидроксиапатита костного матрикса, за счет которых кость приобретает прочность. Следствием хронической недостаточности фосфора является нарушение минерализации костей и развитие рахита.
Основными клиническими признаками ГФР являются задержка роста, прогрессирующие деформации ног с момента начала ходьбы, мышечная слабость, боль в костях, позднее прорезывание зубов или их частый кариес и абсцессы. Во взрослом возрасте заболевание может манифестировать в виде нефрокальциноза, кальцификации связок, артрозов крупных суставов, остеопороза; при некоторых формах ГФР описана нейросенсорная тугоухость. Биохимическими и гормональными маркерами ГФР служат гипофосфатемия на фоне гиперфосфатурии, повышение активности ЩФ, нормальный или умеренно повышенный уровень ПТГ при нормокальциемии. Концентрация 1,25(ОН)2D3 в сыворотке крови и степень кальциурии варьируют и зависят от молекулярной основы заболевания [4].
Классификация ГФР
По типу наследования различают несколько врожденных форм ГФР.
Х-сцепленный доминантный ГФР (X-linked hypophosphatemia, XLH)
Данная форма ГФР имеет наибольшую распространенность, частота которой составляет 1:20 000 новорожденных. Причиной развития заболевания являются инактивирующие мутации в гене PHEX.
Ген PHEX – это фосфатрегулирующий ген с гомологией к эндопептидазам на Х-хромосоме (Phosphate regulating gene with Homology to Endopeptidases located on the X chromosome), экспрессируется в остеобластах.
Первоначально считалось, что продукт гена PHEX принимает участие в деградации фосфотонина FGF23. В большинстве случаев при инактивации гена PHEX концентрация FGF23 в крови возрастает, что приводит к снижению экспрессии генов натрий-фосфорных котранспортеров 2 типа, которые регулируют в проксимальных почечных канальцах реабсорбцию фосфора, снижение их числа и активности ведет к потере фосфора из организма. FGF23 также подавляет активность 1α-гидроксилазы (CYP27B1), участвующей в образовании кальцитриола и, наоборот, активирует 24-гидроксилазу (CYP24A1), превращающую кальцитриол в неактивные метаболиты [5, 6].
Несмотря на то что гены PHEX и FGF23 экспрессируются на остеобластах, исследования на мышах (модель ГФР человека) не смогли подтвердить прямое взаимодействие продуктов данных генов. Предполагается, что существует промежуточный фактор в данной системе, который пока неизвестен (рис. 1).
Х-сцепленный рецессивный ГФР с гиперкальциурией (X-linked recessive hypercalciuric hypophosphataemic rickets)
Х-сцепленный рецессивный ГФР с гиперкальциурией, или болезнь Дента, – редкое наследственное заболевание, характеризующееся дисфункцией проксимальных почечных канальцев в виде низкомолекулярной протеинурии (НМП), гиперкальциурии, фосфатурии, с развитием нефрокальциноза/мочекаменной болезни, прогрессирующей почечной недостаточности и рахита [11–12]. По литературным данным, частота диагностики основных признаков патологии представлена следующим образом: НМП – 100%, гиперкальциурия – 95%, нефрокальциноз – 74%, рахит или остеомаляция – 30%, почечная недостаточность – 64%, аминоацидурия – 76%, глюкозурия – 54%, гипофосфатемия – 50%, аминоацидурия – 17% [7].
Болезнь проявляется у пациентов мужского пола. У мальчиков в возрасте 3–5 лет развивается почечная недостаточность, которая достигает терминальной стадии к 30–40 годам. Как правило, женщины являются бессимптомными носительницами заболевания, в редких случаях могут иметь НМП и гиперкальциурию.
Болезнь Дента имеет генетическую гетерогенность: примерно 50–60% пациентов – носители мутации гена CLCN5 (болезнь Дента 1), около 15% – дефектов гена OCRL1 (болезнь Дента 2), а остальные 25–35% не имеют поломок в этих генах, но, возможно, несут дефекты других генов.
Ген CLCN5 локализован на хромосоме Xp11.22, кодирует белок хлорпроводящего ионного канала-5 (CLCN-5).
Помимо тяжелого поражения почек с прогрессирующим снижением азотовыделительной функции, для пациентов с рецессивным Х-сцепленным ГФР характерно наличие гиперкальциурии и высокого уровня 1,25(ОН)2D3 в крови, в отличие от больных с доминантным Х-сцепленным ГФР.
Гиперкальциурия обусловлена гиперпродукцией кальцитриола при избыточной активации 1α-гидроксилазы паратгормоном. 1,25(ОН)2D3 стимулирует всасывание кальция в кишечнике, и увеличение его концентрации в сыворотке крови может косвенно привести к гиперкальциурии и нефрокальцинозу. Учитывая данную особенность, врачам стоит помнить о применении более низких доз альфакальцидола.
ГФР может манифестировать и в более позднем возрасте и иметь менее выраженные клинические проявления. Таким примером может являться аутосомно-доминантный (АД) ГФР.
Аутосомно-доминантный ГФР (Autosomal dominant hypophosphatemic rickets, ADHR)
АД ГФР – это очень редкая форма ГФР, характеризующаяся клинической картиной рахита с возможно более поздней манифестацией, гипофосфатемией, гиперфосфатурией и неадекватно низким или нормальным уровнем 1,25(ОН)2D3 [8].
Причиной данной формы ГФР являются точковые миссенс-мутации в гене фактора роста фибробластов 23 (fibroblast growth factor-23; FGF23), которые изменяют аминокислотную последовательность в участке белка, что приводит к нарушению расщепления этого белка и увеличению концентрации активной формы FGF23 в крови [9].
FGF23 считается основным фосфотонином, т.е. увеличивает клиренс фосфатов в почках за счет снижения натрий-фосфорных котранспортеров и подавления активности 1α-гидроксилазы.
Важно помнить, что пациенты с АД ГФР демонстрируют неполную пенетрантность заболевания: члены одной семьи с одной и той же мутацией в гене FGF23 могут иметь различную степень тяжести рахита или быть полностью здоровыми носителями генетического дефекта. Клиническая картина данной формы ГФР включает в себя рахитические деформации скелета, нарушение ходьбы вследствие мышечной слабости и болевого синдрома, абсцессы зубов. Возраст манифестации заболевания может быть различным, в некоторых случаях у пациентов возможна спонтанная ремиссия после периода полового созревания. Биохимическими и гормональными маркерами патологии являются гипофосфатемия на фоне гиперфосфатурии, повышение уровня щелочной фосфатазы и нормальный уровень 1,25(OH)2D в крови [ADHR Consortium, 2000].
В своей практике врач также может встретиться с сочетанием ГФР с эпидермальным невусом вдоль линий Блашко, полиоссальной кистозно-фиброзной дисплазией костей и тимомой. Данное заболевание получило название «синдром эпидермального невуса» (Epidermal nevus syndrome), или «кожно-скелетный гипофосфатемический синдром» (Cutaneous skeletal hypophosphatemia syndrome (CSHS)), при котором ГФР вызван гиперпродукцией FGF23 [11]. Генетической основой заболевания являются дефекты в генах RAS-цепи (HRAS, KRAS, NRAS). Столь большое количество пораженных органов обусловлено экспрессией RAS-генов.
Аутосомно–рецессивный (АР) ГФР (Autosomal recessive hypophosphatemic rickets, ARHR)
АР ГФР вызван инактивирующими мутациями в гене, кодирующем белок матрицы дентина 1 (DMP1), или гене эктонуклеотид/пирофосфатазы-фосфодиэстеразы 1 (ENPP1) [24–27].
Отличительными особенностями данных форм ГФР является наличие сенсоневральной глухоты из-за склероза внутреннего слухового канала, повышенная плотность различных костей скелета (черепа, ключиц, ребер) и ярко выраженная энтезопатия.
АР ГФР с гиперкальциурией (Hereditary hypophosphatemic rickets with hypercalciuria, HHRH)
Данная форма ГФР вызвана мутациями гена SLC34A3. Характерными биохимическими признаками данного варианта ГФР являются гиперкальциурия и повышенное содержание 1,25(ОН)2D3 в крови при низком уровне ПТГ [12].
Клинические проявления рахита заболевания могут быть умеренными либо полностью отсутствовать или манифестировать в более старшем возрасте. Точная диагностика ГФР с гиперкальциурией имеет важное терапевтическое значение. В отличие от других форм ГФР, при данном варианте назначение только солей фосфора может привести к полной ремиссии заболевания, в то время как добавление нативных или активных форм витамина D может вызвать развитие осложнений, таких как гиперкальциемия, нефрокальциноз и мочекаменная болезнь.
Гипофосфатемический рахит (ГФР)
Гипофосфатемический рахит (ГФР) — это наследственная группа заболеваний, обусловленная повышенным выведением фосфора из организма и развитием рахита. Причиной развития данной формы рахита могут быть поломки в различных генах, которые участвуют в регуляции обмена фосфора. В результате недостатка фосфора нарушается минерализация костной ткани скелета, зубов, а также нарушено обеспечение мышц энергией, т.к. молекула АТФ (аккумулятор энергии в организме) включает фосфор. Под тяжестью собственного тела с момента начала ходьбы у детей появляются рахитические деформации ног. Некоторые формы ГФР могут манифестировать в более позднем возрасте.
Синонимы названия диагноза: витамин-Д-резистентный рахит; Х-сцепленный доминантный гипофосфатемический рахит; Х-сцепленный рецессивный гипофосфатемический рахит или Х-сцепленный гиперкальциурический нефролитиаз; Аутосомно-доминантный гипофосфатемический рахит; Аутосомно-рецессивный гипофосфатемический рахит 1, 2; Гипофосфатемический рахит с гиперкальциурией; Гипофосфатемический рахит и гиперпаратиреоз; Гипофосфатемический рахит с нефролитиазом/остеопорозом)
Англоязычные названия: Hypophosphatemic rickets; Vitamin D-Resistant Rickets (VDRR); X-Linked Hypophosphatemic rickets (XLHR); X-linked recessive hypophosphatemic rickets (XLR) or X-linked hypercalciuric nephrolithiasis; Autosomal dominant hypophosphatemic rickets (ADHR); Autosomal recessive hypophosphatemic rickets - 1 (ARHR1); Autosomal recessive hypophosphatemic rickets - 2 (ARHR2); Hypophosphatemic rickets with hypercalciuria (HHRH); Hypophosphatemic rickets and hyperparathyroidism; Hypophosphatemic nephrolithiasis/osteoporosis-1 (NPHLOP1).
Генетика: мутации различных генов: фосфатрегулирующего гена с гомологией к эндопептидазам на Х-хромосоме (PHEX); потенциал-зависимого канала хлоридов 5 (CLCN5); фактора роста фибробластов 23 (FGF23); дентин-матричного протеина 1 (DMP1); эктонуклеотид пирофосфатаза/фосфодиэстераза 1 (ENPP1); натрий-фосфорного котранспортера тип 2 (SLC34A3; Npt2c); клото (Klotho, KL); натрий-фосфорного транспортера тип 2 (SLC34A1, Npt2)
Гены картированы на Xp22.11, Хp.11.23-p11.22-хромосоме, 12p13.32, 4q22.1, 6q23.2, 9q34.3, 13q13.1, 5q35.3, соответственно.
Тип наследования: Х-сцепленный доминантный, Х-сцепленный рецессивный, аутосомно-доминантный, аутосомно-рецессивный, соответственно.
Эпидемиология: относятся к редким наследственным заболеваниям. Х-сцепленный доминантный гипофосфатемический рахит встречается с частотой 1:20 000 живых новорожденных. Частота остальных форм неизвестна.
Клинические проявления: как правило, манифестация заболевания отмечается на 1-2 году жизни ребенка, при некоторых формах- начало клинических проявлений отмечается в более позднем возрасте (5-6 лет). Корреляции между генотипом и фенотипом не отмечено. В одной семье тяжесть клинических проявлений между пострадавшими родственниками может быть различной.
Основными клиническими признаками ГФР являются:
- задержка роста,
- прогрессирующие деформации ног с момента начала ходьбы,
- мышечная слабость,
- боль в костях,
- позднее прорезывание зубов или частый кариес и абсцессы.
На первом году жизни у ребенка могут отмечаться характерные признаки рахита: гипотония, гипертрофия лобных бугров, рахитические «браслетки» на лучезапястных суставах, «четки» на ребрах, с началом самостоятельной опоры на ноги или самостоятельной ходьбы появляется варусная (или О-образная деформация ног). Дети начинаются ходить широко расставив ноги, походка переваливающаяся (по типу «утиной»), при этом быстро устают. У некоторых пациентов в раннем детском возрасте может сформироваться краниостеноз.
Прорезывание зубов у детей запаздывает, со 2-3 года жизни отмечается истончение эмали, частый кариес, часто- спонтанные абсцессы зубов по причине формирования прикорневых кист.
На 2-3 году отмечается замедление динамики роста, дети отстают от сверстников в физическом развитии, в последующем отставание в росте наростает, что может быть связано с прогрессирующими деформациями ног.
Во взрослом возрасте - нефрокальциноз, кальцификация связок, артрозы крупных суставов, остеопороз.
Диагностика: к сожалению, ранняя диагностика ГФР часто запаздывает из-за низкой осведомленности врачей о данном заболевании, однако сама патология не является очень редкой (1:20000 новорожденных для Х-сцепленного гипофосфатемического рахита). Это приводит к неправильному началу терапии. Пациент в течении нескольких лет может пройти множество обследований у различных специалистов (у педиатра, эндокринолога, нефролога и ортопеда), прежде чем ему будет установлен диагноз «Гипофосфатемический рахит».
Чем раньше установлен диагноз и начато лечение с использованием препаратов фосфора, тем лучше прогноз для ребенка.
Основные лабораторно-инструментальные критерии в постановке диагноза:
- Низкий уровень фосфора в крови, по сравнению с соответствующей возрастной нормой
- Нормальный уровень кальция в сыворотке крови
- Умеренно или резко повышенный уровень щелочной фосфатазы крови
- Повышенное выделение фосфора с мочой (или низкие показатели тубулярной реабсорбции фосфатов - индекс TmP/GFR- максимум тубулярной реабсорбции фосфата в расчете на креатинин, низкий показатель TRP (%) - тубулярная реабсорбция фосфата).
Следует помнить, что рассчитывать количество фосфора в разовой порции мочи необходимо по отношению к креатинину.
- При одной из форм ГФР может выявляться повышенное выделение кальция в моче (повышение индекса кальций/креатинин- гиперкальциурия)
- Низконормальный уровень биологически активного витамина D (1,25 (OH) 2D3)
- Паратгормон (ПТГ или PTH) может быть нормальным или умеренно повышенным.
Рентгенологические признаки рахита:
расширение и разрушение метафизов длинных трубчатых костей, грубая структура трабекулярной кости. Бокаловидное изменение метафизов происходит в проксимальной и дистальной части большеберцовой кости, лучевой и локтевой костях, кортикальный слой утолщен по медиальной и задней поверхностям костей. В области диафизов- корковый слой истончен, выглядит слоистым, контуры его как со стороны костномозгового канала, так и снаружи становятся нечеткими, «смазанными». Нередко, в области диафизов длинных трубчатых костей возможны поднадкостничные переломы.
Молекулярногенетическая диагностика проводится в НИИ Детской эндокринологии ФГБУ ЭНЦ (г. Москва), ФГБУН Медико-генетический научный центр (г. Москва).
Гипофосфатемический рахит
Гипофосфатемический рахит – заболевание, характеризующееся гипофосфатемией, нарушением всасывания кальция в кишечнике и резистентным к терапии витамином D рахитом или остеомаляцией. Обычно передается по наследству. Симптомами являются боль в костях, переломы и нарушения роста. Диагноз ставят на основании определения в сыворотке крови уровня фосфатов, щелочной фосфатазы и 1,25-дигидроксивитамина D3. Лечение состоит в пероральном применении фосфата и кальцитриола; буросумаб назначается в случае гипофосфатемией, связанной с Х-хромосомой.
Спорадические приобретенные случаи иногда вызывабтся доброкачественными мезенхимальными опухолями, продуцирующими гуморальный фактор, снижающий резорбцию фосфата в проксимальных почечных канальцах (индуцированная опухолью остеомаляция).
Общие справочные материалы
1. Bitzan M, Goodyer PR: Hypophosphatemic rickets. Pediatr Clin N Am 66(1):179–207, 2019. doi: 10.1016/j.pcl.2018.09.004
Патофизиология гипофосфатемических рахитов
Нарушение заключается в снижении реабсорбции фосфатов в проксимальных канальцах, приводящем к потери мочевого фосфата и гипофосфатемии Гипофосфатемия Гипофосфатемия – концентрация фосфата в сыворотке крови 2,5 мг/дл (0,81 ммоль/л). Причинами гипофосфатемии являются расстройство, связанное с употреблением алкоголя, ожоги, голодание и применение. Прочитайте дополнительные сведения . Этот дефект обусловлен циркулирующими факторами, называемыми фосфатонинамы. Основным фосфатонином при наследственных гипофосфатемических рахитах является фактор роста фибробластов-23 (FGF-23). Также происходит снижение абсорбции кальция и фосфатов в кишечнике. Дефект минерализации кости развивается из-за низких уровней фосфата и дисфункции остеобластов, а не из-за низкой концентрации кальция и повышенного уровня паратиреоидного гормона (ПТГ), как при кальципеническом рахите ( Недостаточность витамина D и зависимость Недостаточность витамина D и зависимость Недостаточное пребывание на солнце предрасполагает к дефициту витамина D. Дефицит нарушает минерализацию кости, вызывает рахит у детей и остеомаляцию у взрослых и, возможно, вносит свой. Прочитайте дополнительные сведения ). Поскольку уровень 1,25-дигидроксивитамина D3 в норме или незначительно снижен, предполагается дефект конверсии; гипофосфатемия в норме должна бы приводить к повышению уровня D3.
Существует несколько форм гипофосфатемического рахита (см. таблицу Формы наследственного гипофосфатемического рахита Формы наследственного гипофосфатемического рахита* ). Наследственная форма гипофосфатемического рахита с гиперкальциурией (ГФР), как известно, возникает в результате мутаций натрий-фосфатного котранспортера в проксимальных канальцах типа 2с (NaPi2c). Нарушенный транспорт фосфатов и гипофосфатемия в этом случае приводят к соответственно повышенным уровням 1,25-дигидрокси-витамина D3, тем самым приводя к гиперкальциурии.
Симптомы и признаки гипофосфатемического рахита
Заболевание проявляется спектром нарушений от только гипофосфатемии до замедления роста, низкого роста и в наиболее тяжелых случаях – тяжелого рахита или остеомаляции. У детей, как правило, нарушения выявляют после того, как они начинают ходить, и проявляются в искривление ног и других деформациях костей, псевдофрактурах (т.е., данные рентгенографии при остеомаляции, которые могут отображать области предшествующих переломов напряжения, которые были заменены недостаточно минерализованным остеоидом, против областей костных эрозий), болью в костях и низкорослостью. Костный нарост в месте прикрепления мышц может ограничивать движения.
Рахитические изменения позвоночника или тазовых костей, дефекты зубной эмали и тетании, возникающие при недостаточности в пище витамина D, редко выявляют при гипофосфатемическом рахите.
ГФР у пациентов может проявляться нефролитиазом и/или нефрокальцинозом.
Диагностика гипофосфатемического рахита
Сывороточные уровни кальция, фосфата, щелочной фосфатазы, 1,25-дигидроксивитамина D3, ПТГ, FGF-23 и креатинина
Уровни фосфат и креатинина в моче (для расчета канальцевой реабсорбции фосфата)
Уровни фосфатов в сыворотке крови снижены, но экскреция с мочой фосфатов достаточно высокая. Уровни кальция и ПТГ в сыворотке крови в норме, а щелочная фосфатаза часто повышена. Стимуляции продукции кальцитриола, индуцированной гипофосфатемией, не возникает. Как правило, уровни кальцидиола являются нормальными, в то время как уровни кальцитриола являются нормальными или низкими. При кальципеническом рахите присутствует гипокальциемия, гипофосфатемия имеет легкую степень или отсутствует и уровень фосфата в моче не повышен.
Лечение гипофосфатемического рахита
Перорально препараты фосфора и кальцитриол
Буросумаб - для Х-сцепленной гипофосфатемии
Лечение гипофосфатемического рахита состоит в приеме раствора или таблеток нейтрального фосфата. Начальная доза у детей составляет 10 мг/кг (на основе элементарного фосфора) перорально 4 раза в день. Фосфатные добавки снижают концентрацию ионизированного кальция и дополнительно ингибируют трансформацию кальцитриола, что приводит к вторичному гиперпаратиреозу и усугубляет потерю фосфатов с мочой. Таким образом, пероральный витамин D назначается в виде кальцитриола в начальной дозе 5-10 нг/кг 2 раза в день. Однако это не так при врождённом гипофосфатемическом рахите или с гиперкальцийурией или ГГН (гипофосфатемия, гиперкальциемия и нефрокальциноз), где уровни 1,25-дигидрокси-витамина D3 повышены и прием кальцитриола может быть вредным.
Может потребоваться увеличение дозы фосфата для обеспечения роста костей или облегчения боли в костях. Диарея может ограничить пероральный прием фосфата. Происходит увеличение концентрации фосфатов и снижение щелочной фосфатазы в плазме крови, вылечивание рахита и улучшение темпов роста. Гиперкальциемия, гиперкальциурия и нефрокальциноз с пониженной функцией почек могут осложнить лечение. Пациенты, получающие лечение, нуждаются в постоянном наблюдении.
Буросумаб является анти-FGF-23 моноклональным антителом; он стал препаратом выбора для пациентов с Х-сцепленной гипофосфатемией (XLH) и заменил традиционную терапию, описанную выше ( 1 Справочные материалы по лечению Гипофосфатемический рахит – заболевание, характеризующееся гипофосфатемией, нарушением всасывания кальция в кишечнике и резистентным к терапии витамином D рахитом или остеомаляцией. Обычно передается. Прочитайте дополнительные сведения ). Дозировка для детей весом менее 10 кг начинается с 1 мг/кг (с округлением до 1 мг) подкожно каждые 2 недели. Для детей от 6 месяцев до < 18 лет и массой тела >10 кг начальная доза составляет 0,8 мг/кг (округленная до ближайших 10 мг) подкожно каждые 2 недели. Для взрослых 18 лет и старше начальная доза составляет 1 мг/кг (с округлением до 10 мг) подкожно каждые 4 недели. В соответствии с инструкцией производителя manufacturer’s instructions для нормализации уровня фосфата в сыворотке при необходимости дозу можно постепенно подбирать до максимальной 2 мг/кг или 90 мг.
Дефицит железа Дефицит железа Железо (Fe) представляет собой компонент гемоглобина, миоглобина и многих ферментов в организме. Железо гема, которое содержится, главным образом, в продуктах животного происхождения, усваивается. Прочитайте дополнительные сведения вызывает повышение выработки костной тканью FGF-23 и может усугублять состояния, сопутствующие высокому уровню FGF-23/патологическому расщеплению FGF. Поэтому для пациентов с дефицитом железа в условиях гипофосфатемии с высокими показателями FGF-23 восполнение запасов железа Лечение Дефицит железа является наиболее частой причиной анемии и обычно обусловлен кровопотерей; мальабсорбция, к примеру такая,, как при целиакии, является гораздо менее распространенной причиной. Прочитайте дополнительные сведенияУлучшение состояния взрослых с онкогенным рахитом возможно только при удалении мезенхимальной опухоли, вызывающей данное нарушение. В противном случае онкогенный рахит лечат с применением кальцитриола по 5–10 нг/кг перорально 2 раза в день и элементного фосфора по 250–1 000 мг перорально 3–4 раза в день.
Справочные материалы по лечению
1. Imel EA, Glorieux FH, Whyte MP, et al: Burosumab versus conventional therapy in children with X-linked hypophosphataemia: A randomised, active-controlled, open-label, phase 3 trial. Lancet 393(10189):2416–2427, 2019. doi: 10.1016/S0140-6736(19)30654-3. Clarification and additional information. Lancet 394(10193):120, 2019. doi: 10.1016/S0140-6736(19)31426-6
Ключевые моменты
Сниженная реабсорбция фосфатов почками приводит к потере мочевого фосфата и гиперфосфатемии.
Возникает недостаточная минерализация костей из-за низких уровней фосфата и дисфункции остеобластов.
У детей наблюдается задержка роста, боль в костях и их деформация (например, искривление ноги) и низкорослость.
Гипофосфатемический рахит с гиперкальциурией (ГФР) может проявляться у пациентов нефролитиазом и/или нефрокальцинозом.
Диагноз устанавливают на основании выявленного низкого уровня фосфатов в сыворотке, повышенного уровня фосфатов в моче и нормальных сывороточных показателей кальция и паратгормона.
Следует назначить лечение с применением пероральных фосфатных добавок и, за исключением случаев ГФР, витамина D (в форме кальцитриола).
Применяйте буросумаб для Х-сцепленной гипофосфатемии
Дополнительная информация
Ниже следует англоязычный ресурс, который может быть информативным. Обратите внимание, что The manual не несет ответственности за содержание этого ресурса.
Manufacturer’s instructions: Dosing and administration information for burosumab
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: