Хромосомные аномалии при хроническом лимфолейкозе - прогноз
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Хронический лимфоцитарный лейкоз (ХЛЛ) характеризуется прогрессирующим накоплением фенотипически зрелых злокачественных В-лимфоцитов. Первичные места заболевания включают периферическую кровь, костный мозг, селезенку и лимфоузлы. Симптоматика заболевания может отсутствовать или проявляться лимфаденопатией, спленомегалией, гепатомегалией, утомляемостью, лихорадкой, ночными приливами, непреднамеренной потерей веса и чувством быстрого насыщения. Диагноз ставится на основании результатов проточной цитометрии и иммунофенотипирования периферической крови. Лечение откладывается до появления симптомов и, как правило, включает проведение химиотерапии и иммунотерапии. Тем не менее методы лечения прогрессируют, и схемы терапии первой линии могут включать в себя целевые агенты, такие как ингибиторы тирозинкиназы Брутона (Btk) и регуляторы апоптоза Bcl-2, с химиотерапией или без нее.
Хотя причина ХЛЛ неизвестна, в некоторых случаях отмечается семейный анамнез. ХЛЛ редко встречается в Японии и Китае и, судя по данным, частота заболеваемости не увеличивается у этнических японцев, живущих в Соединенных Штатах Америки, что также подтверждает важность генетических факторов. Наиболее распространен ХЛЛ у евреев из Восточной Европы.
Патофизиология ХЛЛ
Во время хронического лимфолейкоза CD5-положительные B-клетки подвергаются злокачественной трансформации. В-клетки непрерывно активируются путем приобретения мутаций, что приводит к моноклональному В-клеточному лимфоцитозу (MBL). Дальнейшее накопление генетических аномалий и последующая онкогенная трансформация моноклональных В-клеток приводит к развитию ХЛЛ. Лимфоциты сначала накапливаются в костном мозге, а затем распространяются в лимфатические узлы и другие лимфоидные ткани, в конечном итоге вызывая спленомегалию, гепатомегалию, а также системные проявления, такие как утомляемость, лихорадка, ночная потливость, чувство быстрого насыщения и необъяснимое снижение массы тела.
По мере прогрессирования ХЛЛ аномальный гемопоэз приводит к анемии, нейтропении, тромбоцитопении и снижению выработки иммуноглобулина. Гипогаммаглобулинемия может развиться у двух третей пациентов, повышая риск развития инфекционных осложнений. Пациенты имеют повышенную восприимчивость к аутоиммунным гемолитическим анемиям (с прямой положительной антиглобулиновой реакцией) и аутоиммунной тромбоцитопении.
ХЛЛ может прогрессировать в В-клеточный пролимфоцитарный лейкоз, а также может трансформироваться в высоко дифференцированную неходжкинскую лимфому Неходжкинские лимфомы Неходжкинские лимфомы представляют собой гетерогенную группу заболеваний, развивающихся по причине злокачественной моноклональной пролиферации лимфоидных клеток в лимфоретикулярной ткани в лимфоузлах. Прочитайте дополнительные сведенияСимптомы и признаки ХЛЛ
На ранних стадиях часто протекает бессимптомно с незаметным появлением неспецифических симптомов (например, усталости, слабости, анорексии, потери веса, лихорадки и/или ночной потливости), которые могут побуждать к проведению диагностики. У более половины пациентов наблюдается лимфаденопатия. Она может быть локализованной (чаще всего поражены шейные и надключичные узлы) или генерализованной. Спленомегалия и гепатомегалия встречаются реже, чем лимфаденопатия. Поражение кожи (лейкоз кожи) встречается редко.
Хромосомные аномалии при хроническом лимфолейкозе - прогноз
Хромосомные аномалии при хроническом лимфолейкозе - прогноз
По классификации ВОЗ, хронический лимфолейкоз (ХЛЛ) относится к опухолям из зрелых В-клеток. Клиническое течение и длительность болезни сильно варьируют: на одном полюсе находятся формы болезни с длительным, относительно спокойным течением, не требующим лечения в течение многих лет, на другом — агрессивные варианты лейкоза, нуждающиеся в интенсивном лечении сразу после постановки диагноза. Нередко встречаются «промежуточные» формы.
Прогнозирование течения хронического лимфолейкоза в момент его выявления может вызывать определенные затруднения. Существует две основные клинические системы определения стадии болезни и прогнозирования ее течения, однако ни одна из этих систем не позволяет определить прогноз в каждом конкретном случае.
Постоянно ведется поиск новых, более надежных прогностических показателей, которые оказались бы полезными в решении практических вопросов. В частности, важно решить, нужна ли интенсивная терапия больным с малой массой опухоли, но с лабораторными признаками плохого прогноза? Какой конкретный протокол лечения предпочтителен конкретному больному при прогрессии болезни?
Известно, что хронический лимфолейкоз неоднороден не только в клиническом отношении; имеются существенные биологические и молекулярно-генетические отличия между популяциями лейкозных клеток у разных больных. В результате клинико-лабораторных сопоставлений удалось провести определенные параллели между длительностью течения болезни (прогнозом) и отдельными особенностями лейкозных клеток. Полученные данные уже используются в клинической практике, в частности с прогностическими целями.
Как и при других гемобластозах, при хроническом лимфолейкозе важное клиническое значение приобрели особенности кариотипа лейкозных клеток. Еще несколько лет назад считали, что неоплазированные клетки при этом заболевании имеют нормальный кариотип. Позже удалось выяснить, что без специальных методических приемов хромосомному анализу подвергались не лейкозные, а нормальные лимфоциты больных.
Изучение особенностей кариотипа хронического лимфолейкоза стало возможным только после введения веществ, стимулирующих митотическую активность В-клеток. Обычно в каждом случае применяется несколько стимуляторов, т. е. производится посадка нескольких культур из клеток крови или других пораженных органов с использованием различных стимуляторов. В отдельных культурах удается выявить клоны аномальных клеток. Иногда в разных клеточных культурах, полученных от одного больного и подвергнутых действию разных стимуляторов, обнаруживают клоны клеток с разными нарушениями кариотипа.
Наиболее характерны следующие нарушения: дополнительная хромосома 12, делеции длинного плеча хромосом 6 и 13, различные перестройки длинного плеча хромосомы 11. В ряде случаев наблюдаются характерные для В-клеточных новообразований транслокации с участием района 14q32 (область локализации IgH), среди которых преобладают t(11;14)(q13;q32), t(2;14)(p13;q32) и t(14;19)(q32;p13).
На протяжении нескольких лет для изучения хромосомных изменений при хроническом лимфолейкозе с успехом применяется FISH, нередко — интерфазный анализ, который позволяет выявлять нарушения кариотипа клеток вне митоза, на стадии интерфазы. При характерном для хронического лимфолейкоза низком митотическом индексе лейкозных клеток использование интерфазной FISH резко расширяет возможности анализа. С помощью этого подхода нарушения кариотипа обнаруживают у 80 % больных, тогда как обычное цитогенетическое исследование выявляет их только у 40—50 % больных.
Некоторые авторы приводят другие цифры. Так, в работе D. G. Oscier и соавт. обычный хромосомный анализ позволил обнаружить клоны аномальных клеток у 141(69 %) из 206 больных хроническим лимфолейкозом при изучении краткосрочных клеточных культур, стимулированных ТПА. Использование FISH практически мало изменило эти результаты, но было одно исключение — нарушения хромосомы 13. При стандартном цитогенетическом исследовании эти нарушения удалось выявить у 17 %, а с помощью FISH — у 45% больных. Частота этой аномалии (13q-), по данным других авторов, еще выше.
Так, G. W. Dewald и соавт., обследовав 113 больных хроническим лимфолейкозом, обнаружили делецию одной хромосомы из 13-й пары у 64 % больных, а делецию обоих гомологов — у 28 % обследованных.
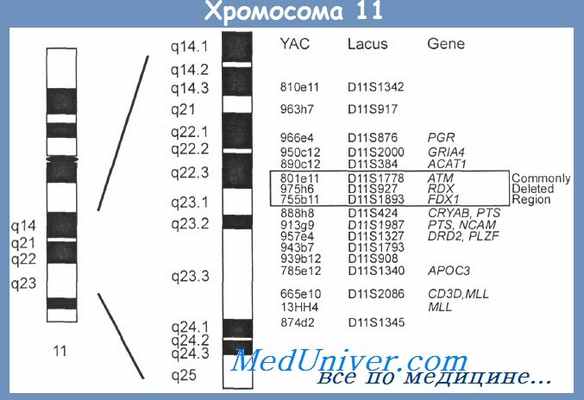
Участок длинного плеча хромосомы 11, который утрачивается при хроническом лимфолейкозе у всех больных с делециями 11q.
Слева — схема хромосомы 11; правее — увеличенный участок длинного плеча хромосомы 11, делеции которого находят у больных хроническим лимфолейкозом с помощью хромосомного анализа; в рамке — молекулярные зонды, позволившие идентифицировать генетические последовательности, которые утрачиваются у всех больных с делециями (commonly deleted regions).
Американские авторы представили частоту отдельных хромосомных перестроек по результатам интерфазной FISH. Так, делеция 13ql4, по их данным, составила 64%, делеция 11q—15%, трисомия 12 - 25 %, делеция 17р — 8 %. Частоты, приводимые в других публикациях, сходны, но не точно такие же. Это может быть обусловлено возрастными и географическими различиями в частоте неслучайных хромосомных аномалий, стадией болезни, на которой производили исследование, а также особенностями В-клеточных стимуляторов и других методических приемов, применявшихся конкретными авторами.
Интересно, что с помощью FISH клоны клеток с делециями хромосомы 13 были выявлены у 50 % обследованных без гематологической патологии, но с так называемым синдромом CLUS (MBCL). CLUS (моноклональный ХЛЛ-фенотип в небольшом количестве клеток крови) наблюдается среди людей старше 40 лет с частотой примерно 3,5 %. Обычно это люди из семей, где есть больные хроническим лимфолейкозом. Таким образом, невозможно без применения FISH сделать заключение о наличии или отсутствии прогностически значимых изменений кариотипа при хроническом лимфолейкозе.
Характерные изменения кариотипа отличаются друг от друга в прогностическом отношении. Так, делеции 17р и 11q, наличие дополнительной хромосомы 12 прогностически неблагоприятны, а присутствие делетированной хромосомы 13 как единственной аномалии считают прогностически благоприятным. По данным D. G. Oscier и соавт., продолжительность жизни (медиана) при хроническом лимфолейкозе с моносомией или делецией длинного плеча хромосомы 13 составила 292 мес, при обнаружении трисо-мии 12 — 122 мес, делеций 11q23 — 117 мес, изменений короткого плеча хромосомы 17 или мутаций гена р53 — всего 47 мес.
В литературе приводятся сведения о клинико-морфологических особенностях хронического лимфолейкоза, ассоциированного с определенными аномалиями кариотипа. В частности, существует точка зрения, что хронический лимфолейкоз с трисомией 12 представляет собою отдельный вариант болезни, при котором часто наблюдаются атипичная морфология лимфоцитов и атипичный иммунофенотип, отсутствие мутации в вариабельном районе гена Н-цепей иммуноглобулина и неблагоприятное течение.
Крайне неблагоприятны в прогностическом отношении делеции длинного плеча хромосомы 11 района 11q22.3-q23.1. Это сравнительно частая аномалия — до 20 % хронического лимфолейкоза.
По вопросу о том, является ли трисомия 12 неблагопрятным прогностическим признаком, существуют разногласия, но все исследователи единодушны, считая делении 11q прогностически неблагоприятными. Больные с этими нарушениями сравнительно молоды, у них отмечается выраженный опухолевый рост периферических, абдоминальных и медиастинальных лимфатических узлов. Медиана выживаемости составляет 64 мес против 209 мес в группе хронического лимфолейкоза без делеций 11q.
Интересные данные сообщили немецкие авторы на международном рабочем совещании по хроническому лимфолейкозу. Они изучали особенности кариотипа клеток крови больных хроническим лимфолейкозом (84 пациента) с помощью FISH. Предварительно проводили совместное культивирование клеток от больных с клетками стабильных линий, экспрессирующих специальные стимуляторы. После культивирования в большинстве проб (87 %) были обнаружены аномальные клоны с нарушениями кариотипа, характерными для хронического лимфолейкоза.
У большинства больных еще до лечения хромосомные нарушения были множественными, т. е. клетки лейкозного клона содержали по 3—4 и более изменений. Частота клонов со сложными перестройками не зависела от стадии болезни. Анализ материала позволил авторам сделать вывод, что клоны клеток, содержащих более 3 аномалий, характерны для быстро прогрессирующего хронического лимфолейкоза. Большинство больных со сложными перестройками были сравнительно молоды. Авторы высказали предположение, что для прогноза хронического лимфолейкоза важно не количество, а качество хромосомных аномалий. Например, перестройки короткого плеча хромосомы 17 однозначно ассоциированы с плохим прогнозом.
Другие авторы сравнили особенности кариотипа в двух группах хронического лимфолейкоза: со стабильным течением (без прогрессии по меньшей мере в течение полугода) и с более агрессивным течением. У больных обеих групп обнаружены как прогностически благоприятные, так и прогностически неблагоприятные аномалии кариотипа, но отмечена следующая тенденция: при более агрессивной форме доля клеток с аномалиями была выше и чаще наблюдались случаи со сложными перестройками.
В последние годы при прогнозировании хронического лимфолейкоза большое значение придается гиперэкспрессии маркера CD38 на поверхности лимфоцитов крови. Получены убедительные данные, что у больных с гиперэкспрессией CD38 гораздо худший прогноз, чем у больных без этого признака. Однако существует и противоположное мнение. Есть также противоречия по вопросу о том, какой процент клеток с гиперэкспрессией CD38 является прогностически неблагоприятным показателем. Большинство исследователей называют 30 %, а некоторые 7 %.
Тем не менее подавляющее большинство специалистов считают, что гиперэкспрессия CD38 — четкий маркер плохого прогноза, обычно сочетающийся с другими плохими прогностическими признаками, в частности, цитоге-нетическими. Кроме того, гиперэкспрессия CD38 нередко коррелирует с так называемым VH-статусом. Нередко повышенная экспрессия CD38 наблюдается на лимфоцитах, которые не претерпели мутации вариабельного участка гена Н-цепей иммуноглобулинов (отрицательный, или «немутированный», VH-статус).
По VH-статусу вся группа хронического лимфолейкоза делится примерно пополам. Различия в продолжительности жизни больных с «немутированным» и «мутированным» VH-статусом очень значительны: медианы составляют 6—8 лет и 24—25 лет. Показано, что «немутированный» VH-статус нередко ассоциирован с такими прогностически неблагоприятными генетическими маркерами, как делеции 11q и 17р, трисомия хромосомы 12 и нарушения р53. Эти нарушения позволяют прогнозировать невысокую эффективность терапии. Сочетание «немутированного» VH-статуса с неблагоприятными изменениями кариотипа (делеции 11q и 17р) ухудшает прогноз: общая выживаемость составляет соответственно 98 и 58 мес.
Обнаружен новый важный прогностический маркер при хроническом лимфолейкозе — белок ZAP70 (zeta-chain associated protein 70). Его выявляют у больных с «немутированным» Vн-статусом. Этот белок — тоже четкий маркер «плохого» прогноза. Вероятно, определение ZAP70 войдет в широкую клиническую практику, поскольку методика его выявления более проста, чем идентификация VH-статуса.
Обследование больших групп больных хроническим лимфолейкозом с одновременным изучением разных прогностических маркеров (хромосомный анализ, FISH, VH-статус, CD38, мутации гена ТР53) позволило увидеть, что данные этих тестов нередко совпадают. Высказано предположение, что за этим совпадением стоят общий биологический смысл, общее происхождение группы хронического лимфолейкоза, при котором наблюдается сочетание отрицательного VH-статуса с одной (или двумя) из таких прогностически неблагоприятных аномалий кариотипа, как 17р- и 11q-. Даже на ранних клинико-гематологических стадиях болезни сочетание этих признаков предсказывает сравнительно невысокую эффективность терапии.
Обобщая данные об особенностях хронического лимфолейкоза в зависимости от отсутствия или наличия мутаций VH, Т. Hamblin приводит следующие данные: в группе с «мутированным» VH-статусом 90 % больных имеют стабильную форму болезни, а в группе с отсутствием мутации доля таких больных всего 15 %. При цитогенетическом исследовании в первой из этих групп у большинства больных обнаружен нормальный кариотип или 13q-; мутации гена ТР53 и аномалии 11q23 практически всегда совпадают с «немутированным» статусом, как и трисомия хромосомы 12; продолжительность жизни при диагнозе на стадии В или С по Binet в 2 раза выше в группе с мутациями. Т. Hamblin высказывает предположение, что это две болезни, а не две формы одного заболевания.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Хромосомные аномалии при хроническом лимфолейкозе - прогноз
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Молекулярно-генетические нарушения при острых лейкозах как основа разработки диагностических тестов (обзор литературы)
Журнал: Лабораторная служба. 2020;9(4): 26‑45
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Острые лейкозы представляют собой гетерогенную группу онкологических заболеваний крови, обусловленных генетическими дефектами и дисбалансом экспрессии генов, отвечающих за пролиферацию и дифференцировку клеток на ранних этапах кроветворения. Основными методами лабораторной диагностики лейкозов являются морфологический, иммуноцитометрический и цитогенетический анализы клеток крови и костного мозга. В задачи современной лабораторной диагностики также входит дифференцировка различных молекулярно-генетических особенностей лейкозных клеток с целью оценки индивидуального прогноза и выбора оптимальной схемы терапии. При этом ряд молекулярно-генетических маркеров рассматривается как перспективные инструменты мониторинга эффективности терапии и оценки минимальной остаточной болезни. В настоящем обзоре рассмотрены отдельные молекулярно-генетические маркеры: транскрипты химерных генов и мРНК эмбриональных генов в качестве потенциальных мишеней разработки, основанных на полимеразной цепной реакции тест-систем с целью диагностики острых лейкозов, в том числе для скрининговых тестов на первичном амбулаторном уровне наряду с рутинным гематологическим анализом.
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Красноярский филиал ФГБУ «Национальный медицинский исследовательский центр гематологии» Минздрава России;
ФГБНУ Федеральный исследовательский центр «Красноярский научный центр Сибирского отделения РАН»
Дата принятия в печать:
Острые лейкозы (ОЛ) — гетерогенная группа клональных заболеваний системы кроветворной ткани, возникающих вследствие мутаций и (или) эпигенетических нарушений в кроветворных клетках, приводящих к блоку дифференцировки, апоптоза и бесконтрольной пролиферации. По морфологическим и иммунофенотипическим критериям атипичных «бластных» клеток ОЛ подразделяют на лимфоидные, миелоидные и (ОЛСФ).
Заболеваемость острыми миелоидными лейкозами (ОМЛ) в России составляет около 3—5 человек на 100 тыс. населения в год, причем среди населения старше 60 лет она резко возрастает и достигает 12—13 человек на 100 тыс. В среднем 5-летняя общая выживаемость пациентов в возрасте до 60 лет, по данным больших кооперативных исследовательских групп, составляет 35—50%, варьируя от 10 до 90% в зависимости от молекулярно-генетических особенностей лейкемии.
Острые лимфобластные лейкозы/острые лимфобластные лимфомы (далее — ОЛЛ) обусловлены онкогенной трансформацией клеток-предшественниц гемопоэза преимущественно лимфоидной направленности дифференцировки. ОЛЛ может встречаться у лиц любого возраста, при этом 60% пациентов с ОЛЛ моложе 20 лет. ОЛЛ является самой распространенной опухолью кроветворной ткани у детей, составляя 30% всех злокачественных опухолей детского возраста.
Классификация ОЛ, предложенная в 1976 г. (FAB), была основана исключительно на морфологических и цитохимических критериях и в связи с бурным накоплением знаний о молекулярно-генетических основах канцерогенеза в 2008 г. была пересмотрена и дополнена генетическими, иммунологическими и эпидемиологическими маркерами. В современную классификацию включены хромосомные транслокации, играющие важную роль в патогенезе заболеваний: BCR-ABL t(9;22)(q34;q11), TEL-AML1 t(12;21)(p33;q22), MLL-AF4 t(4;11)(q21;q23), E2A-PBX1 t(l;19)(q23;p33), MLL-ENL t(11;19)(q23;p33), SIL-TALI, PICALM-AF10 t(10;11)(p33;q14-21); RUNX1-RUNX1T1 t(8;21)(q22;q22.1), CBFB-MYH11 inv(16)(p13.1;q22) и t(16;16)(p13.1;q22), PML-RARA t(15;17) (q24;q21), MLLT3-MLL t(9;11)(p22;q23). В классификации ВОЗ 2016 г. было дополнительно уделено внимание мутациям генов KIT, FLT3-ITD, FLT3-TKD, NPM1, CEBPA, IDH1/IDH2, RUNX1, ASXL1 и TP53 — для которых доказаны важное прогностическое значение при отдельных вариантах ОЛ и возможность создания эффективных таргетных лекарственных препаратов [1, 2].
На сегодняшний день спектр выявленных молекулярно-генетических аномалий при ОЛ очень широк: от точечных однонуклеотидных замен до крупных хромосомных транслокаций. В табл. 1 представлена частота встречаемости отдельных молекулярно-генетических дефектов при различных формах ОЛ у детей и взрослых. Следует отметить, что опухолевые клетки с выявленными отдельными генетическими аномалиями могут появляться в кровотоке и исчезать в процессе клональной эволюции и давления со стороны терапевтических воздействий. Отдельные генетически детерминированные субклоны лейкозных клеток, существуя по отдельности или в сочетании, обусловливают индивидуальное течение заболевания. Поэтому для повышения эффективности диагностики и мониторинга течения ОЛ актуальным является обеспечение доступного и быстрого исследования, охватывающего максимальное количество известных генетических аномалий.
Таблица 1. Частота встречаемости отдельных молекулярно-генетических дефектов при ОЛ у детей и взрослых
Публикации в СМИ
Хронический лимфолейкоз (ХЛЛ) характеризуется резким увеличением количества зрелых лимфоидных клеток в крови, лимфатических узлах, селезёнке, печени. Источник опухоли — клетка-предшественник лимфопоэза.
Генетические аспекты. Заболевание имеет наследственный характер (*151400, соматическая мутация гена BCL1, 11q13.3 или *109543, мутация генов D13S25, DB, 13q14).
Патогенетические особенности • Отсутствие признаков опухолевой прогрессии • Отсутствие клеточного атипизма (за исключением волосатоклеточного лейкоза) • Отсутствие хромосомных аномалий • Склонность к сопутствующим аутоиммунным синдромам.
Стадии. Опухолевая природа ХЛЛ связана с определёнными клиническими проявлениями, имеющими прогностическое значение • Стадия 0 ограничена лимфоцитозом, прогноз в целом положительный (средняя продолжительность жизни — 10–12 лет) • Стадии 1 и 2. Присоединение клинических проявлений. Стадия 1 — лимфаденопатия; стадия 2 — спленомегалия; прогноз хуже (пациенты обычно живут 4–7 лет) • Стадии 3 и 4. Возникновение аутоиммунной патологии — аутоиммунная гемолитическая анемия; аутоиммунная тромбоцитопения — утяжеляет прогноз (продолжительность жизни пациентов составляет менее 18 мес).
Диагностика. Достаточно выявления абсолютного лимфоцитоза, представленного зрелыми клетками в мазке периферической крови. Подтверждающие данные — инфильтрация костного мозга зрелыми лимфоцитами, увеличение селезёнки и лимфаденопатия.
Лечение консервативное — не изменяет продолжительность жизни • В ранних стадиях ХЛЛ (1 и 2), как правило, нет необходимости в химиотерапии • На поздних стадиях ХЛЛ — алкилирующие средства (например, хлорамбуцил) в сочетании со ГК или без них; тотальное облучение организма в низких дозах. При недостаточности или отсутствии эффекта при лечении алкилирующими цитостатиками — флударабин.
Прогноз относительно благоприятный при стадиях 0, 1. Продолжительность жизни в отдельных случаях может достигать 15–20 лет.
Сокращение. ХЛЛ — хронический лимфолейкоз.
МКБ-10 • C91.1 Хронический лимфоцитарный лейкоз.
Код вставки на сайт
Хронический лимфолейкоз
Хронический лимфолейкоз (ХЛЛ) характеризуется резким увеличением количества зрелых лимфоидных клеток в крови, лимфатических узлах, селезёнке, печени. Источник опухоли — клетка-предшественник лимфопоэза.
Генетические аспекты. Заболевание имеет наследственный характер (*151400, соматическая мутация гена BCL1, 11q13.3 или *109543, мутация генов D13S25, DB, 13q14).
Патогенетические особенности • Отсутствие признаков опухолевой прогрессии • Отсутствие клеточного атипизма (за исключением волосатоклеточного лейкоза) • Отсутствие хромосомных аномалий • Склонность к сопутствующим аутоиммунным синдромам.
Стадии. Опухолевая природа ХЛЛ связана с определёнными клиническими проявлениями, имеющими прогностическое значение • Стадия 0 ограничена лимфоцитозом, прогноз в целом положительный (средняя продолжительность жизни — 10–12 лет) • Стадии 1 и 2. Присоединение клинических проявлений. Стадия 1 — лимфаденопатия; стадия 2 — спленомегалия; прогноз хуже (пациенты обычно живут 4–7 лет) • Стадии 3 и 4. Возникновение аутоиммунной патологии — аутоиммунная гемолитическая анемия; аутоиммунная тромбоцитопения — утяжеляет прогноз (продолжительность жизни пациентов составляет менее 18 мес).
Диагностика. Достаточно выявления абсолютного лимфоцитоза, представленного зрелыми клетками в мазке периферической крови. Подтверждающие данные — инфильтрация костного мозга зрелыми лимфоцитами, увеличение селезёнки и лимфаденопатия.
Лечение консервативное — не изменяет продолжительность жизни • В ранних стадиях ХЛЛ (1 и 2), как правило, нет необходимости в химиотерапии • На поздних стадиях ХЛЛ — алкилирующие средства (например, хлорамбуцил) в сочетании со ГК или без них; тотальное облучение организма в низких дозах. При недостаточности или отсутствии эффекта при лечении алкилирующими цитостатиками — флударабин.
Прогноз относительно благоприятный при стадиях 0, 1. Продолжительность жизни в отдельных случаях может достигать 15–20 лет.
Хромосомные аномалии при хроническом лимфолейкозе - прогноз
Лимфолейкоз – это злокачественное заболевание, при котором костный мозг вырабатывает большое количество незрелых, неспособных выполнять свои функции лимфоцитов. Лимфоциты являются разновидностью лейкоцитов и отвечают за иммунитет. Злокачественные лейкоциты не справляются с защитной функцией, при этом подавляют образование нормальных клеток крови и нарушают работу других органов.
Существует много разновидностей этого заболевания. Лимфолейкозы делят в зависимости от скорости развития патологического процесса, степени зрелости лимфоцитов, типа поврежденных лимфоцитов (Т- и В-лимфоциты). Однако чаще всего выделяют 2 основных типа: острый лимфобластный лейкоз и хронический лимфоцитарный лейкоз.
Лечение лейкозов обычно комплексное. На данный момент существует несколько вариантов терапии, и каждый год появляются новые, все более эффективные методы лечения лимфолейкозов. Прогноз при остром лимфолейкозе благоприятный – 95 % пациентов полностью излечиваются. Прогноз при хроническом лимфолейкозе зависит от скорости развития заболевания и сопутствующих патологий, он неуклонно прогрессирует, однако соответствующее лечение часто приводит к ремиссии и значительному улучшению состояния пациента.
Синонимы русские
Острый лимфолейкоз, острый лимфоцитарный лейкоз, острый лимфобластный лейкоз, хронический лимфолейкоз, хронический лимфоцитарный лейкоз.
Синонимы английские
Pediatric acute lymphoblastic leukemia, childhood acute lymphoblastic leukemia, acute lymphocytic leukemia, ALL, chronic lymphocytic leukemia, CLL.
Симптомы
Острый лимфобластный лейкоз развивается достаточно быстро – обычно в течение нескольких недель. Симптомами его являются:
- лихорадка,
- слабость, недомогание,
- кровоточивость десен, частые носовые кровотечения,
- боли в животе,
- боли в костях,
- головные боли,
- увеличение лимфатических узлов шеи, подмышечной и паховой области,
- бледность.
Хронический лимфоцитарный лейкоз обычно никак не проявляется на начальной стадии. Он развивается годами, и постепенно возникают следующие симптомы:
- частые инфекционные заболевания,
- повышенная потливость, особенно ночью,
- кровоизлияния в кожу и слизистые,
- тяжесть в животе,
- слабость, недомогание,
- беспричинная потеря веса,
- бледность,
- одышка.
Общая информация о заболевании
Лимфоциты – это разновидность лейкоцитов. Как и все остальные клетки крови, лимфоциты образуются из единой стволовой клетки, которая находится в костном мозге и дает начало лимфоидной и миелоидной стволовым клеткам. От лимфоидной стволовой клетки происходят лимфоциты, от миелоидной – другие виды лейкоцитов, эритроциты и тромбоциты.
Для того чтобы из лимфоидной стволовой клетки развились зрелые Т- и В-лимфоциты, она должна пройти через ряд последовательных делений. Сначала из лимфоидной стволовой клетки образуются лимфобласты, которые затем дают начало 2 типам клеток – предшественникам Т-лимфоцитов и предшественникам В-лимфоцитов. В процессе деления клетки становятся все более зрелыми и специализированными. Последние этапы созревания лимфоцитов проходят уже не в костном мозге, а в лимфоидных органах: тимусе, лимфатических узлах и селезенке. В результате формируются зрелые Т- и В-лимфоциты.
Лимфоциты, по сути, – клетки иммунитета. Это значит, что они участвуют в распознавании и уничтожении чужеродных тел (вирусов, бактерий) или патологически измененных тканей собственного организма (например, клеток опухоли). Т- и В- лимфоциты делают это по-разному. В-лимфоциты борются с чужеродными клетками (антигенами) с помощью иммуноглобулинов – белков, которые связывают антигены и запускают процесс их разрушения специальными белками. Т-лимфоциты распознают антигены, разрушают их самостоятельно или при взаимодействии с другими клетками крови, активируют выработку иммуноглобулинов В-лимфоцитами.
При лимфолейкозе нарушается формирование лимфоцитов. Появляется большое количество незрелых клеток, которые не способны выполнять свою работу. Это приводит к серьезным сбоям иммунитета. Человек становится более подвержен таким инфекционным заболеваниям, как туберкулез, кандидоз. Могут возникать серьезные осложнения от плановых прививок. Часто появляются так называемые аутоиммунные реакции – то есть иммунитет борется с нормальными клетками организма, например эритроцитами. В результате возникает анемия.
Злокачественные лимфоциты проникают в лимфатические узлы и селезенку, вызывая их увеличение, могут повреждать печень, легкие, головной мозг, кости.
При остром лимфолейкозе в костном мозге преобладают лимфобласты. Они очень быстро делятся, вытесняют из костного мозга и крови другие клетки, активно заселяют лимфатические узлы, селезенку. Чаще всего встречаются острые В-клеточные лейкозы, при которых образуется большое количество незрелых В-лимфоцитов. Острый В-клеточный лейкоз – самый распространенный вид лейкозов среди детей.
При хроническом лимфолейкозе в крови находят более зрелые формы лимфоцитов, которые способны некоторое время выполнять свои функции. Этот тип лейкоза характерен для людей старше 50-55 лет.
Кто в группе риска?
При остром лимфолейкозе в группе риска:
- люди, подвергавшиеся радиоактивному облучению,
- люди, подвергавшиеся химиотерапии или лучевой терапии в связи с другой формой рака,
- больные синдромом Дауна и другими генетическими нарушениями,
- люди, у братьев или сестер которых был диагностирован острый лимфолейкоз.
При хроническом лимфолейкозе в группе риска:
- представители европеоидной расы,
- люди старше 60 лет,
- люди, у родственников которых наблюдались случаи лейкоза.
Диагностика
- с лейкоцитарной формулой. Это исследование дает врачу информацию о количестве, соотношении и степени зрелости элементов крови. . При остром лимфолейкозе лейкоциты могут быть повышены, в норме или понижены. Лейкоцитарную формулу (соотношение отдельных видов лейкоцитов) определяют по мазку крови. Для этого тонкий мазок наносится на предметное стекло, окрашивается специальными красителями, а затем исследуется под микроскопом. Таким образом врач может не только определить соотношение лейкоцитов, но и выявить патологические, незрелые клетки, которые внешне отличаются от нормальных.
- Тромбоциты могут быть снижены.
- Эритроциты и гемоглобин. Тоже могут быть снижены.
- Проточная цитометрия, иммунофенотипирование. При сложных вариантах лимфолейкоза эти методики позволяют точно определить тип злокачественных клеток. При проточной цитометрии измеряют параметры клетки с помощью лазерного луча. Иммунофенотипирование заключается в обнаружении специфических для разных типов клеток белков на поверхности мембраны лимфоцита.
- Цитогенетические исследования. Обычно берут венозную кровь. Клетки крови фиксируют и окрашивают, после чего специалист под микроскопом исследует их кариотип – полный набор хромосом, который идентичен в любой клетке человека. Используется для выявления хромосомных нарушений, характерных для лимфолейкоза. При лимфолейкозе могут быть повреждены 11-я, 13-я, 17-я хромосомы – выпадает определенный их участок, а так же может появляться лишняя 12-я хромосома (трисомия). Прогноз заболевания во многом зависит от вида хромосомных аномалий. Например, выпадение участка 13-й хромосомы или трисомия по 12-й при отсутствии других изменений в хромосомах являются благоприятным признаком, а выпадение участка 11-й или 17-й хромосом определяют более плохой прогноз.
- Биопсия костного мозга – взятие образца костного мозга из грудины или костей таза с помощью тонкой иглы. Проводится после предварительной анестезии. Затем под микроскопом выявляют лейкозные клетки.
- Спинномозговая пункция для выявления в спинномозговой жидкости, омывающей спинной и головной мозг, лейкозных клеток. Образец спинномозговой жидкости берется тонкой иглой, которая вводится между 3-м и 4-м поясничными позвонками после местной анестезии.
- Рентгенография грудной клетки. Может показать увеличение лимфатических узлов.
- УЗИ органов брюшной полости. Помогает выявить увеличение печени и селезенки.
- Химиотерапия – это использование специальных препаратов, которые разрушают лейкозные клетки или препятствуют их делению.
- Лучевая терапия – разрушение лейкозных клеток с помощью ионизирующего излучения.
- Таргетированная терапия – назначение препаратов, имеющих направленное действие на определенные виды злокачественных клеток. Эти препараты взаимодействуют с определенными белками на поверхности лейкозных клеток и вызывают их разрушение.
- Пересадка костного мозга – пациенту пересаживают нормальные клетки костного мозга от подходящего донора. Предварительно проводят курс химиотерапии или лучевую терапию в высоких дозах, чтобы уничтожить все патологические клетки.
- Лучевая терапия – разрушение лейкозных клеток с помощью ионизирующего излучения. Может быть использована при остром лимфолейкозе для полного разрушения лейкозных клеток перед пересадкой костного мозга.
- При остром лимфолейкозе в крови могут быть обнаружены лимфоциты разной степени зрелости – от лимфобластов до зрелых клеток.
При хроническом лимфолейкозе лимфобласты обычно отсутствуют в крови. Характерным признаком хронического лейкоза является обнаружение в мазке крови клеток (или теней) Боткина – Гумпрехта. Они представляют собой остатки разрушенных лимфоцитов. Тени Боткина – Гумпрехта отсутствуют в жидкой крови и образуются в процессе приготовления мазка. Их количество определяет интенсивность разрушения лимфоцитов в крови.
Лечение
При остром лимфобластном лейкозе прогноз благоприятный, особенно у детей. Большинство пациентов полностью излечиваются. Прогноз хронического лимфолейкоза зависит от скорости прогрессирования болезни и чувствительности к химиотерапии. Средняя продолжительность жизни пациентов с хроническим лимфолейкозом составляет 3-5 лет.
Профилактика
Специфической профилактики лимфолейкозов нет. Необходимо своевременно проходить профилактические осмотры, в ходе которых нередко выявляют заболевания крови.
Читайте также: