Глухота с врожденной спондило-эпифизарной дисплазией
Добавил пользователь Алексей Ф. Обновлено: 29.01.2026
Врожденная спондилоэпифизарная дисплазия (DEEC) редкое генетическое заболевание, которое приводит к низкорослости и аномалиям скелета, в первую очередь поражающим позвоночник и длинные кости рук и ног.
Это форма карликовости, дети с этим расстройством часто имеют проблемы со зрением и слухом. Состояние присутствует при рождении.
Люди с CES маленькие; Рост взрослого человека колеблется от 34 до 57 дюймов в высоту. Его ноги, руки и голова нормального размера, но его ноги, руки и позвоночник укорачиваются.
Дети с DEEC могут иметь ряд проблем с опорно-двигательным аппаратом., включая шатающуюся шею, связки свободный, отсутствие мышечного тонуса, заболевания спины и позвоночника, деформации бедра, проблемы со стопами и дегенеративные заболевания тазобедренных суставов, колени или плечи.
Дети с этим заболеванием могут иметь черепно-лицевые деформации, включая расщелину неба., плоское лицо и глазной гипертелоризм (большее расстояние между глазами).
Врожденная спондилоэпифизарная дисплазия встречается редко., так как это происходит менее чем 1 десятилетие 100 000 рождения. Встречается в равной степени у мужчин и женщин.
Причина врожденной спондилоэпифизарной дисплазии
DEEC вызывается мутацией хромосомы 12 в COL2A1 (альфа-коллаген II типа 1 нить). Мутация влияет на коллаген и соединительные ткани в костях., глаза и другие части тела.
Большинство случаев DEEC являются результатом новых мутаций этого гена и возникают в семьях без истории болезни.. тем не менее, может ли это расстройство передаваться по наследству.
Состояние обычно следует аутосомно-доминантному типу., то есть, если один из родителей болеет, у каждого из их детей есть 50 процент наследования.
Сообщалось также о некоторых случаях аутосомно-рецессивного наследования у детей с DEEC..
Симптомы врожденной спондилоэпифизарной дисплазии
Признаки DEEC могут варьироваться от ребенка к ребенку., но может включать:
- костные деформации, в том числе низкорослые, короткая туловище, длинные конечности относительно их тела, деформации бедра, как палочка для бедра, и дряблая шея
- Заболевания спины и позвоночника, как сколиоз, Цифосис, О цифоз.
- скелетно-мышечная боль, включая остеоартрит, боль в бедре и тазобедренном суставе суставы.
- Проблемы с мышцами и суставами, такие как отсутствие мышечного тонуса и боли в суставах.
- деформации стопы, включая плоскостопие и ноги в.
- проблемы с подвижностью, например утиный марш, тугоподвижность суставов, рыхлые связки и снижение способности ходить.
- Черепно-лицевые деформации, например, расщелина неба, плоское лицо и глазной гипертелоризм.
- Проблемы со слухом и зрением, в том числе снижение зрения, нарушение слуха и глухота.
- Проблемы с пищеварением и желудком, такие как паховая грыжа и выступающий живот.
- Проблемы с дыханием, связанные с аномальным развитием грудной клетки, такие как проблемы с дыханием и апноэ во сне.
Диагностика врожденной спондилоэпифизарной дисплазии (DEEC)
Диагностическая оценка обычно начинается с полной истории болезни и медицинского осмотра вашего ребенка..
- рентген, создавать изображения костей.
- Ваш врач проведет медицинский осмотр и заполнит историю болезни. (ИМР), в котором используется комбинация больших магнитов, радиочастоты и компьютер для получения подробных изображений органов и структур внутри тела.
- генетические тесты, в котором образец слюны или крови вашего ребенка используется для идентификации ДНК вашего ребенка.
- артрография тазобедренного сустава, который использует рентгеновские лучи и краситель, введенный в сустав, чтобы показать мягкие ткани (связки, сухожилия, хрящи и мышцы) сустава.
- эос изображения, недавно одобренная FDA технология визуализации, которая создает модели в 3 размеры из двух плоских изображений. В отличие от КТ, Изображения EOS делаются, когда ребенок находится в вертикальном или стоячем положении., что позволяет улучшить диагностику за счет положения опоры.
- Проверка зрения и слуха, которые оценивают уровни функции глаз и слуха.
- Легочные функциональные тесты, которые проверяют, насколько хорошо работают легкие.
- Все эти тесты позволяют клиницистам составить полную картину состояния здоровья вашего ребенка и помочь принять решение о индивидуальном плане ухода..
Лечение врожденной спондилоэпифизарной дисплазии (DEEC)
Лечение DEEC различается, поскольку это состояние влияет на многие системы организма., и случай каждого ребенка индивидуален. Некоторым детям потребуется только тщательное наблюдение.
Другим потребуется нехирургическое или хирургическое лечение для решения конкретных аспектов их состояния..
Как правило, это практика совместного ухода., семейный центр. Команда клинических экспертов – включая хирургов-ортопедов и врачей, детские медсестры, физиотерапевты и эрготерапевты, психологи и другие специалисты – будет сотрудничать с вами в уходе за вашим ребенком.
У многих детей с DEEC также диагностированы различные ортопедические заболевания., включая: сколиоз, тазик вара, аномалии стопы и проблемы с суставами. Во многих случаях, эти условия становятся очевидными или проявляются только в процессе развития ребенка..
В зависимости от потребностей вашего ребенка, могут потребоваться специалисты по позвоночнику, заболевания бедра и голени или стопы, для лечения ортопедических заболеваний и заболеваний опорно-двигательного аппарата.
Детей с другими последствиями DEEC будут обследовать и лечить специалисты-генетики., гастроэнтерология, Общая хирургия, неврология, офтальмология (глаза), Оториноларингология (ухо, нос и горло) и пластическая хирургия.
- Поддержки и/или операции при проблемах с позвоночником
- поддерживает и / или операции по поводу заболеваний тазобедренного сустава
- поддерживает и / или операция по поводу нестабильности шеи
- поддерживает и / o хирургия нестабильности колена и аномалий стопы
- Этапные реконструктивные операции при черепно-лицевых патологиях
- Очки при проблемах со зрением.
- Слуховые аппараты при проблемах со слухом; трубки для уменьшения ушных инфекций
- Лекарства или обезболивающие при боли в суставах
- Физиотерапия, чтобы помочь ребенку оставаться подвижным
- Последующий уход за DEEC.
Ваш ребенок с врожденной спондилоэпифизарной дисплазией (DEEC) на протяжении всего развития должен находиться под наблюдением врача-ортопеда, и во взрослом возрасте.
Спондилоэпифизарная дисплазия (SEDT), Col2A1 м.
Ген COL2A1 (COLLAGEN, TYPE II, ALPHA-1) расположен на хромосоме 12 в локусе 12q13.11-q13.2. Содержит 54 экзона.
Мутации в данном гене приводят также к развитию ахондрогенеза II типа; дисплазии Книста; аваскулярному некрозу головки бедренной кости; чешской дисплазии; множественной эпифизарной дисплазии с миопией и глухотой; болезни Легга-Кальве-Пертеса; остеоартриту с мягкой хондродисплазией; отоспондиломегаметафизарной дисплазии; платиспондилической скелетной дисплазии тип Торранса; врождённой спондилоэпифизарной дисплазии тип Намакваланда; врождённой спондилометаэпифизарной дисплазии тип Струдвика; спондилопериферической дисплазии; синдрому Стиклера тип 1, несиндромальному глазному; витреоретнопатии с эпифизарной дисплазией фаланг.
Спондилоэпифизарная дисплазия (SEDT, SEDC, СЭД, OMIM313400) представляют собой гетерогенную группу системных костных заболеваний (остеохондропатий) с преимущественным поражением тел позвонков и эпифизов трубчатых костей с формированием диспропорционально низкого роста за счет укорочения туловища и развитием дегенеративных заболеваний суставов. Формируется бочкообразная грудная клетка с укороченным позвоночным столбом. Руки, имея нормальный размер, кажутся относительно длинными; появляются боли в спине, ногах, нарушается походка, приобретая вид утиной. Известны врождённая и поздняя формы. В зависимости от возраста манифестации и тяжести клинических проявлений, описаны все типы наследования для данного заболевания.
В ортопедической практике хорошо известна врождённая аутосомно-доминантная форма СЭД (SED congenita, OMIM 183900). Заболевание проявляется с двухлетнего возраста, однако рентгенологические признаки могут выявляться уже в младенчестве: наблюдается замедленная оссификация головок бедренных костей, неправильные волнистые контуры позвонков. С возрастом тела позвонков уплощаются, отмечается расширение и разрыхление ростковых зон эпифизов в коленных, голеностопных и лучезапястных суставах. Помимо основных признаков заболевания у пациентов наблюдается плоское лицо, иногда расщелина нёба. У половины больных возникает миопия, отслоение сетчатки; рост взрослых пациентов не превышает 128 см. Данная форма СЭД представляет собой один из множества аллельных вариантов нарушений в гене коллагена второго типа (COL2A1). Ген COL2A1 состоит из 54-х экзонов, кодирует alpha1-цепь коллагена II типа, входящего в состав внеклеточного матрикса хряща и стекловидного тела глаз. Помимо врождённой СЭД, мутации в данном гене приводят к развитию целого ряда доминантных остеохондропатий разной тяжести: ахондрогенез II типа (Лангера - Салдино), гипохондрогенез, дисплазии Книста, синдром Стиклера I типа, спондилоэпиметафизарная дисплазия Страдвика, спондилопериферическая дисплазия.
Поздняя Х-сцеплённая рецессивная СЭД (X-linked spondyloepiphyseal dysplasia tarda, SEDL, OMIM 313400) -- редкая остеохондропатия, встречающаяся с частотой 1 на 150-200 тысяч человек. От врождённой формы СЭД данное заболевание отличается более мягким клиническим течением: не выявляется при рождении, до 5-летнего возраста физическое развитие не отличается от сверстников. К 10 годам становится заметным отставание в росте, появляются боли в спине, формируется кифосколиоз в грудном отделе позвоночника, характерное телосложение. У взрослых пациентов рост редко превышает 150 см. Рано развивающиеся артроз и остеохондроз нередко сопровождаются болями и приводят к ограничению подвижности суставов. При Х-сцеплённой СЭД не выявляются нарушения слуха, зрения, интеллекта. Характерным рентгенологическим признаком является распространенная платиспондилия с особой формой тел позвонков в грудном отделе позвоночника в виде возвышающейся центральной части верхней поверхности тела и скошенными участками по периметру, в целом, придающая позвонку вид горба, который формируется после 10-летнего возраста. У всех больных значительно снижается высота тел позвонков грудного и поясничного отделов, в большей степени их передних, вентральных отделов.
Преобладающее большинство, если не всегда, Х-сцеплённой СЭД обусловлены мутациями в гене SEDL (или TRAPPC2, Tracking Protein Particle Complex, subunit 2), который состоит из шести экзонов, кодирует белок Sedlin, выполняющий регуляторную функцию в различных белок-белковых взаимодействиях, в том числе в процессе транспорта от эндоплазматического ретикулума к аппарату Гольджи. Установлено, что ген SEDL избегает инактивации на хромосоме X. Это предполагает невозможность проявления фенотипа Х-сцепленной СЭД у гетерозиготных носительниц заболевания в результате эффекта гаплонедостаточности.
Описаны некоторые сходные со спондилоэпифизарными дисплазиями заболевания, например, синдром Моркио (мукополисахаридоз IV типа - гены GALNS и GLB1) и множественная эпифизарная дисплазия (гены COMP, COL9A1, COL9A2, COL9A3 и MATN3).
Ген COL2A1 кодирует коллаген II типа класса альфа-1. Этот белок является одним из основных компонентов межклеточного вещества соединительной ткани (костной ткани, хрящевой, сухожилий) и стекловидного тела глаз. Повреждения в гене COL2A1 приводят к экспрессии дефектного коллагена и нарушению структуры соответствующих тканей.
Заболевание проявляется с двухлетнего возраста, однако рентгенологические признаки могут выявляться уже в младенчестве: наблюдается замедленная оссификация головок бедренных костей, неправильные волнистые контуры позвонков. С возрастом тела позвонков уплощаются, отмечается расширение и разрыхление ростковых зон эпифизов в коленных, голеностопных и лучезапястных суставах. Помимо основных признаков заболевания у пациентов наблюдается плоское лицо, иногда расщелина неба, в 50% случаев – миопия, отслоение сетчатки, рост взрослых пациентов не превышает 128 см.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование: Атлас-справочник-3-е изд., - Авторская академия, - 2007, - 448 с.
- Федотов В.П., Близнец Е.А., Кочанова Т.И., Коробов А.В., Плотко И.С., Поляков А.В. , Молекулярно-генетическая диагностика поздней X-сцепленной рецессивной спондилоэпифизарной дисплазии, обусловленной дефектом в гене SEDL // Медицинская генетика, 2009, т.8, N4, с.311-314.
- Fedotov, E. A. Bliznetz, I. S. Plotko, A. V. Korobov, A. V. Polyakov., Novel mutation in SEDL gene in three sibling of Russian family with X-linked spondyloepiphyseal dysplasia tarda // Europ Journal of Human Genetic/ ESHG, May 23-26, 2009, Vienna, Austria, P02.166.
- Murray, L. W., Bautista, J., James, P. L., Rimoin, D. L. Type II collagen defects in the chondrodysplasias. I. Spondyloepiphyseal dysplasias. Am. J. Hum. Genet. 45: 5-15, 1989.
- OMIM
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование: Атлас-справочник-3-е изд., - Авторская академия, - 2007, - 448 с.
- Федотов В.П., Близнец Е.А., Кочанова Т.И., Коробов А.В., Плотко И.С., Поляков А.В. , Молекулярно-генетическая диагностика поздней X-сцепленной рецессивной спондилоэпифизарной дисплазии, обусловленной дефектом в гене SEDL // Медицинская генетика, 2009, т.8, N4, с.311-314.
- Fedotov, E. A. Bliznetz, I. S. Plotko, A. V. Korobov, A. V. Polyakov., Novel mutation in SEDL gene in three sibling of Russian family with X-linked spondyloepiphyseal dysplasia tarda // Europ Journal of Human Genetic/ ESHG, May 23-26, 2009, Vienna, Austria, P02.166.
- Murray, L. W., Bautista, J., James, P. L., Rimoin, D. L. Type II collagen defects in the chondrodysplasias. I. Spondyloepiphyseal dysplasias. Am. J. Hum. Genet. 45: 5-15, 1989.
- OMIM
- Характерная клиническая картина.
- Наличие в семье или у ближайших родственников этого заболевания.
Литература
- Кеннет Л. Джонс «Наследственные синдромы по Девиду Смиту» Атлас-справочник. Москва, Практика, 2011.
- Козлова С.И., Демикова Н.С. Наследственные синдромы и медико-генетическое консультирование: Атлас-справочник-3-е изд., - Авторская академия, - 2007, - 448 с.
- Федотов В.П., Близнец Е.А., Кочанова Т.И., Коробов А.В., Плотко И.С., Поляков А.В. , Молекулярно-генетическая диагностика поздней X-сцепленной рецессивной спондилоэпифизарной дисплазии, обусловленной дефектом в гене SEDL // Медицинская генетика, 2009, т.8, N4, с.311-314.
- Fedotov, E. A. Bliznetz, I. S. Plotko, A. V. Korobov, A. V. Polyakov., Novel mutation in SEDL gene in three sibling of Russian family with X-linked spondyloepiphyseal dysplasia tarda // Europ Journal of Human Genetic/ ESHG, May 23-26, 2009, Vienna, Austria, P02.166.
- Murray, L. W., Bautista, J., James, P. L., Rimoin, D. L. Type II collagen defects in the chondrodysplasias. I. Spondyloepiphyseal dysplasias. Am. J. Hum. Genet. 45: 5-15, 1989.
- OMIM
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Глухота с врожденной спондило-эпифизарной дисплазией
Глухота с врожденной спондило-эпифизарной дисплазией
Врожденная спондило-эпифизарная дисплазия, описанная впервые Spranger и Wiedemann, представляет собой дисплазию, распознаваемую при рождении. Заболевание характеризуется укорочением туловища и проксимальных отделов конечностей, а также задержанным окостенением и различными скелетными аномалиями. Другими частыми симптомами являются резкая близорукость п отслойка сетчатки, расщепление неба и нейросенсорная глухота. Вероятно, наблюдения таких заболеваний были опубликованы ранее Uhlig, а также Braun и Meythaler. Врожденная спондило-эпифизарная дисплазия составляет 10% среди случаев карликовости (исключая рахит, несовершенное костеобразоваиие и артрогрипоз), направленных в детскую ортопедическую клинику (Bailey).
Клинические данные. Рост при рождении достигает 40—48 см. Постоянно наблюдаются лобные бугры, легкий гипертслоризм, слегка монголоидный косой разрез глаз, седловидный нос и короткая шея. Рост взрослых больных достигает 85—125 см (Spranger, Langer). В периоде новорожденности может наступить смерть в связи с легочной недостаточностью (Holthusen).
Костно-мышeчная система. Уже при рождении видно легкое укорочение конечностей. Отмечаются также снижение мышечного тонуса и задержка развития моторики, что вместе с резко выраженной парусной деформацией бедер приводит к переваливающейся (утиной) походке. Больные дети начинают ходить около 30 мес или позже. В детстве наблюдается затрудненное глотание жидкости (Michaelis et al.). Выражен рано развивающийся поясничный лордоз. Грудная клетка бочковидной формы с кнлевидной грудью. Постоянными чертами являются варусная и вальгусная деформация коленей и сгибательные контрактуры локтей, бедер, коленей (Bailey, Michaelis et al.). Иногда наблюдаются конская стопа, приведение плюсны и вывих бедер и коленей, а также рекурвация коленей и латеральное смещение надколенника.
У некоторых больных отмечается чрезмерное разгибание н суставах пальцев. В детстве или подростковом возрасте может развиться кифосколиоз. Описаны грыжи (Bach et al.).
Орган зрения. Почти в 60% случаев в подростковом возрасте становится актуальной проблема зрения; отмечается близорукость от 10 до 20 диоптрий, первичные и/или вторичные катаракты, буфтальм И вторичная глаукома (Kozlowski et al., Fraser et al., Rupprecht, Spranger, Langer).
Полость рта. Почти в 40% наблюдений отмечается расщепление неба (Spranger, Wiedemann, Fraser et al., Bach et al., Spranger, Langer, Holthusen, Michaelis et al.).
Нервная система. Примерно у 10% больных обнаружена умственная отсталость (Fraser et al., Bach et al., Michaelis et al.).
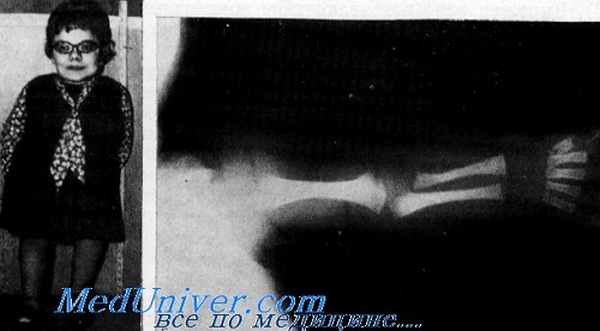
Орган слуха. Умеренно тяжелая нейросенсорпая глухота (30— 60 дБ), выраженная резче на высоких тонах, была описана многими авторами (Fraser et al., Michaelis et al.). Глухота не является постоянной чертой болезни. Согласно нашим наблюдениям, она встречается примерно у 30% больных.
Вестибулярная система. Результаты исследования не опубликованы.
Лабораторные данные. Рентгенологическая патология определяется при рождении. Принципиальные изменения встречаются в тазу и позвонках. Отмечается выраженная задержка окостенения костей таза, эпифизов коленей, таранной и пяточной костей. Крылья подвздошной кости короткие с уплощенными боковыми краями и гризонтально расположенной крышей вертлужной впадины. Крестцово-седалищная вырезка не сужена. Зубовидный отросток гипопластичен. Тела грудных позвонков уплощены сзади, что придает им грушевидную форму (Spranger, Langer).
У детей старшего возраста наблюдается задержка окостенения головки бедра и резко выраженная варусная деформация бедра. Бедренная кость слегка укорочена.
Наследственность. Наследование отчетливо аутосомно-доминантное. Большая часть случаев обусловлена новыми мутациями. Демонстративен эффекта возраста отца (Spranger, Wiedemann, Fraser et al., Michaelis et al.).
Диагноз. Необходимо исключить синдром Моркио, который характеризуется отставанием в росте и скелетными деформациями, становящимися очевидными на 2-м году жизни, облаковидным помутнением роговицы, кератосульфатурией и аутосомно-рецессивным наследованием.
Лечение. Терапия заключается в ранней коррекции конской стопы и хирургическом закрытии расщепленного неба. Чрезвычайно важно направление к офтальмологу для оценки и коррекции миопии, а также для лечения отслойки сетчатки.
Прогноз. Заболевание не ведет к укорочению жизни. С возрастом становится все более затрудненной ходьба и все более выраженный сколиоз. У многих больных наблюдается резкая близорукость и/или отслойка сетчатки.
Выводы. Характеристика этого заболевания включает: 1) аутосомно-доминантное наследование; 2) укорочение туловища и проксимальных отделов конечностей; 3) различные аномалии скелета; 4) резкую близорукость и со временем отслойку сетчатки; 5) расщепление неба примерно у 40% больных; 6) нейросенсорную глухоту у 30% больных.
Дисплазия Книста, ген Col2A1 м
Дифференциальная диагностика:
Спондилоэпифизарная дисплазия, мукополисахаридоз IV типа. Результат исследования:
• Мутация не выявлена.
• Мутация выявлена в гетерозиготном состоянии.
Описание
Метод определения Секвенирование. Выдаётся заключение врача-генетика.
Исследуемый материал Венозная кровь (ЭДТА).
Исследование мутаций в гене Col2A1.
Тип наследования.
Аутосомно-доминантный.
Гены, ответственные за развитие заболевания.
COL2A1 (COLLAGEN, TYPE II, ALPHA-1).
Расположен на длинном плече хромосомы 12 (12q13.11-q13.2) и содержит 54 экзона. Ген кодирует коллаген II типа класса альфа-1 (COL2A1). Этот белок является одним из основных компонентов межклеточного вещества соединительной ткани (костной ткани, хрящевой, сухожилий).
Мутации в данном гене приводят также к развитию ахондрогенеза 2 типа или гипохондрогенезу, аваскулярному некрозу головки бедренной кости, чешской дисплазии, множественной эпифизарной дисплазии с миопией и глухотой, болезни Легга-Кальве-Пертеса, остеоартриту с мягкой хондродисплазией, отоспондиломегаметафизарной дисплазии, платиспондилической скелетной дисплазии тип Торранса, врождённой спондилоэпифизарной дисплазии, врождённой спондилоэпифизарной дисплазии тип Намакваланда, врождённой спондилометаэпифизарной дисплазии тип Струдвика, спондилопериферической дисплазии, синдрому Стиклера, витреоретинопатии с эпифизарной дисплазией фаланг.
Определение заболевания.
Наследственное заболевание, характеризующееся карликовостью, аномальным строением скелета и специфическим лицом с седловидным носом и пучеглазием.
Патогенез и клиническая картина.
Повреждения в гене COL2A1 приводят к экспрессии дефектного коллагена и нарушению структуры соответствующих тканей. Заболевание проявляется с рождения и характеризуется генерализованным нарушением эпи- и метафизальной оссификации. Туловище при этом укорочено, отмечается прогрессирующий кифоз грудного отдела позвоночного столба и резкий лордоз поясничного отдела позвоночного столба. Типичны короткая и широкая грудная клетка с вдавленной грудиной, короткие конечности с утолщением в области суставов, кости таза широкие. Движения в суставах ограничены. В 50% случаев отмечается миопия, косолапость, проводящая и нейросенсорная глухота; нередки расщелина нёба, паховые и пупочные грыжи. Речевое и моторное развитие замедлены, дети поздно начинают ходить и испытывают затруднения при ходьбе; интеллект обычно не страдает. При рентгенологическом исследовании находят уплощение тел позвонков, укороченные длинные трубчатые кости с короткими уплощенными эпифизами, расширенными, неравномерно разреженными метафизами, укороченные и расширенные кости таза с задержкой окостенения головок бедренных костей, расширение и разряжение костей кисти. Срок жизни больных укорочен.
Частота встречаемости:
Не установлена.
Перечень исследуемых мутаций может быть предоставлен по запросу.
Список литературы
• Козлова С. И., Демикова Н. С. Наследственные синдромы и медико-генетическое консультирование. – М.: КМК, 2007 – 448 с.
• Winterpacht, A., Hilbert, M., Schwarze U., Mundlos, S., Spranger, J., Zabel, R. U. Kniest and Stickler dysplasia phenotypes caused by collagen type II gene (COL2A1) defect. Nature Genet. 3: 323-326, 1993.
• OMIM.
Подготовка
Специальной подготовки к исследованию не требуется. Обязательны к заполнению:
• анкета генетического исследования *.
•.
• информированное согласие.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Спондилоэпифизарная дисплазия (SEDT)
(СЭД) представляют собой гетерогенную группу системных костных заболеваний (остеохондропатий) с преимущественным поражением тел позвонков и эпифизов трубчатых костей с формированием диспропорционально низкого роста за счет укорочения туловища и развитием дегенеративных заболеваний суставов. У больных формируется бочкообразная грудная клетка с укорочением позвоночного столба, при этом руки, имея нормальный размер, кажутся относительно длинными, появляются боли в спине, ногах, нарушается походка, приобретая вид «утиной». Известны врожденная и поздняя формы СЭД в зависимости от возраста манифестации и тяжести клинических проявлений, описаны все типы наследования для данного заболевания.
В ортопедической практике хорошо известна врожденная аутосомно-доминантная форма СЭД (SED congenita, OMIM 183900). Заболевание проявляется с двухлетнего возраста, однако рентгенологические признаки могут выявляться уже в младенчестве: наблюдается замедленная оссификация головок бедренных костей, неправильные волнистые контуры позвонков. С возрастом тела позвонков уплощаются, отмечается расширение и разрыхление ростковых зон эпифизов в коленных, голеностопных и лучезапястных суставах. Помимо основных признаков заболевания у пациентов наблюдается плоское лицо, иногда расщелина неба, в 50% случаев – миопия, отслоение сетчатки, рост взрослых пациентов не превышает 128 см.
Данная форма СЭД представляет собой один из множества аллельных вариантов нарушений в генеколлагена второго типа COL2A1. Ген COL2A1 состоит из 54-х экзонов, кодирует α1-цепь коллагена II типа, входящего в состав внеклеточного матрикса хряща и стекловидного тела глаз. Помимо врожденной СЭД, мутации в данном гене приводят к развитию целого ряда доминантных остеохондропатий разной тяжести:ахондрогенез II типа (Лангера-Салдино), гипохондрогенез, дисплазии Книста, синдром Стиклера I типа, спондилоэпиметафизарная дисплазия Страдвика, спондилопериферическая дисплазия.
Поздняя Х-сцепленная рецессивная СЭД (X-linked spondyloepiphyseal dysplasia tarda, SEDL, OMIM 313400) – редкая остеохондропатия, встречающаяся с частотой 1 на 150-200 тысяч человек. От врожденной формы СЭД данное заболевание отличается более мягким клиническим течением: не выявляется при рождении, до 5 летнего возраста физическое развитие не отличается от сверстников. К 10 годам становится заметным отставание в росте, появляются боли в спине, формируется кифосколиоз в грудном отделе позвоночника, характерное телосложение. У взрослых пациентов рост редко превышает 150 см. Рано развивающиеся артроз и остеохондроз нередко сопровождаются болями и приводят к ограничению подвижности суставов. При Х-сцепленной СЭД не выявляются нарушения слуха, зрения, интеллекта. Характерным рентгенологическим признаком является распространенная платиспондилия с особой формой тел позвонков в грудном отделе позвоночника в виде возвышающейся центральной части верхней поверхности тела и скошенными участками по периметру, в целом, придающая позвонку вид «горба», который формируется после 10-летнего возраста. У всех больных значительно снижается высота тел позвонков грудного и поясничного отделов, в большей степени их передних, вентральных отделов.
Преобладающее большинство, если не все случаи Х-сцепленной СЭД обусловлены мутациями в гене SEDL. SEDL (или TRAPPC2, Tracking Protein Particle Complex, subunit 2) состоит из шести экзонов, содирует белок Sedlin, выполняющий регуляторную функцию в различных белок-белковых взаимодействиях, в том числе в процессе транспорта от эндоплазматического ретикулума к аппарату Гольджи. Установлено, что ген SEDL избегает инактивации на хромосоме X. Это предполагает невозможность проявления фенотипа Х-сцепленной СЭД у гетерозиготных носительниц заболевания в результате эффекта гаплонедостаточности.
Описаны некоторые сходные со спондилоэпифизарными дисплазиями заболевания, например, синдром Моркио (мукополисахаридоз IV типа) (гены GALNS и GLB1) и множественная эпифизарная дисплазия (гены COMP, COL9A1, COL9A2, COL9A3 и MATN3).
В Центре Молекулярной Генетики в целях диагностики доминантной и Х-сцепленной СЭД возможен анализ мутаций методом прямого секвенирования кодирующей последовательности генов COL2A1 и TRAPPC2.
При проведении пренатальной (дородовой) ДНК-диагностики в отношении конкретного заболевания, имеет смысл на уже имеющемся плодном материале провести диагностику частых анеуплоидий (синдромы Дауна, Эдвардса, Шерешевского-Тернера и др), пункт 4.54.1. Актуальность данного исследования обусловлена высокой суммарной частотой анеуплоидий - около 1 на 300 новорожденных, и отсутствием необходимости повторного забора плодного материала.
Читайте также: