Хромосомные аномалии при миелодиспластическом синдроме - прогноз
Добавил пользователь Дмитрий К. Обновлено: 08.01.2026
Миелодиспластический синдром — это несколько сходных гематологических заболеваний, которые развиваются, как правило в старшем возрасте (в основном старше 60 лет). Основным проявлением МДС является цитопения. В костном мозге при этом обнаруживаются диспластические изменения. В некоторых случаях миелодиспластический синдром может переходить в острый лейкоз.
Несколько причин лежит в основе миелодиспластического синдрома. И хотя конкретные факторы могут быть разными, все они приводят к генетическим повреждениям клетки, сложным хромосомным нарушениям или гиперметилированию ДНК. В конечном итоге это вызывает нарушение пролиферации миелоидных клеток (появлению миелобластных клеток) как костного мозга, так и крови. Это приводит к их неправильному функционированию и соответствующей клинической симптоматике.
При миелодиспластическом синдроме генетические исследования необходимы не только для выявления конкретных генетических аномалий, но и для мониторинга прогрессирования заболевания.
К сожалению, более чем половина пациентов с миелодиспластическим синдромом имеют нормальный кариотип., что делает мониторинг заболеваний у этих пациентов затруднительным.
Пациенты с нормальным кариотипом, но агрессивным заболеванием могут иметь отклонения, которые не могут быть идентифицированы FISH или обычным цитогенетическим методом (кариотипирование). Хромосомный микроматричный анализ является методом выбора для этих пациентов.
С помощью этого метода можно обнаружить делеции, дупликации, несбалансированные транслокации, а также потерю гетерозиготности, которая часто является результатом мутаций и последующего отбора мутантных опухолевых генов-супрессоров и онкогенов.
Сравнительная характеристика обычного цитогенетического метода, FISH и хромосомного микроматричного анализа.
В сочетании с обычной цитогенетикой, повышает точность диагностики. FISH является более чувствительным, чем обычный цитогенетический анализ при обнаружении клональных аномалий.
Тест выбора для больных миелодиспластическим синдромом с нормальным кариотипом Позволяет обнаруживать очень мелкие изменения, которые могут определять прогноз и тактику лечение пациентов с миелодиспластическим синдромом, которые имеют нормальный кариотип..
Острый миелоидный лейкоз
Обычная классификация острого миелоидного лейкоза с помощью цитогенетического анализа не позволяет точно определить прогноз заболевания и, соответственно выбрать правильную тактику лечения у более чем 60% пациентов. AML-профайлер, тест на основе молекулярный микроматричной технологии, позволяет дать более точную классификацию у половины этих пациентов и отнести их к группе с благоприятным и неблагоприятным прогнозом.
AML-профайлер заменяет 7 отдельных тестов на основе 3-х различных технологий: цитогенетики, анализа мутаций и анализа экспрессии.
С помощью AML-профайлера можно определить транслокации t(8, 21),t(15, 17), инверсии inv(16), мутации NPM1 (типа A, B и D), а также двойную мутацию CEBPA. Кроме того, определяются уровни экспрессии 2 прогностических генов, EVI1 и BAALC. Мутация гена FLT3 не входят в AML-профайлер и может быть выполнена в нашей лаборатории отдельно.
Хромосомные аномалии при миелодиспластическом синдроме - прогноз
Хромосомные аномалии при миелодиспластическом синдроме - прогноз
Обнаружение клонов клеток с аномальным кариотипом при миелодиспластическом синдроме (МДС) имеет важное теоретическое и клиническое значение, поскольку свидетельствует о принадлежности этой группы заболеваний к новообразованиям.
Цитогенетические изменения весьма разнообразны, спектр их близок к спектру хромосомных аномалий, наблюдаемых при остром нелимфобластном лейкозе, особенно вторичных.
Наиболее характерны моносомии 5 и 7, а также делеции длинного плеча этих хромосом, появление дополнительной хромосомы 8 и делеции длинного плеча хромосомы 20.
Установлено, что частота обнаружения клонов анеуплоидных клеток нарастает по мере прогрессирования болезни: на относительно ранних этапах она составляет 20—30 %, при появлении начальных признаков трансформации в острый лейкоз — до 40—60 %, при трансформации в острый миелобластный лейкоз — 80—90 %.
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
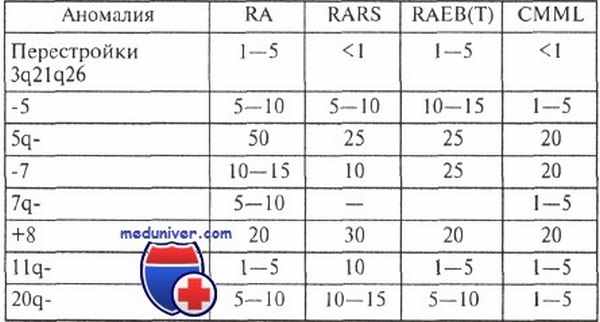
Основные хромосомные аномалии, характерные для миелодисплазий:
-7 или 7q-
-5 или 5q-
t(1;7)(q10;p10)
del(12)(р12-р13)
t(2;ll)(p13;q23)
del(13)(обязательно с включением 3q14)
t(6;9)(p23;q34)
del(20)(q11ql3)
+8
t(1;3)(p36;q21)
Перечисленные хромосомные аномалии наблюдаются при различных формах миелодисплазий, но частота их несколько различается.
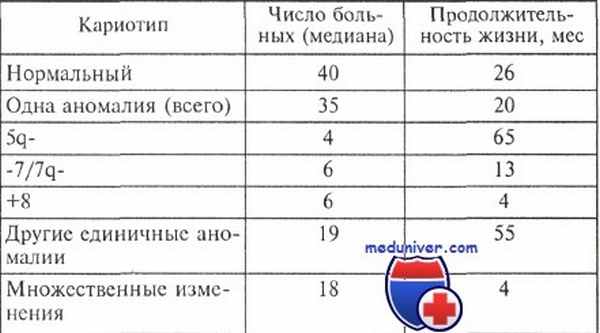
Опыт большинства исследователей свидетельствует о том, что существует корреляция между особенностями кариотипа и продолжительностью жизни больных с миелодиспластическим синдромом (МДС). Относительно благоприятным считается прогноз, если выявлены клоны клеток с единственной перестройкой 5q- или 20q; в то же время при любом варианте миелодиспластического синдрома обнаружение клона с множественными хромосомными аномалиями является крайне неблагоприятным.
Остановимся подробнее на отдельных нарушениях кариотипа, характерных для миелодиспластического синдрома (МДС).
Синдром 5q — рефрактерная сидеробластная анемия у пожилых больных, преимущественно женщин. В новой классификации ВОЗ этот синдром выделен как самостоятельный вариант миелодиспластического синдрома (МДС). Характерна макроцитарная анемия, резистентная к лечению, в костном мозге — признаки миелодисплазии клеток красного ряда и мегакариоцитов. Число тромбоцитов нормально или повышено, в костном мозге наблюдается гиперплазия гиполобулярных микромегакариоцитов. Клиническое течение сравнительно медленное. Трансформации в острый лейкоз наблюдаются приблизительно в 10 % случаев. Синдром впервые описан van den Berghe и соавт. в 1974—1985 гг..
Делеции длинного плеча хромосомы 5 наблюдаются и при других гематологических заболеваниях.
Предполагают, что делетирующийся участок содержит один или более генов-супрессоров. В этом направлении ведутся интенсивные исследования. До настоящего времени не подтверждена важная роль в патогенезе рефрактерной анемии ни одного из изучавшихся кандидатов на роль гена- супрессора.
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
Синдром моносомии хромосомы 7 встречается преимущественно у мальчиков до 4 лет. Характерна спленомегалия, часто наблюдаются лейкоцитоз с моноцитозом, тромбоцитопения, анемия. Прогноз плохой.
Как отмечалось, утрата одной из хромосом 7-й пары (моносомия 7) наблюдается при самых различных гемобластозах, включая острый нелимфобластный лейкоз, при этом она обычно ассоциирована с неблагоприятным прогнозом.
Делеции короткого плеча хромосомы 17 (17р-) обычно входят в число сложных изменений кариотипа. Как правило, 17р- сочетается с двумя или более хромосомными аномалиями и имеет неблагопрятное прогностическое значение.
В 75 % случаев в присутствии маркера 17р- наблюдается своеобразный дисгранулоцитопоэз в виде псевдопельгеровских гиподольчатых ядер и вакуолизации цитоплазмы. Этот маркер обнаружен не только при миелодисплазиях, но и при самых разнообразных злокачественных новообразованиях, включая солидные опухоли, его присутствие — плохой прогностический признак.
В 1997 г. опубликованы материалы международного совещания, посвященного диагностике и прогнозированию миелодиспластического синдрома. На основании ретроспективной оценки длительности заболевания до перехода в острый лейкоз и общей продолжительности жизни больных результат цитогенетического анализа был расценен как важнейший прогностический признак. В группу с благоприятным прогнозом отнесены случаи с единичными хромосомными аномалия ми: -Y, 5q- и 20q-. Неблагоприятное течение наблюдали при множественных (сложных) нарушениях (три или более перестройки кариотипа) и изменениях хромосомы 7 (делеции длинного плеча, моносомии).
Другие аномалии определяли «промежуточный» прогноз. Средняя длительность заболевания до перехода в острый лейкоз составила по группам 9,4; 0,4 и 1,1—3,3 года соответственно. Эти данные используются при оценке эффективности новых схем лечения миелодисплазии и зарекомендовали себя как одна из лучших прогностических систем при миелодиспластическом синдроме.
Важное диагностическое значение может иметь метод FISH в тех случаях, когда стандартное цитогенетическое исследование неинформативно или обнаруживаются только единичные клетки с нарушением кариотипа, которое по формальным критериям нельзя считать клоном. Для диагностики наиболее характерных хромосомных нарушений при миелодиспластическом синдроме разрабатывается панель FISH-зондов.
Попытки выделить цитогенетические особенности каждой из клинико-морфологических субъединиц, входящих в общую гетерогенную группу миелодиспластических синдромов, не увенчались успехом. В то же время ХММЛ, рассматриваемый как миелопролиферативное заболевание с морфологическими признаками миелодисплазии, нередко ассоциируется со специфической хромосомной аномалией t(5;12)(q33;p13), однако в большинстве случаев ХММЛ эта хромосомная аномалия не выявляется.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Миелодиспластический синдром (МДС)
Миелодиспластический синдром (МДС) представляет собой группу заболеваний, характеризующихся цитопенией в периферической крови, дисплазией гемопоэтических клеток-предшественников, гиперклеточностью или гипоклеточностью костного мозга и высоким риском развития острого миелолейкоза Острый миелолейкоз (ОМЛ) При остром миелолейкозе (ОМЛ) злокачественная трансформация и неконтролируемая пролиферация аномально дифференцированных, долго живущих клеток-предшественниц миелоидного ряда вызывает появление. Прочитайте дополнительные сведенияЕжегодное число людей в Соединенных Штатах с диагнозом миелодиспластический синдром (МДС) неизвестно. Согласно некоторым оценкам, это число составляет около 10 000, в то время как по другим оценкам оно намного выше. МДС чаще всего диагностируется у пациентов в возрасте 70 лет.
Патофизиология МДС
Миелодиспластические синдромы представляют собой группу заболеваний клональных гемопоэтических стволовых клеток, объединенных наличием различных мутаций гематопоэтических стволовых клеток, чаще всего в генах, участвующих в сплайсинге РНК. Миелодиспластические синдромы характеризуются неэффективным и диспластическим гемопоэзом и включают в себя следующее:
Рефрактерная анемия: анемия с ретикулоцитопенией; нормальный или гиперклеточный костный мозг с эритроидной гиперплазией и дизэритропоэзом; содержание бластных клеток составляет ≤ 5% ядросодержащих клеток костного мозга
Рефрактерная анемия с кольцевыми сидеробластами: то же, что и рефрактерная анемия с ретикулоцитопенией, за исключением того, что кольцевые сидеробласты составляют > 15% ядросодержащих клеток костного мозга
Рефрактерная цитопения с мультилинейной дисплазией: цитопения не определяется только эритроцитами;имеет место выраженная дисплазия предшественников лейкоцитов и мегакариоцитов
Рефрактерная цитопения с мультилинейной дисплазией и кольцевыми сидеробластами с наличием кольцевых сидеробластов, которые составляют > 15% ядросодержащих клеток костного мозга
Рефракторная анемия с избытком бластов (РАИБ) (RAEB en.): цитопения ≥ 2 клеточных линий с морфологическими аномалиями гематопоэтических клеток; гиперцеллюлярный костный мозг с дизэритропоэзом и дисгранулопоэзом; разрушает от 5 до 9% (RAEB-I) или от 10 до 19% (RAEB-II) ядросодержащих клеток костного мозга.
Миелодиспластический синдром неклассифицированный: МДС, который не попадает ни в одну из определенных категорий
МДС с изолированной делецией 5q: обычно тяжелая анемия и тромбоцитоз с делецией длинного плеча пятой хромосомы.
Хронический миеломоноцитарный лейкоз (ХММЛ) и ювенильный миеломоноцитарный лейкоз (ЮММЛ): смешанные миелодиспластические/миелопролиферативные новообразования; абсолютный моноцитоз (> 1000/мкл [> 1/л]) крови; значительное увеличение количества предшественников моноцитов в костном мозге
Хронический нейтрофильный лейкоз: характеризуется нейтрофилией, гибридным геном BCR-ABL1 и отсутствием филадельфийской хромосомы.
Этиология миелодиспластического синдрома неизвестна. Риск повышается с возрастом из-за приобретенных соматических мутаций, которые могут способствовть клональной экспансии и доминированию определенных гемопоэтических стволовых клеток, и, возможно, посредством воздействия внешних токсинов, таких как бензин, ионизирующие излучение и химиотерапевтические препараты (особенно продолжительные или интенсивные курсы лечения, а также с использованием алкилирующих агентов, гидроксимочевины или ингибиторов топоизомеразы). Часто присутствуют хромосомные аномалии (например, делеции, дупликации, структурные аномалии).
Костный мозг может быть гиперклеточным или гипоклеточным Неэффективный гемопоэз приводит к анемии (встречается наиболее часто), нейтропении, тромбоцитопении, или к комбинации этих патологий, вплоть до аплазии костного мозга. У пациентов со значительной рефрактерной или хронической анемией в конечном итоге развивается перегрузка железом ввиду переливания крови и/или повышенной абсорбции железа с кишечника.
Нарушение клеточной продукции также сопровождается изменениями морфологии клеток в костном мозге и крови. Иногда развивается экстрамедуллярный гемопоэз, приводящий к гепатомегалии и спленомегалии. Во время МДС может развиваться миелофиброз Первичный Миелофиброз Первичный миелофиброз (ПМФ) – это хроническое миелопролиферативное новообразование, которое характеризуется фиброзом костного мозга, спленомегалией и анемией с наличием ядросодержащих и каплевидных. Прочитайте дополнительные сведения . Классификация основана на данных общего анализа крови и исследований костного мозга, а также учитывается кариотип и мутация. Клон МДС имеет тенденцию к трансформации в острый миелолейкоз Острый миелолейкоз (ОМЛ) При остром миелолейкозе (ОМЛ) злокачественная трансформация и неконтролируемая пролиферация аномально дифференцированных, долго живущих клеток-предшественниц миелоидного ряда вызывает появление. Прочитайте дополнительные сведенияСимптомы и признаки МДС
Симптомы миелодиспластического синдрома зависят от наиболее пораженной клеточной линии и могут включать бледность, слабость и утомляемость (анемия), лихорадку и инфекции (нейтропения), повышенную склонность к кровоизлияниям, петехиям и кровоточивости из слизистых оболочек (тромбоцитопения). Спленомегалия и гепатомегалия не редкость.
Миелодиспластические синдромы
Миелодиспластические синдромы (МДС) — группа заболеваний, которые характеризуются нарушениями кроветворения миелоидной линии. В результате этих нарушений возможность выработки зрелых клеток крови частично сохраняется, но наблюдается дефицит тех или иных их видов, а сами клетки при этом изменены и плохо функционируют.
У значительной части больных МДС через некоторый промежуток времени, обычно от нескольких месяцев до нескольких лет, развивается острый миелоидный лейкоз.
МДС в обиходе иногда называют «предлейкемией», ранее применялись также термины «малопроцентный лейкоз», «тлеющий лейкоз» или «дремлющий лейкоз». Это связано с содержанием бластных клеток в костном мозге: если их более 20% (согласно классификации Всемирной организации здравоохранения) или более 30% (согласно франко-американо-британской классификации FAB), то речь уже идет о миелоидном лейкозе, если же их уровень ниже порогового значения, то может быть диагностирован МДС.
Под термином «миелодиспластический синдром» в настоящее время подразумевается целая группа заболеваний, различающихся по частоте встречаемости, клиническим проявлениям, а также по вероятности и ожидаемым срокам трансформации в лейкоз. Специалисты используют две классификации МДС: франко-американо-британскую (FAB) и классификацию Всемирной организации здравоохранения (ВОЗ). Рассмотрим классификацию FAB:
Рефрактерная анемия (РА). Термин «рефрактерная» здесь означает, что анемия не поддается лечению препаратами железа и витаминами. В костном мозге менее 5% миелобластов, аномалии в основном касаются предшественников эритроцитов.
Рефрактерная анемия с кольцевыми сидеробластами (РАКС): миелобластов в костном мозге менее 5%, но не менее 15% предшественников эритроцитов представлены особыми аномальными клетками — так называемыми кольцевыми сидеробластами. Это клетки с кольцеобразными «отложениями» железа, которые не могут обеспечивать эффективный транспорт кислорода.
Рефрактерная анемия с избытком бластов (РАИБ): миелобластов в костном мозге 5–20%. В классификации ВОЗ дополнительно подразделяется на РАИБ-I (5–9% бластов) и РАИБ-II (10-19% бластов).
Рефрактерная анемия с избытком бластов на стадии трансформации (РАИБ-T): миелобластов 21–30% (по классификации ВОЗ это уже острый миелоидный лейкоз).
Хронический миеломоноцитарный лейкоз, ХММЛ (по классификации ВОЗ относится к миелодиспластическим-миелопролиферативным заболеваниям).
Частота встречаемости, факторы риска
Общая частота МДС составляет 3-5 случаев на 100 000 населения. Однако у детей и молодых взрослых это заболевание встречается во много раз реже: более 80% случаев МДС фиксируется после 60 лет, причем несколько чаще у мужчин, чем у женщин.
В большинстве случаев МДС возникает без какой-либо известной причины, но иногда его развитие может быть спровоцировано предшествующей химиотерапией или лучевой терапией по поводу какой-либо опухоли - например, лимфомы Ходжкина или неходжкинской лимфомы. В этих случаях говорят о вторичном МДС.
Частота МДС (как и острого миелоидного лейкоза) повышена у людей с определенными генетическими аномалиями, такими как синдром Дауна, анемия Фанкони, нейрофиброматоз и некоторые другие.
Изучается роль и других факторов в возникновении этого заболевания — например, воздействия некоторых вредных химических веществ. Но у детей и молодых взрослых эти факторы не играют существенной роли.
Признаки и симптомы
Симптомы и степень их выраженности могут различаться в зависимости от разновидности МДС и конкретного случая. Но большинство наблюдаемых проявлений возникает в результате цитопении, то есть дефицита нормальных клеток крови.
Анемия (недостаток эритроцитов, сниженный уровень гемоглобина) характерна для подавляющего большинства случаев МДС; она проявляется бледностью, утомляемостью, плохой переносимостью физических нагрузок; могут также возникнуть одышка, головокружения, боли в груди и т.п.
Примерно у половины больных встречается нейтропения (то есть пониженное содержание зрелых функциональных нейтрофилов) и поэтому снижена сопротивляемость инфекциям. Нередко наблюдается и тромбоцитопения, то есть недостаточное содержание тромбоцитов, что ведет к возникновению кровотечений, синяков, петехий (мелкоточечных подкожных кровоизлияний). Возможны и другие симптомы.
Впрочем, в некоторых случаях пациенты с МДС долгое время не замечают существенного ухудшения самочувствия, и тогда проблемы обнаруживаются только в ходе обычного медосмотра по отклонениям от нормы в клиническом анализе крови.
Диагностика
Как правило, поводом к медицинскому осмотру служат жалобы на симптомы, связанные с анемией, причем эта анемия не поддается обычному лечению препаратами железа и витаминами. В клиническом анализе крови снижено количество эритроцитов, может быть также снижено количество лейкоцитов (нейтрофилов) и/или тромбоцитов. Характерно, что при уменьшении числа эритроцитов их размеры и цветовой показатель крови могут быть увеличены. Полезен также подсчет числа ретикулоцитов — незрелых эритроцитов, так как он дает информацию об интенсивности образования новых красных клеток крови.
Для точной диагностики и определения конкретного варианта МДС необходимо детальное исследование образца костного мозга, взятого в ходе трепанобиопсии: анализируются характер расположения различных клеток («архитектоника» костного мозга), число бластных клеток, содержание кольцевых сидеробластов и других аномальных клеток, степень изменений во всех ростках кроветворения — то есть среди предшественников эритроцитов, гранулоцитов и тромбоцитов, изменения стромы — соединительной ткани костного мозга. Выявленные нарушения могут быть очень разнообразными.
Так как развитие МДС нередко связано с известными хромосомными аномалиями, определенную роль в диагностике и прогнозе играют цитогенетические исследования.
Лечение
Лечение МДС зависит от его конкретной формы. Так, если речь идет об относительно «доброкачественных» разновидностях МДС с небольшим числом бластных клеток, то больные из групп низкого риска могут длительное время сохранять нормальное качество жизни, просто время от времени получая заместительную терапию компонентами крови — эритроцитами и, возможно, тромбоцитами. При перегрузке железом после множественных переливаний необходима соответствующая терапия. Иногда для стимуляции кроветворения используют факторы роста. При инфекционных осложнениях требуется антибактериальная и противогрибковая терапия. В ряде случаев применяют и другие лекарственные средства.
Если же речь идет о формах болезни, связанных с более высоким риском, то вопрос о лечении таких пацентов достаточно сложен. Химиотерапия с использованием обычных цитостатиков (цитарабин и т.п.) малоэффективна и не приводит к долговременной ремиссии. Общепринятых стандартов химиотерапии при МДС практически не существует. Есть и новые лекарства; в частности, обнадеживающие результаты показало применение гипометилирующих препаратов, таких как «Дакоген» (децитабин) и «Вайдаза» (азацитидин). Иногда может применяться иммуносупрессивная терапия и другие методы.
Единственным радикальным методом, позволяющим в случае успеха рассчитывать на полное излечение больных с МДС, является аллогенная трансплантация костного мозга — особенно у молодых пациентов, которые лучше переносят эту процедуру и связанные с ней осложнения. Однако аллогенная трансплантация по поводу МДС, как и по поводу других заболеваний, связана с проблемой поиска совместимого донора и с опасностью жизнеугрожающих осложнений.
При трансформации МДС в острый миелоидный лейкоз проводится химиотерапия этого лейкоза. Однако вторичный лейкоз, развившийся из МДС, обычно плохо поддается терапии. В этой ситуации, как правило, также показано проведение аллогенной трансплантации костного мозга, особенно у молодых больных.
Прогноз
Развитие МДС происходит с разной скоростью в зависимости от конкретной разновидности болезни. Если при некоторых формах МДС пациенты могут, особенно при наличии поддерживающей терапии (переливания компонентов крови и т. д.), жить достаточно долго и полноценно, то при более «активных» и злокачественных разновидностях заболевания средняя продолжительность жизни составляет не более года. Особенно плохой прогноз при вторичном МДС. Пациенты могут погибнуть как от проявлений самого МДС, так и от развившегося на его основе острого миелоидного лейкоза.
При использовании аллогенной трансплантации костного мозга можно добиться нормализации кроветворения и стойкой ремиссии болезни у большинства молодых пациентов. Иными словами, в случае успеха трансплантация приводит к выздоровлению. Однако надо помнить о высокой вероятности жизнеугрожающих осложнений (таких как реакция «трансплантат против хозяина», инфекции и др.) и рецидивов после трансплантации.
Хромосомные аномалии при миелодиспластическом синдроме - прогноз
ККМ — клетки костного мозга
КМ — костный мозг
МСК — мезенхимальные стромальные клетки
ОМЛ — острый миелоидный лейкоз
РА — рефрактерная анемия
РАИБ — рефрактерная анемия с избытком бластов
РАКС — рефрактерная анемия с кольцевыми сидеробластами
FISH — флюоресцентная in situ гибридизация

Оценка цитогенетических аномалий в костном мозге (КМ) у больных с миелодиспластическим синдромом (МДС) имеет большое значение для подтверждения клональной природы заболевания, определения прогноза заболевания и выбора тактики лечения больных. Цитогенетические аномалии в клетках костного мозга (ККМ) выявляют у 40—70% пациентов с МДС, и их разнообразие характеризует заболевание. Так, изолированная делеция (5q) выявляется у пациентов с отдельной нозологической формой МДС, 5q-синдромом, характеризующейся своеобразной клинической картиной, хорошим ответом на иммуномодулирующую терапию и благоприятным прогнозом. Изолированная трисомия 8 позволяет отнести пациента к группе промежуточного риска и является прогностическим фактором эффективности иммуносупрессивной терапии. Аномалии 7-й хромосомы (моносомия или делеция q-плеча) определяют крайне неблагоприятный прогноз и диктуют необходимость выбора агрессивной тактики лечения, в том числе выполнения трансплантации аллогенных гемопоэтических стволовых клеток в качестве первой линии терапии. Комплексные хромосомные аномалии (3 и более) могут включать –5/del(5q), –7/del(7q), del(17p) и чаще всего ассоциируются с вторичным МДС и неблагоприятным течением заболевания. Разнообразие выявляемых генетических аномалий при МДС представлено в табл. 1 [1].
Клиническое течение МДС различно и зависит от варианта заболевания. Рефрактерные цитопении (рефрактерная анемия — РА, рефрактерная тромбоцитопения, рефрактерная нейтропения), как правило, имеют благоприятный прогноз. При увеличении количества бластных клеток в КМ (варианты РА с избытком бластов — РАИБ-1 и РАИБ-2) вероятность трансформации в острый лейкоз увеличивается, и при определении более 20% бластных клеток диагностируют острый миелоидный лейкоз (ОМЛ) с предшествующей миелодисплазией [1—5].
Развитие МДС обусловлено клональным повреждением стволовой кроветворной клетки. Происходит ли это нарушение на уровне истинно стволовой клетки или коммитированных миелоидных предшественников, остается неясным. В последние годы в литературе опубликованы данные о выявлении хромосомных аберраций в популяции гемопоэтических клеток—предшественниц CD34+ у пациентов с МДС и ОМЛ [6—10]. Указывается также на определение патологического клона не только среди миелоидных клеточных линий, но и лимфоидных [11—13], что может быть обусловлено повреждением мультипотентной стволовой клетки.
Кроветворение в КМ обеспечивается постоянным взаимодействием гемопоэтических клеток-предшественниц и клеток стромального микроокружения. Мезенхимальные стромальные клетки (МСК) являются главным компонентом микроокружения и удобной моделью для изучения in vitro. В последнее время опубликованы работы о нарушенной функции стромы при МДС, в частности нарушен механизм миграции гемопоэтических клеток-предшественниц в костномозговые ниши. При сочетанном культивировании гемопоэтических и стромальных предшественников показано, что МСК, полученные от больных с МДС, демонстрируют сниженную способность к поддержанию нормальных гемопоэтических предшественников, у них существенно снижена пластичность по сравнению с таковой у МСК, полученных из КМ здоровых лиц, что может обусловливать неэффективный гемопоэз при МДС [11, 14—18].
Рядом исследовательских групп получены данные, согласно которым в МСК не определяются характерные для МДС хромосомные аберрации, и было сделано предположение, что строма не вовлечена в клональный процесс при данном заболевании и кариотип МСК не изменен [19—21]. Однако в работах E. Flores-Figueroa и соавт. [22, 23], O. Blau и соавт. [24, 25] показано наличие хромосомных нарушений в МСК, в том числе клональных, у отдельных пациентов с МДС. Исследование экспрессии генов с помощью микрочипов демонстрирует наличие генетических аномалий МСК у пациентов с МДС, в частности с 5q-синдромом [26]. Кроме того, на непосредственное участие стромального микроокружения в развитии МДС указывают данные, полученные M. Raaijmakers и соавт. [27], об индуцированном появлении признаков дисплазии в кроветворных клетках мышей при специфическом повреждении стромы.
С учетом несомненной тесной взаимосвязи гемопоэтических и стромальных клеток-предшественниц при развитии МДС представляется важным изучение цитогенетических особенностей этих клеточных популяций у пациентов с МДС.
Материалы и методы
В работе представлены результаты обследования 35 первичных больных, полученные в момент диагностики, в период с июля 2011 г. по май 2012 г.: 29 больных, их которых с МДС (РА) — 4, с рефрактерная цитопенией с мультилинейной дисплазией (РЦМД) — 13, с рефрактерной анемией с кольцевыми сидеробластами (РАКС) — 2, с РАИБ — 7, с 5q-синдром — 3; 6 больных в стадии трансформации в ОМЛ (1 пациент с вторичным ОМЛ) и 7 здоровых доноров КМ. Диагноз устанавливали в соответствии с классификацией ВОЗ (2008) [1]. Соотношение мужчин и женщин 18/17. Медиана возраста составила 60 лет (19—77 лет).
Цитогенетическое исследование ККМ проводили прямым методом и после краткосрочного (16—24 ч) культивирования клеток. Фиксацию и приготовление препаратов хромосом выполняли по общепринятой методике. G-дифференциальную окраску хромосом (G-band) осуществляли по методике M. Seabright [28] в модификации с применением красителя Wright. Цитогенетическое исследование МСК выполняли по методу, описанному Z. Zhang и соавт. [29]. Хромосомный анализ проводили в соответствии с Международной цитогенетической номенклатурой хромосом человека (ISCN 2005) [30]. Анализировали в среднем 20 метафаз. Клональной считали аномалию при обнаружении структурной перестройки и трисомии минимум в 2 метафазах и моносомии минимум в 3. При обнаружении структурной перестройки в одной метафазе перестройку обозначали как неклональную (спонтанную). Единичные неполные митозы не учитывали ввиду возможности потерь хромосом при обработке клеточного осадка. Флюоресцентную in situ гибридизацию (FISH) выполняли на ККМ и изолированных клетках CD34+ с использованием набора ДНК-зондов LSI (5q33—q34) EGR-1 SpectrumOrange/D5S721/D5S23 SpectrumGreen Probe, LSI (7q31) SpectrumOrange/CEP 7 SpectrumGreen Probe, CEP 8 SpectrumOrange DNAProbeKit, LSIAML1/ETO Dual Color Fusion Translocation Probe, CEP X SpectrumOrange/CEPY SpectrumGreen DNA Probe, LSI D20S108 (20q12) SpectrumOrange Probe («Vysis Abbott», США). Гибридизацию осуществляли согласно методике фирмы-производителя. Анализ сигналов проводили под флюоресцентным микроскопом Zeiss-Axioscope с использованием тройного фильтра Orange/Green/Dapi. Анализировали 200 интерфазных ядер.
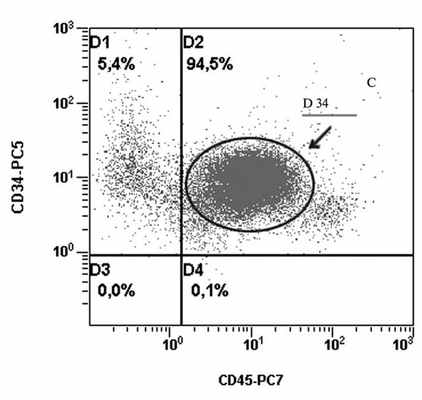
Гемопоэтические клетки—предшественницы СD34+ получали из КМ и периферической крови. Мононуклеарные клетки выделяли, центрифугируя в градиенте плотности фиколла, и метили CD34 MicroBeads human. Изолированную фракцию клеток CD34+ получали, используя MACS колонки для магнитной сепарации, в соответствии с инструкциями фирмы-производителя («Miltenyi Biotec», «Bergisch Gladbach», Германия). Чистота полученной фракции проверена с помощью проточной цитометрии и составила 94,5% (рис. 1). Рисунок 1. Контроль чистоты популяции клеток CD34+ после магнитной сепарации с помощью проточной цитометрии. Выделенную фракцию клеток концентрировали с помощью цитоспина на адгезивном стекле для выполнения FISH-исследования.
Культуру МСК получали из КМ. Фракцию мононуклеаров выделяли из аспирата КМ в градиенте плотности фиколла (1,077 г/см 3 ). Для получения МСК мононуклеары культивировали в пластиковых матрасах с площадью дна 25 и 75 см 2 в концентрации 1—6·10 6 клеток на флакон в среде α-МЕМ («HyClone», США) с 10% эмбриональной телячьей сывороткой («HyClone», США), 2 мМ L-глутамина («BioWest»), 100 ед/мл пенициллина и 50 ед/мл стрептомицина («Ферейн», Россия). После образования конфлюэнтного монослоя из фибробластов клетки снимали 0,05% раствором трипсина и пассировали. В большинстве исследований использовали клетки после 2—3 пассажей.
Результаты
Цитогенетическое исследование ККМ. Характеристика больных и результаты цитогенетического исследования КМ представлены в табл. 2. Нормальный кариотип определен у 19 из 35 больных, у одного из них выявлена конституциональная инверсия inv(9)(p13q21); у 13 (МДС у 11 и ОМЛ у 2) — аномалии кариотипа и у 3 делящиеся клетки не обнаружены. Спектр выделенных аномалий следующий: у 7 больных (МДС у 6 и ОМЛ у 1) выявлены изолированные del(5q), –7, i(14), inv(3); у 1 (МДС) — 2 аномалии — del(5q) и –7, и у 5 (МДС — у 3 и ОМЛ — у 2) комплексный кариотип.
У 5 (14%) из 35 больных методом FISH выявлены скрытые хромосомные аномалии, которые не определены при кариотипировании: у 2 из них (МДС) при цитогенетическом исследовании не получены митозы и методом FISH выявлены хромосомные аномалии: в первом случае — одновременно del(5q) и del(7q) в 84% клеток, во втором — трисомия 8 в 67% клеток. Еще у 2 больных (ОМЛ у 1 и МДС у 1) методом G-band определен нормальный кариотип, однако с помощью метода FISH выявлена del(5q) в обоих случаях. У 1 больного МДС, у которого в кариотипе определена изолированная del(5q), FISH-анализ позволил выявить дополнительно трисомию 21 в 52% клеток (исследование выполнено прицельно, так как имелись анамнестические данные предыдущего цитогенетического анализа у этого пациента, выявившего 2 аномалии). Таким образом, после применения метода FISH дополнительно к исследованию кариотипа число больных с цитогенетическими аномалиями в ККМ увеличилось с 13 до 17 (49%).
FISH-исследование выделенных гемопоэтических клеток—предшественниц CD34+ из КМ и периферической крови. Методом FISH проанализированы гемопоэтические клетки—предшественницы CD34+, полученные из КМ и периферической крови, у 24 из 35 пациентов (у 3 ОМЛ, у 21 МДС). У 10 из 24 пациентов в КМ определены хромосомные аномалии. Мы подтверждали эти аномалии в популяции клеток CD34+ с помощью FISH-исследования.
Цитогенетическое исследование МСК. Кариологический анализ МСК выполнен у 23 из 35 пациентов (МДС у 19 и ОМЛ у 4) и у 7 здоровых доноров КМ (см. табл. 2). У 12 больных из 35 не удалось получить рост МСК в культуре вследствие ограниченной способности стромальных клеток больных с МДС к пролиферации. Нормальный кариотип МСК определен у всех больных ОМЛ и у 6 больных с МДС, у одного из которых в кариотипе МСК определена конституциональная инверсия inv(9). У 2 (11%) из 19 пациентов с МДС выявлены аномалии кариотипа.
У одного из них с конституциональной inv(9) (пациент №12) выявлена неклональная транслокация в одной метафазе: 46ХY,t(2;22)(p10;q11), inv(9)(p13q21)[1]/46ХY, inv(9) (p13q21)[19]. У второго (пациент №21) в кариотипе МСК выявлена клональная перестройка в 7 метафазах из 20: 46ХУ,add(2q)[7]/46ХУ[13]. В КМ у этого больного определен комплексный кариотип (рис. 2). Рисунок 2. Кариотип ККМ и МСК пациента №21 И., 58 лет, диагноз: МДС, РА. а — кариотип ККМ: 45—46,XY,–5,–13,der(19),add(q13?or p13),–20,+2mar,+mar del(13q21)?,+d min. Рисунок 2. Кариотип ККМ и МСК пациента №21 И., 58 лет, диагноз: МДС, РА. б — кариотип МСК: 46,XY,add (2)(q36). У обоих пациентов с инверсией inv(9) в ККМ и МСК ее конституциональный характер был подтвержден результатами исследования кариотипа стимулированной фитогемагглютинином культуры лимфоцитов периферической крови. При кариотипировании МСК от здоровых доноров КМ во всех исследованных образцах получен нормальный кариотип.
В настоящей работе представлен анализ цитогенетических изменений в популяциях гемопоэтических и стромальных клеток-предшественниц у пациентов с различными вариантами МДС, а также в стадии трансформации заболевания в ОМЛ. Интерес к изучению этой области определяется тем, что до сих пор остается неуточненным вопрос об истинном уровне генетического повреждения при МДС, а также участии стромального микроокружения в патогенезе клональной эволюции миелодисплазии.
Определение кариотипа ККМ в настоящее время входит в перечень стандартных исследований, необходимых для верификации диагноза МДС. Выявленные хромосомные аномалии определяют прогноз заболевания и соответственно тактику лечения больных. Современные прогностические шкалы, в частности шкала IPSS-R, помимо количества бластных клеток в КМ, степени цитопении и зависимости от гемотрансфузий, включают данные цитогенетического анализа [31]. В нашем исследовании цитогенетические аномалии в КМ обнаружены у 17 (49%) из 35 обследованных, что коррелирует с данными литературы [1—5]. Стоит еще раз отметить, что не во всех случаях метод стандартного цитогенетического исследования явился достаточным для обнаружения аномалий кариотипа, и у около 10% пациентов не удалось получить делящиеся клетки для анализа. По меньшей мере в 14% случаев (по нашим данным, у 5 из 35 больных) методом FISH могут быть выявлены дополнительные (скрытые) хромосомные аномалии. У всех 5 больных размер выявленных клонов был довольно велик (от 52 до 84%); такие клоны не могут быть незамеченными при хромосомном анализе. По-видимому, все эти клетки находятся в стадии G0, не выходят в митоз и поэтому не обнаруживаются при кариотипировании. У 1 из 5 пациентов с выявленной del(5q) неразделившиеся клетки содержали вторую аномалию — трисомию 21. Можно предположить, что у этого больного наблюдалось 2 клона клеток в КМ — один только с del(5q) и второй одновременно с del(5q) и трисомией 21. Выявление дополнительных хромосомных аномалий у ряда пациентов может иметь принципиальное значение, так как от этого зависят и прогноз заболевания, и терапевтический подход. Так, при определении нормального кариотипа в ККМ может быть выбрана тактика динамического наблюдения с последующими исследованиями КМ, тогда как выявление del(5q) позволяет эффективно применять иммуномодулирующую терапию. Следовательно, в дебюте заболевания всем пациентам целесообразно сначала проводить стандартное цитогенетическое исследование, и в случаях определения нормального кариотипа или отсутствия делящихся клеток необходимо выполнять FISH-исследование с целью определения клинически значимых хромосомных аномалий. Всем пациентам, не дожидаясь получения результатов кариотипирования, проводить FISH-исследование нецелесообразно [32].
В клетках CD34+, выделенных из КМ и периферической крови, определены те же клональные нарушения, что и в ККМ пациентов с аномалиями кариотипа, что подтверждает поражение гемопоэтических предшественников при МДС. По данным литературы, количество клеток—предшественниц CD34+ в КМ больных МДС значительно больше, чем у здоровых лиц, пациентов с апластической анемией и вторичными цитопеническими синдромами. Процент клеток CD34+ неоднороден у пациентов категорий низкого (1,7%) и высокого (10,5%) риска и максимальный у пациентов с ОМЛ (35%), он коррелирует с процентом бластных клеток и немного превышает его [33]. Количество циркулирующих клеток CD34+ у пациентов с МДС также значительно больше, чем у здоровых лиц, и зависит от процентного содержания их в КМ [34]. Наряду с увеличением количества бластных клеток, увеличение количества клеток CD34+ тоже может свидетельствовать о прогрессии заболевания. В нашем исследовании выявлено, что в среднем процент клонально измененных клеток среди гемопоэтических предшественников не отличается от такового в общей популяции ККМ пациентов с аномалиями кариотипа. Однако у 4 из 10 больных отличия выявлены. Согласно нашим наблюдениям размер патологического клона в клетках CD34+ может быть как значительно больше, так и меньше размера клона в общей популяции ККМ. Возможно, выраженность клональных изменений в гемопоэтических клетках-предшественницах может свидетельствовать о динамике развития заболевания и иметь прогностическое значение. Ввиду того что результаты FISH-исследования циркулирующих клеток СD34+ в среднем сопоставимы с результатами исследования ККМ, цитогенетический контроль динамики заболевания в процессе лечения при наличии маркеров для FISH-исследования может осуществляться с использованием изолированной фракции клеток CD34+ или мононуклеарной фракции периферической крови, что поможет снизить кратность пункций КМ у пациентов с МДС. У пациентов с нормальным кариотипом хромосомные перестройки не обнаружены и в популяции клеток CD34+. Таким образом, детальное цитогенетическое исследование (стандартное цитогенетическое исследование и FISH) не выявило у пациентов с нормальным кариотипом в КМ «скрытых» хромосомных аномалий в гемопоэтических предшественниках CD34+.
Почти у 90% пациентов с МДС и у всех пациентов с ОМЛ в нашем исследовании определен нормальный кариотип МСК. Хромосомные аномалии МСК выявлены у 2 (11%) из 19 пациентов с МДС. У обоих выявлены структурные аномалии: у одного — 35% клон add(2q), у другого — с конституциональной хромосомной нестабильностью, inv(9)(p13;q21) — единичный митоз с t(2;22)(p10;q11). Учитывая, что у пациента с клональными изменениями кариотипа МСК в ККМ обнаружен комплексный кариотип, можно предположить, что генетическая нестабильность стромы обусловлена множественными цитогенетическими аномалиями в кроветворных клетках. Численные аномалии кариотипа МСК не выявлены ни в одном случае.
Как упоминалось выше, в литературе есть публикации, согласно которым хромосомные аномалии в МСК у пациентов с МДС не определяются [19, 20]. По-видимому, причина заключается в использовании разных методов цитогенетического анализа, так как авторы с целью определения аномалий кариотипа МСК применяли только FISH-исследование с выявленными в ККМ цитогенетическими маркерами и не исследовали кариотип МСК, тогда как выявленные нами аномалии МСК определяются только при кариотипировании. Таким образом, в МСК у пациентов с МДС, действительно, не определяются характерные для ККМ цитогенетические аномалии, однако возможны отличные от них аберрации. В работах E. Flores-Figueroa и соавт. [22, 23] и Lu-Xi Song и соавт. [35] описаны аномалии кариотипа МСК более чем у 50% обследованных пациентов с МДС, в основном за счет неклональных потерь отдельных хромосом. Однако встречающиеся отдельные неполные митозы нами не учитывались, так как подобные ситуации могут встречаться вследствие технических особенностей обработки клеточного осадка. O. Blau и соавт. [25] описывают клональные структурные аномалии МСК у 16% пациентов с МДС и ОМЛ. Результаты нашего исследования сопоставимы с результатами O. Blau, однако мы не выявили аномалии кариотипа МСК у пациентов в стадии трансформации МДС в ОМЛ.
Заключение
Таким образом, несмотря на небольшое число проанализированных больных, на основании проведенного исследования можно сделать заключение, что в гемопоэтических и мезенхимальных клетках-предшественницах у пациентов с МДС определяются разные цитогенетические аномалии. В клетках CD34+, выделенных из КМ и периферической крови, выявлены те же аномалии, что и в общей популяции ККМ. У 11% пациентов с МДС выявлены аномалии кариотипа МСК, что указывает на генетическую нестабильность стромы при этом заболевании и подверженность возникновению хромосомных поломок. Различия хромосомных аномалий, определяемых в гемопоэтических и мезенхимальных клетках-предшественницах, подтверждают, что клетки стромального микроокружения в отличие от гемопоэтических предшественников не являются частью патологического клона при МДС, однако, вероятно, играют важную роль в патогенезе развития заболевания.
Читайте также: