Хромосомные аномалии при злокачественных лимфомах - прогноз
Добавил пользователь Алексей Ф. Обновлено: 27.01.2026
Лимфома неходжкенская — гетерогенная группа заболеваний, характеризующаяся неопластической пролиферацией незрелых лимфоидных клеток, накапливающихся вне костного мозга.
Лимфосаркоматоз (Кундрата болезнь) — генерализованная форма неходжкенской лимфомы, характеризующаяся множественным поражением лимфатических узлов, а в последующем — поражением печени и селезёнки.
Частота. Ежегодно в США диагноз неходжкенской лимфомы устанавливают примерно у 35000 больных.
Патогистологическая классификация. Известно множество гистологических классификаций заболевания. Для устранения противоречий между ними в 1982 г. принята классификация Национального Института Рака: • Лимфома низкой степени злокачественности •• Мелкоклеточная лимфоцитарная •• Преимущественно фолликулярная (мелкие клетки с расщеплёнными ядрами) •• Фолликулярная — смешанного типа (мелкие клетки с расщеплёнными ядрами и крупные клетки) • Лимфома средней степени злокачественности •• Преимущественно фолликулярная крупноклеточная •• Диффузная мелкоклеточная с расщеплёнными ядрами •• Диффузная смешанная (мелко- и крупноклеточная) •• Диффузная крупноклеточная • Лимфома высокой степени злокачественности •• Крупноклеточная •• Лимфобластная с изогнутыми ядрами •• Мелкоклеточная с нерасщеплёнными ядрами (Беркетта).
Этиология • Иммунодефициты • Длительный приём иммунодепрессантов (например, после пересадки почки или сердца) • Вирус Эпстайна–Барр связан с развитием лимфом Беркетта • Цитогенетические аномалии (например, хромосомные транслокации).
Клиническая картина • Пролиферативный синдром: лимфаденопатия (увеличение поражённых лимфатических узлов); опухолевый синдром: увеличение печени, селезёнки • Интоксикационный синдром: лихорадка, повышенная утомляемость, снижение массы тела и ночные поты • Клинические проявления зависят от локализации лимфосаркомы (кишечная непроходимость при абдоминальной локализации; синдром сдавления трахеи при поражении внутригрудных лимфатических узлов).
Стадии заболевания и диагностика • Принципы стадирования аналогичны таковым при лимфогранулематозе. 4-я стадия заболевания выставляется при вовлечении в патологический процесс костного мозга (лейкемизация) и ЦНС • Установление стадии •• Биопсия лимфатического узла и анализ биопсийного материала •• Гематологическое исследование, в т.ч. подсчёт лейкоцитарной формулы, тромбоцитов, определение содержания мочевой кислоты. Электрофорез белков крови позволяет исключить гипогаммаглобулинемию и/или болезнь тяжёлых цепей •• Сбор полного анамнеза и врачебный осмотр с акцентом на все группы лимфатических узлов (в первую очередь, кольцо фон Вальдейера–Пирогова), а также на размеры печени и селезёнки •• Двусторонняя биопсия и аспирация костного мозга •• Радиологические исследования — рентгенография органов грудной клетки, брюшной полости и таза, реже — двусторонняя лимфангиография нижних конечностей и таза •• Другие процедуры — диагностическая лапаротомия, сцинтиграфия или рентгенография костей, эндоскопия и биопсия печени.
ЛЕЧЕНИЕ обычно комбинированное. Как и при лечении лейкозов используют различные протоколы химиотерапии.
• Химиотерапия •• Лимфомы I и II стадии промежуточной и высокой степени злокачественности часто хорошо реагируют на комбинированную химиотерапию (большие дозы циклофосфамида с метотрексатом, винкристином и часто с доксорубицином) с облучением или без лучевой терапии (излечение наблюдают в 80–90% случаев) •• При поражении ЦНС цитостатики вводят эндолюмбально или в желудочки головного мозга.
• Облучение. Неходжкенские лимфомы чрезвычайно радиочувствительны •• При локализованном процессе облучение должно быть направлено на поражённую область (в дозе 40 Гр) •• При диссеминированной лимфоме облучение имеет паллиативный эффект, а также усиливает лечебный эффект химиотерапии •• I стадия вялотекущих лимфом. Длительное наблюдение за больными с локализованными I и II стадиями лимфомы низкой степени злокачественности, получившими общее облучение лимфатических узлов, выявило наличие 10-летнего безрецидивного периода в 50% случаев (особенно у молодых пациентов).
Особенности у детей
• Преобладающий возраст — 5–9 лет, соотношение мальчики/девочки — 2–2,5/1.
• Особенности течения •• Быстрое прогрессирование опухоли •• Преобладание экстранодальной локализации •• Первично-генерализованная опухоль.
• Локализация •• В-клеточные лимфомы — кишечник (35%), носоглотка (20%) •• Т-клеточные лимфомы — средостение (25%), периферические лимфатические узлы (15%).
• Лечение •• Основной метод — комбинированная полихимиотерапия •• Лучевую терапию применяют только при поражении ЦНС (местно).
• Течение и прогноз. 5-летняя выживаемость при лечении достигает 80%.
Синонимы • Лимфосаркома • Лимфобластома • Лимфома злокачественная.
МКБ-10 • C82 Фолликулярная [нодулярная] неходжкинская лимфома • C83 Диффузная неходжкинская лимфома
Примечание. Повышение заболеваемости у пожилых людей происходит за счёт диффузной крупноклеточной лимфомы.
Код вставки на сайт
Лимфома неходжкенская
Лимфома неходжкенская — гетерогенная группа заболеваний, характеризующаяся неопластической пролиферацией незрелых лимфоидных клеток, накапливающихся вне костного мозга.
Лимфосаркоматоз (Кундрата болезнь) — генерализованная форма неходжкенской лимфомы, характеризующаяся множественным поражением лимфатических узлов, а в последующем — поражением печени и селезёнки.
Частота. Ежегодно в США диагноз неходжкенской лимфомы устанавливают примерно у 35000 больных.
Патогистологическая классификация. Известно множество гистологических классификаций заболевания. Для устранения противоречий между ними в 1982 г. принята классификация Национального Института Рака: • Лимфома низкой степени злокачественности •• Мелкоклеточная лимфоцитарная •• Преимущественно фолликулярная (мелкие клетки с расщеплёнными ядрами) •• Фолликулярная — смешанного типа (мелкие клетки с расщеплёнными ядрами и крупные клетки) • Лимфома средней степени злокачественности •• Преимущественно фолликулярная крупноклеточная •• Диффузная мелкоклеточная с расщеплёнными ядрами •• Диффузная смешанная (мелко- и крупноклеточная) •• Диффузная крупноклеточная • Лимфома высокой степени злокачественности •• Крупноклеточная •• Лимфобластная с изогнутыми ядрами •• Мелкоклеточная с нерасщеплёнными ядрами (Беркетта).
Этиология • Иммунодефициты • Длительный приём иммунодепрессантов (например, после пересадки почки или сердца) • Вирус Эпстайна–Барр связан с развитием лимфом Беркетта • Цитогенетические аномалии (например, хромосомные транслокации).
Клиническая картина • Пролиферативный синдром: лимфаденопатия (увеличение поражённых лимфатических узлов); опухолевый синдром: увеличение печени, селезёнки • Интоксикационный синдром: лихорадка, повышенная утомляемость, снижение массы тела и ночные поты • Клинические проявления зависят от локализации лимфосаркомы (кишечная непроходимость при абдоминальной локализации; синдром сдавления трахеи при поражении внутригрудных лимфатических узлов).
Стадии заболевания и диагностика • Принципы стадирования аналогичны таковым при лимфогранулематозе. 4-я стадия заболевания выставляется при вовлечении в патологический процесс костного мозга (лейкемизация) и ЦНС • Установление стадии •• Биопсия лимфатического узла и анализ биопсийного материала •• Гематологическое исследование, в т.ч. подсчёт лейкоцитарной формулы, тромбоцитов, определение содержания мочевой кислоты. Электрофорез белков крови позволяет исключить гипогаммаглобулинемию и/или болезнь тяжёлых цепей •• Сбор полного анамнеза и врачебный осмотр с акцентом на все группы лимфатических узлов (в первую очередь, кольцо фон Вальдейера–Пирогова), а также на размеры печени и селезёнки •• Двусторонняя биопсия и аспирация костного мозга •• Радиологические исследования — рентгенография органов грудной клетки, брюшной полости и таза, реже — двусторонняя лимфангиография нижних конечностей и таза •• Другие процедуры — диагностическая лапаротомия, сцинтиграфия или рентгенография костей, эндоскопия и биопсия печени.
ЛЕЧЕНИЕ обычно комбинированное. Как и при лечении лейкозов используют различные протоколы химиотерапии.
• Химиотерапия •• Лимфомы I и II стадии промежуточной и высокой степени злокачественности часто хорошо реагируют на комбинированную химиотерапию (большие дозы циклофосфамида с метотрексатом, винкристином и часто с доксорубицином) с облучением или без лучевой терапии (излечение наблюдают в 80–90% случаев) •• При поражении ЦНС цитостатики вводят эндолюмбально или в желудочки головного мозга.
• Облучение. Неходжкенские лимфомы чрезвычайно радиочувствительны •• При локализованном процессе облучение должно быть направлено на поражённую область (в дозе 40 Гр) •• При диссеминированной лимфоме облучение имеет паллиативный эффект, а также усиливает лечебный эффект химиотерапии •• I стадия вялотекущих лимфом. Длительное наблюдение за больными с локализованными I и II стадиями лимфомы низкой степени злокачественности, получившими общее облучение лимфатических узлов, выявило наличие 10-летнего безрецидивного периода в 50% случаев (особенно у молодых пациентов).
Особенности у детей
• Преобладающий возраст — 5–9 лет, соотношение мальчики/девочки — 2–2,5/1.
• Особенности течения •• Быстрое прогрессирование опухоли •• Преобладание экстранодальной локализации •• Первично-генерализованная опухоль.
• Локализация •• В-клеточные лимфомы — кишечник (35%), носоглотка (20%) •• Т-клеточные лимфомы — средостение (25%), периферические лимфатические узлы (15%).
• Лечение •• Основной метод — комбинированная полихимиотерапия •• Лучевую терапию применяют только при поражении ЦНС (местно).
• Течение и прогноз. 5-летняя выживаемость при лечении достигает 80%.
Синонимы • Лимфосаркома • Лимфобластома • Лимфома злокачественная.
МКБ-10 • C82 Фолликулярная [нодулярная] неходжкинская лимфома • C83 Диффузная неходжкинская лимфома
Примечание. Повышение заболеваемости у пожилых людей происходит за счёт диффузной крупноклеточной лимфомы.
Лимфома желудка
Лимфома желудка составляет 1/95-99 всех злокачественных процессов в органе, где на лидирующей позиции — рак. Внешне и клинически лимфома желудка схожа с аденокарциномой или большой язвой. Раньше такие лимфомы лечили хирургически, современная терапия ориентирована на консервативные методы.
Общие сведения
Редкая опухоль лимфома желудка относится к лимфопролиферативным процессам, развивающимся не в лимфатическом узле, а в тканях желудка. В очень разнообразной по клеточному строению группе неходжкинских лимфом доля желудочной локализации составляет менее 8%.
Нет статистики заболеваемости, но из 100 тысяч взрослых лимфома желудка развивается едва ли у 5-6 человек, преимущественно у зрелых и пожилых. Замечено, что мужчины болеют чаще.
Лечат лимфомы онкогематологи, а процесс в желудочно-кишечном тракте диагностируют онкологи, что объясняется стандартами маршрутизации пациента с подозрением на опухоль.
Причины лимфомы желудка
Причины развития лимфом желудка неизвестны, и не определены мутации генов, ведущие к перерождению нормальных клеток. В семейной истории страдавших лимфомами желудка часто имеются родственники со злокачественными заболеваниями.
В инициации опухоли предполагается соучастие нарушений иммунитета:
- избыточной защиты при аллергии и аутоиммунных болезнях;
- иммунодефицита при ВИЧ и наследственных синдромах;
- нарушений толерантности при противоопухолевой терапии и хроническом воспалении;
- угнетении из-за повышенного радиационного воздействия.
Определенная этиологическая роль принадлежит хеликобактерной инфекции, у большинства пациентов в желудочном секрете находят Helicobacter pylori (Н.pylori). Скопления В-лимфоцитов в желудочной стенке, из которых развивается опухоль, возникают при вызываемом и поддерживаемом бактериями хроническом воспалении. Выявление хеликобактерной инфекции и её лечение — основополагающий подход при MALT (мукозоассоциированном) варианте опухоли.
Классификация лимфом желудка
По клеточному составу лимфомы неоднородны:
- Подавляющее большинство в 80% составляют опухоли, развивающиеся из В-лимфоцитов маргинальной зоны, или MALT-лимфомы, у части больных находят инфекцию Н.pylori, что в пользу более благоприятного течения.
- Остальные 20% — агрессивные лимфомы из В-лимфоцитов или В-лимфомы, требующие тяжелого лечения при незавидных перспективах на долгую жизнь. Некоторые исследователи считают, что этот тип опухоли возникает в результате трансформации давно существующей лимфомы MALT, так как у некоторых пациентов одновременно обнаруживают оба вида опухоли.
- Редчайший тип доброкачественной опухоли с возможной трансформацией в агрессивную В-лимфому — псевдолимфома, процесс локализован только в желудке, не выходит за его пределы, хороший прогноз на излечение.
Стадирование лимфом осуществляется не по привычной онкологической классификации TNM или гематологической Ann-Arbor, а по Lugano, где стадия:
- IE — небольшое поражение только стенки органа;
- IIE — локальная опухоль органа и ближайших лимфоузлов;
- IIIE — вовлечение лимфоузлов за пределами брюшной полости;
- IV — диссеминация в организме.
Симптомы лимфомы желудка
Клинические проявления желудочной лимфомы на ранних стадиях неспецифичны и неотличимы от симптомов карциномы и язвы. В разных комбинациях возможны: боли «под ложечкой», дискомфорт, изжога, отрыжка, чувство тяжести после еды, ухудшение аппетита, снижение веса.
При распространенном процессе увеличиваются лимфатические узлы, поражаются легкие, кишечник, печень, селезенка.
Вне зависимости от размеров опухоли при агрессивном варианте лимфомы возможна высокая температура с ночными «обливными» потами, слабость и быстрая потеря веса — так называемые «В-симптомы», указывающие на неблагоприятный прогноз лимфомы.
Диагностика лимфомы желудка
Диагностика лимфомы на первом этапе не отличается от выявления других злокачественных процессов желудка: эндоскопия с биопсией, УЗИ, КТ брюшной полости, КТ грудной клетки.
Как при всех злокачественных процессах крови и лимфатической ткани обязательно выполняется трепанобиопсия костного мозга.
После морфологической верификации проводится уточняющая терапию диагностика:
- тесты на выявление в желудочном содержимом Helicobacter Pylori;
- определяется чувствительность хеликобактера к антибиотикам;
- цитогенетический анализ на обнаружения переноса участка t(11;18) с одной хромосомы на другую, при выявлении транслокации проводится уточняющий FISH-анализ.

Лечение лимфомы желудка
Лечение лимфомы MALT и В-клеточных лимфом существенно различается, последние подлежат активной химиотерапии.
Вариант терапии MALT-лимфомы ранних стадий определяет хеликобактерная инфекция, если она присутствует, то на первом этапе проводится антибактериальное лечение, аналогично эрадикации H.pilory при язвенной болезни.
Второй этап зависит от результатов первичного лечения:
- При устойчивости H.pilory к антибиотикам изменяют схему лечения, при неблагоприятном прогностическом варианте с t(11;18) параллельно подключают таргетную терапию ритуксимабом или облучение.
- При очищении желудочного секрета от хеликобактерной инфекции и полной регрессии лимфомы пациент наблюдается с регулярными обследованиями по графику.
- При доказанном отсутствии инфекции и остаточной опухоли проводится лучевая на желудок или монотерапия ритуксимабом.
Развившиеся без участия хеликобактера, ограниченные желудочными стенками MALT-омы, особенно при наличии хромосомной аномалии, на первом этапе облучают, при невозможности лучевой терапии проводят курс таргетного препарата. Операция по удалению желудка проводится только при осложненных кровоточащими язвами лимфомах.
При распространенной MALT-лимфоме предпочтение отдано лекарственной терапии.

Диета при лимфоме желудка
Питание при злокачественных лимфомах на этапе эрадикации хеликобактерной инфекции аналогично диете при язвенной болезни. Расширение меню до «общего стола» возможно в периоды ремиссии заболевания, предварительно и обязательно режим питания обсуждается с лечащим врачом.
Диетический режим во время лучевой и лекарственной терапии должен разрабатываться нутрициологом, поскольку высока вероятность развития мукозита.
Восстановление после лечения
Восстановительные мероприятия у больных лимфомой желудка разрабатываются только индивидуально и в соответствии с опухолевым статусом, единого стандарта реабилитации не существует.
В «Евроонко» проводится комплексная медицинская реабилитация с использованием лечебной физкультуры и физиопроцедур, обязательно с нутритивной поддержкой и психологической помощью. На каждом лечебном этапе — отдельный комплекс мероприятий.
Осложнения
Для любой опухоли в желудке характерны осложнения:
- болевой синдром,
- изъязвление с острым или хроническим кровотечением,
- прободение стенки органа с развитием перитонита.
Кроме того, при лимфоме возможно снижение вырабатываемого нормальными иммунными клетками специфического гамма-глобулина IgG, что способствует частым инфекциям и воспалительным процессам. Недостаток компенсируется внутривенными введениями иммуноглобулина.
Во всех случаях перед началом химиотерапии проводится профилактика синдрома лизиса опухоли, возникающего при высокой чувствительности злокачественных клеток к лекарственным препаратам.
Рецидивы
MALT-лимфома желудка отличается медленным течением, тем не менее с увеличением стадии вероятность рецидива возрастает. Начиная со IIЕ стадии возврат опухоли возможен у каждого второго пациента, но средние сроки рецидива не определены из-за редкой встречаемости болезни. Отмечена способность некоторых рецидивных узлов к уменьшению без лечения — самокупирование.
При агрессивной лимфоме рецидивы возникают чаще, а ремиссии короче. Во всех ситуациях при рецидиве прибегают к химиотерапии.
Прогноз
Прогноз определяется морфологией, изначальным распространением и чувствительностью к лечению. Считается, что MALT-омы редко метастазируют, ассоциированные с H.Pylori более благоприятны по течению, нежели формы с генетическими аномалиями.
Выживаемость в зависимости от стадии не определена из-за редкости заболевания, но уменьшившаяся уже на антибиотиках лимфома IE стадии в 95% случаев позволит прожить много больше 5 лет.
Профилактика лимфомы желудка
Профилактика заболеваний желудка основана на раннем выявлении и эффективном лечении инфицирования H. Pylori, а также на ежегодной профилактической эндоскопии.
Список литературы:
- Capelle L.G., de Vries A.C., Looman C.W. et al. /Gastric MALT lymphoma: epidemiology and high adenocarcinoma risk in a nation-wide study// Eur J Cancer 2008; 44.
- Ferrucci P.F., Zucca E. /Primary gastric lymphoma pathogenesis and treatment: what has changed over the past 10 years// Brit J Haematol 2007; 136.
- Koch P., Probst A., Berdel W.E. et al. /Treatment results in localized primary gastric lymphoma: data of patients registered within the German multicenter study (GIT NHL 02/96)// J Clin Oncol 2005; 23.
- Levy M., Copie-Bergman C., Traulle C. et al. /Conservative treatment of primary gastric low-grade B-cell lymphomaof mucosa-associated lymphoid tissue: predictive factors of response and outcome// Am J Gastroenterol 2002; 97: 292–297.
Наши врачи


Главный врач сети "Евроонко", онколог, кандидат медицинских наук

Эндоскопист, доктор медицинских наук

По моему мнению Евроонко - это лучшая клиника в Москве на сегодня. После посещения с моей мамой нескольких платных клиник и многочасовых! ожиданий в очередях, регистратурах и пр., ответ из Евроонко.
Здесь продлили жизнь моей маме. Слов для благодарности нет. Когда в 4х больницах отказали в госпитализации (даже в платной) - ответ из Евроонко: «Приезжайте в любое время суток» - был просто Чудом.
Пациент 70 лет осенью 2020 года отметил появление болей в животе и прошёл обследование, установившее наличие у него опухоли желудка с метастатическим поражением регионарных лимфатических узлов. По .
Лечение пациентов проводится в соответствии со стандартами и рекомендациями наиболее авторитетных онкологических сообществ. «Евроонко» является партнёром Фонда борьбы с раком. ВНИМАНИЮ ПАЦИЕНТОВ: Рекомендации по лечению даются только после консультации у специалиста. Ваши персональные данные обрабатываются на сайте в целях его корректного функционирования. Если вы не согласны с обработкой ваших персональных данных, просим вас покинуть сайт. Оставаясь на сайте, вы даёте согласие на обработку ваших персональных данных.
Сведения и материалы, размещенные на сайте , подготовлены исключительно в информационных целях и не являются медицинской консультацией или заключением. Авторы информационных материалов сайта не могут гарантировать применимость такой информации для целей третьих лиц и не несут ответственности за решения третьих лиц и связанные с ними возможные прямые или косвенные потери и/или ущерб, возникшие в результате использования информации или какой-либо ее части, содержащейся на сайте.
Хромосомные аномалии при злокачественных лимфомах - прогноз
Флуоресцентная гибридизация in situ (FISH) – новейший молекулярно-цитогенетический метод исследования, в процессе которого детектируется наличие и локализация специфических ДНК-последовательностей на хромосомах. В данном исследовании, основанном на методе FISH, выявляются генетические аберрации, характерные для следующих онкогематологических заболеваний: хронический лимфолейкоз, MALT-лимфома и лимфома Беркитта.
Синонимы русские
Флуоресцентная гибридизация in situ, молекулярная диагностика онкогематологических заболеваний, хронический лимфолейкоз, лимфома Беркитта, лимфопролиферативные заболевания.
Синонимы английские
Analysis of all specific aberrations on paraffin slides (FISH Histology, quantitative), Lymphoproliferative disorders, Chronic lymphocytic leukemia (CLL), MALT lymphoma, Burkitt's lymphoma.
Метод исследования
Флуоресцентная гибридизация in situ (FISH)
Какой биоматериал можно использовать для исследования?
Образец ткани в парафиновом блоке
Как правильно подготовиться к исследованию?
Специальной подготовки не требуется. Для исследования используется уже предварительно подготовленный биологический материал (парафиновый блок с образцом биоматериала).
Преимущества исследования
- Является чувствительным методом для идентификации хромосомных аберраций при количествах лейкозных клеток менее 10 9 , обеспечивая при этом быстрый анализ большого (>500) числа клеток. Метод обладает высокой точностью для идентификации неизвестных фрагментов хромосомной ДНК.
- Для исследования могут быть использованы различные биоматериалы – аспираты тонкоугольной аспирационной биопсии, костного мозга, мазки крови, биоптаты, полученные на различных стадиях заболевания;
- Исследование FISH может быть применено как к метафазным, так и к интерфазным ядрам, то есть к неделящимся клеткам;
- Позволяет определить даже самые небольшие генетические аномалии, которые нельзя рассмотреть при помощи обычного микроскопа, при использовании соответствующих ДНК-зондов.
Общая информация об исследовании
Анализ с помощью флюоресцентной in situ гибридизации (fluorescence in situ hybridization, FISH) – молекулярно‐цитогенетический метод для идентификации генетических аберраций (отклонений от нормы). Изначально данный метод использовался как исследовательский для выявления наличия или отсутствия специфической ДНК последовательности в хромосомах, но благодаря прогностической и предсказательной ценности был внедрен в клиническую практику.
Метод основан на использовании флуоресцентно меченых ДНК-зондов, которые представляют собой искусственно синтезированные фрагменты ДНК (олигонуклеотиды), последовательность которых комплементарна последовательности ДНК исследуемых аберрантных хромосом. ДНК-зонды различаются по специфичности: для каждой хромосомной аномалии используются свои ДНК-зонды. Также зонды различаются по размеру: одни могут быть направлены к целой хромосоме, другие – к конкретному локусу (фрагменту хромосомы или гена).
После специальной процедуры – денатурации – молекула ДНК приобретает вид одноцепочечной нити. ДНК-зонд гибридизуется (связывается) с комплементарной ему нуклеотидной последовательностью и может быть обнаружен при помощи флуоресцентного микроскопа. Данное состояние интерпретируется как положительный результат FISH-теста. При отсутствии аберрантных хромосом несвязанные ДНК-зонды в ходе реакции "отмываются", что при исследовании с помощью флуоресцентного микроскопа определяется как отсутствие флуоресцентного сигнала (отрицательный результат FISH-теста). Метод позволяет оценить не только наличие флуоресцентного сигнала, но и его интенсивность и локализацию. Таким образом, FISH-тест – это еще и количественный метод.
FISH имеет широкие возможности в клинической онкологии для обнаружения хромосомных аномалий в опухолевых клетках. Метод позволяет исследовать генетический состав клетки, как во время митоза, так и в интерфазе. FISH имеет высокую чувствительность – позволяет обнаружить индивидуальные гены, кроме того, в одном препарате могут быть использованы несколько зондов с различными красителями.
FISH-анализ широко применяется при лимфопролиферативных заболеваниях, являясь в ряде случаев определяющим фактором для подтверждения диагноза.
Хронический лимфолейкоз (ХЛЛ) – самый частый вид лейкозов у взрослых. Характеризуется пролиферацией и увеличением в периферической крови количества морфологически зрелых лимфоцитов на фоне лимфоцитарной инфильтрации костного мозга, лимфатических узлов, селезенки и других органов. Клеточный субстрат хронического лимфолейкоза представлен чаще В-популяцией (около 30 %) и значительно реже — Т-лимфоцитами (около 70 %). В-лимфоциты при ХЛЛ не развиваются до плазматических клеток вследствие изменений в клеточном геноме. Это ведет к резкому уменьшению выработки иммуноглобулинов, к которым относятся все антитела. Заболевание чаще возникает у лиц старше 65 лет, у 10-15 % больных в возрасте чуть старше 50 лет. До 40 лет хронический лимфолейкоз возникает крайне редко. Мужчины болеют примерно в 2 раза чаще, чем женщины.
Симптомы обычно развиваются медленно, чаще выявляется случайно при обследовании по поводу других причин. При ХЛЛ наблюдаются следующие симптомы: кровоподтеки (если тромбоциты снижены), увеличение лимфатических узлов, печени или селезенка, чрезмерное потоотделение, ночная потливость, усталость, лихорадка, реинфекции, потеря аппетита, потеря веса.
Лимфома маргинальной зоны, ассоциированная со слизистыми (MALT, mucosa-associated lymphoid tissue) является третьим по распространенности подтипом неходжкинской лимфомы и составляет ~ 7 % всех неходжкинских лимфом. Средний возраст, характерный для выявления MALT-лимфомы - 61 год. Это одна из немногих неходжкинских лимфом, которая чаще поражает женщин (соотношение 1,1:1).
Показано, что приблизительно 90 % случаев МАLT-лимфом желудка связано с инфицированием H. рylori. У 70–80 % больных под влиянием эрадикационной (антихеликобактерной) терапии наблюдается регрессия MALT- лимфомы. Дифференциальная диагностику проводят с H. pylori-ассоциированным гастритом.
Лимфома Беркитта — агрессивная форма неходжкинской лимфомы. Развивается из В-лимфоцитов, характерна экстранодальная локализация опухоли, имеет склонность прорастать и распространяться за пределами лимфатической системы – в процесс вовлекается спинномозговая жидкость, кровь и костный мозг. Этиология до сих пор не выявлена. Одним из провоцирующих факторов считается длительная персистенция вируса Эпштейна-Барр в организме. Наиболее часто поражаются органы брюшной полости: тонкая кишка (чаще ее терминальный отдел), брыжейка, а также желудок, толстая кишка, брюшина, печень, селезенка. Специфическое поражение костного мозга наблюдается в 25-35 % случаев, центральной нервной системы - в 20-25 % случаев. Типично вовлечение почек, яичников, яичек, абдоминальных и забрюшинных лимфатических узлов, реже периферических лимфатических узлов. Чаще развивается у детей (в 30-50 % случаев), подростков и молодых людей (средний возраст 20-25 лет). Выделяют три основных типа лимфомы Беркитта:
- Африканский тип (эндемическая) - встречается в основном в Африке. Установлена связь африканского типа лимфомы с инфекцией вирусом Эпштейна-Барра.
- Европейский тип (спорадическая) - встречается во всем мире, поражает в основном детей и молодежь. Чаще всего опухоль образуется в кишечнике.
- Лимфома Беркитта, связанная с иммунодефицитом - встречается у пациентов с ослабленным иммунитетом, прежде всего у больных СПИДом и у людей после трансплантации органов, которые принимают иммуносупрессоры.
Симптомы варьируются в зависимости от типа: лихорадка, потеря веса, вздутие живота, искажение лицевых костей, ночная потливость, кишечная непроходимость, увеличенная щитовидная железа, увеличенные миндалины.
Для чего используется исследование?
- Для уточнения диагноза при подозрении на злокачественное заболевание крови, в том числе при Ph-отрицательных комплексных кариотипах, когда присутствует ген BCR/ABL, определяемый только методом FISH;
- Для повторного консультирования при подозрении на наличие злокачественного заболевания крови;
- Для выбора тактики лечения и прогноза заболевания, которые зависят от хромосомного состава опухоли;
- Чтобы определить наличие или отсутствие конкретной хромосомной аберрации.
Когда назначается исследование?
- При подозрении на наличие злокачественного заболевания крови, для выбора тактики лечения и оценки прогноза, который зависит от хромосомного состава опухоли.
- Для подтверждения диагноза при наличии клинических предпосылок: клиническая картина заболевания, изменения гемограммы, наличие специфических синдромов – гиперпластический, геморрагический, анемический и др.);
- Для контроля "минимальной остаточной болезни" после химиотерапии или пересадки костного мозга.
Что означают результаты?
Отсутствие аберрантных хромосом в исследуемом образце
Хромосомные аномалии, характерные для лимфопролиферативных заболеваний:
- перестройки гена ATM;
- трисомия 12 хромосомы (+12);
- моносомия, делеция 13 хромосомы –(del(13),-13);
- делеция ТР53 гена.
Прогностически значимыми являются следующие аберрации: делеция длинного плеча хромосомы 13 (13q-), трисомия 12 хромосомы, делеция длинного плеча хромосомы 11 (11q-). Делеция короткого плеча хромосомы 17 (17p-) является главным цитогенетическим маркером, непосредственно влияющим на терапевтическую тактику. Рекомендуется проводить скрининг на делецию 17p у всех пациентов, имеющих показания к началу терапии и/или при неэффективности стандартной терапии, особенно пациентам моложе 55 лет, которым может быть проведена аллогенная трансплантация.
- транслокация t(11;18)(q21;q21).
- транслокация t (14; 18) (q32; q21)
t(11;18) (q21;q21) - наиболее распространенная хромосомная транслокация, связанная с MALT-лимфомой, которую не отмечают при других вариантах лимфом. Транслокация t(11;18) ассоциируется с более агрессивным течением.
Второй наиболее частой транслокацией, идентифицированной с MALT-лимфомой, является t (14;18)(q32; q21).Приблизительно в 4 % MALT-лимфомы желудка и 8 % MALT-лимфомы легких выявляется t(1;14) (p22;q32).Характерным для MALT-лимфом является также нарушение нормальной активности важного супрессора опухоли — гена BCL10, что наблюдается при t(1;14)(p22;q32).
Важно отметить, что лимфомы MALT не несут транслокацию t (11; 14) (q13; q32), типичную для лимфомы мантийных клеток.
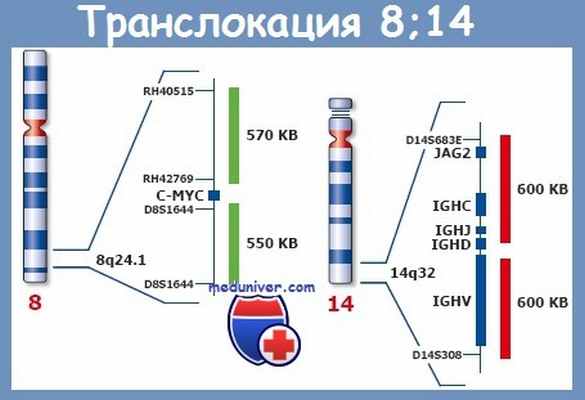
- перестройки С-MYC гена (t(8;14)(q24;q32)t(2;8)(p11;q24)t(8;22)(q24;q11)).
Выявление цитогенетического маркера лимфомы Беркитта – перестройки локуса гена C-MYC и транслокации t(8,14)(q24,q32) или ее вариантов t(2,8)(pl2,q24) или t(8,22)(q24,q11) позволяет диагностировать лимфому Беркитта. Перестройки гена C-MYC выявляется в 100 % случаев лимфомы Беркитта и является одним из главных диагностических критериев этого заболевания. В 80 % случаев встречается t(8;14)(q24;q32) перестройка локусов генов c-myc (8q24) и тяжелых цепей иммуноглобулинов Ig (14q32).
Изменения кариотипа являются независимым прогностическим фактором. При выявлении аберрации (13q-) можно прогнозировать стабильное состояние или медленное течение болезни и благоприятный ответ на терапию, если она является единственной (медиана выживаемости – 11 лет), в то время как остальные аберрации, в особенности (11q- ) и (17p- ) крайне неблагоприятны в прогностическом отношении (медианы выживаемости больных с трисомией 12 – 9,5 лет, ( 11q-) – 6,5 лет, (17p- ) – меньше 3 лет.
Кто назначает исследование?
Также рекомендуется
Литература
1. Иммуногистохимические методы: Руководство / Ed. by George L. Kumar, Lars Rudbeck.: DAKO / Пер. с англ. под ред. Г.А.Франка и П.Г.Малькова. – М., 2011. – 224 с.
2. Wan TS, Ma ES. Molecular cytogenetics: an indispensable tool for cancer diagnosis. Chang Gung Med J. 2012. Mar-Apr: 35(2): 96-110. Review. PubMed PMID: 22537925.
3. Steven H. Swerdlow Diagnosis of ‘double hit’ diffuse large B-cell lymphoma and B-cell lymphoma, unclassifiable, with features intermediate between DLBCL and Burkitt lymphoma: when and how, FISH versus IHC. Hematology . December 5, 2014 vol. 2014 no. 1 90-99
4. E. Zucca & Dreyling. Минимальные клинические рекомендации ESMO по диагностике, лечению и наблюдению при MALT - лимфоме желудка. Москва. 2010г.
5. М. Ж. Алексанян, Е. А. Асеева, А. И. Удовиченко, Е. В. Домрачева. Цитогенетические исследования в гематологии. Организационные аспекты. Гематология и трансфузиология, 2012, т. 57, № 4. С.23 – 27.
6. Хронический лимфолейкоз у взрослых. Клинические рекомендации. Национальное гематологическое общество Российское профессиональное общество онкогематологов. 2016г.
7. И.А. Крячок, Е.О. Ульянченко, Т.В. Кадникова, И.Б. Титоренко и др. MALT-лимфома: причины возникновения, патогенез, классификация, клиническая картина. Клиническая онкология, № 1 (25), 2017.
Хромосомные аномалии при злокачественных лимфомах - прогноз
Транслокации (8;14)(q24;q32), t(8;22)(q24;q11) и t(2;8)(p12;q24) при лимфоме
Транслокация (8;14)(q24;q32), t(8;22)(q24;q11) и t(2;8)(p12;q24) характерны для лимфомы Беркитта, но обнаруживается и при так называемых неберкиттовских лимфомах, однако со значительно меньшей частотой. При лимфоме Беркитта частота t(8;14) составляет 75—85 %, а вариантные транслокации — t(8;22) и t(2;8) — наблюдаются в 15—25 % случаев. Частота t(8;14)(q24;q32) при остальных лим-фопролиферативных заболеваниях в нашей стране не превышает 5 % .
При трех названных транслокациях происходит образование химерных генов за счет слияния участка протоонкогена с-MYC (хромосома 8) с регуляторными участками генов иммуноглобулинов. В результате изменяется экспрессия гена с-MYC, в норме регулирующего клеточное деление и апоптоз. Экспериментально доказана прямая связь между активацией с-MYC и развитием В-клеточных опухолей.
Для выявления t(8;14), кроме хромосомного анализа, можно использовать FISH и ПЦР. Необходимо отметить, что район 14q32, где расположены гены Н-цепей иммуноглобулинов (IgH), — самая частая область перестроек кариотипа, обнаруживаемых при В-клеточных опухолях.
Стандартное цитогенетическое исследование и FISH позволяют выявить t(11;14)(q13;q32) в злокачественных клетках у 50—75 % больных при лимфоме мантийной зоны и гораздо реже при хроническом лимфолейкозе, миеломе и некоторых других лимфомах. В результате этой перестройки регуляторные последовательности гена IgH (хромосома 14) перемещаются в район гена BCL-1 (хромосома 11), что приводит к аномальной экспрессии белка cyclin D1, дерегуляции клеточного цикла и неконтролируемой пролиферации лимфоидных клеток.
В настоящее время для определения t(11;14) используют FISH и ПЦР. Не исключено, что данные, опубликованные ранее, отражают не истинную частоту t(11;14) при отдельных лимфопролиферативных процессах, а трудности в диагностике лимфомы мантийной зоны. Истинную частоту t(11;14) при разных лимфоидных опухолях устанавливают сейчас, когда приобретен опыт в диагностике лимфомы мантийной зоны и дифференциальной диагностике этой и других морфологически сходных новообразований.
Транслокация (11;14)(q13;q32) на цитогенетических препаратах выглядит абсолютно одинаково при таких разных заболеваниях, как множественная миелома и лимфома мантийной зоны, однако точки разрыва, определяемые молекулярными методами, оказались разными. При миеломе они локализуются в районе JH или switch, а при лимфоме мантийной зоны — вблизи участков, которые являются мишенями для VDJ-рекомбиназ.
Самым надежным диагностическим методом при лимфоме мантийной зоны считается иммуногистохимическое обнаружение гиперэкспрессии белка cyclin D1. Нельзя забывать, что для 50—75 % больных с волосатоклеточным лейкозом тоже характерен этот маркер. В последнем случае гиперпродукция белка-маркера имеет другую причину (транслокации нет).
Лимфому мантийной зоны условно делят на 2 подгруппы: с гиперэкспрессией белка cyclin D1 и без гиперэкспрессии. Для 1-й подгруппы характерны агрессивное течение и небольшая продолжительность жизни, а у больных 2-й подгруппы прогноз относительно благоприятный. Есть предложение относить в группу лимфом мантийной зоны только те опухоли, в которых характерные морфологические особенности сочетаются с гиперэкспрессией белка cyclin D1.
При отсутствии последнего признака рекомендуется название «cyclin D1-негативная лимфома, морфологически сходная с лимфомой мантийной зоны».
Гиперэкспрессия белка cyclin D1 может наблюдаться при различных лимфомах и раках, поэтому данный маркер может считаться диагностическим признаком только в определенном контексте.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Молекулярные маркеры

Классическим примером гена-супрессора, осуществляющего негативную регуляцию клеточного цикла, является ген RB1. Наследственные формы рака молочной железы составляют около 5%. Мутации двух генов, BRCA1 и BRCA2, участвующих в репарации двухцепочечных разрывов, имеют практически равноценный вклад. Кроме рака молочной железы, герминальные мутации BRCA1 повышают риск возникновения рака яичников, а мутации BRCA2 предрасполагают к раку молочной железы у мужчин и раку поджелудочной железы. В то же время соматические мутации этих генов довольно редки при указанных новообразованиях. Спектр мутаций показывает, что их основная масса приводит к потери функции гена и представлена небольшими делециями и инсерциями, в том числе и в регуляторных областях генов. Две мутации BRCA1, 185delAG и 5382insC, составляют 20%, а мутации 617delT и 997del5 гена BRCA2 составляют почти 33%. Все мутации BRCA2, определяющие высокий риск развития рака яичников, локализуются в 3,3 т.п.н. экзона 11.
Наследственная форма неполипозного рака толстой кишки развивается в результате мутаций в генах MSH2 и MLH1, участвующих в образовании репарационных комплексов. Наиболее характерным признаком заболевания является нестабильность микросателлитных последовательностей. У женщин, имеющих герминальную мутацию в одном из этих генов, чаще всего возникают рак яичников и эндометрия. Среди мутаций MSH2 наиболее часто обнаруживается замена A→T в донорном сайте сплайсинга, приводящая к делеции 5 экзона. Для MLH1 преимущественного типа мутаций не показано, но установлено, что основным механизмом инактивации гена в опухолях является метилирование его промоторного района.
Предрасположенность к возникновению ретинобластомы обусловлена герминальной мутацией в одном из аллелей гена RB1, передающейся потомству по аутосомно-доминантному типу наследования с варьированием экспрессивности и неполной (90%) пенетрантностью. Опухоль развивается при инактивации неповрежденного аллеля гена, происходящей с высокой вероятностью в клетках сетчатки у детей в раннем возрасте или даже внутриутробно. Так как мишенью возникновения второй мутации могут быть и другие соматические клетки, то у пациентов, несущих герминальную мутацию в RB1, существует большой риск развития таких опухолей, как остеосаркома, лимфолейкоз, МРЛ, РМЖ, опухоли половых органов, поэтому за больными с наследственной формой заболевания необходимо наблюдение. Соматические мутации гена вызывают только ретинобластому, хотя нарушения функции RB1 обнаруживают во многих других опухолях, но уже как вторичные, являющиеся признаком дестабилизации генома опухолевой клетки.
Наследственные формы меланомы, синдрома диспластического невуса и атипичных родинок в ряде случаев вызываются герминальными мутациями гена-супрессора CDKN2A/p16. В семьях, несущих мутацию CDKN2A, кроме меланомы обнаруживаются опухоли поджелудочной железы, ОГШ. В то же время функциональная и структурная инактивация гена, обеспечивающего негативную регуляцию клеточного цикла путем ингибирования циклин-зависимых киназ, обнаруживается в различных типах опухолей, но преимущественно в мезотелиомах, спорадических меланомах и глиобластомах. Почти треть соматических мутаций гена представлена делециями, в то время как среди герминальных мутаций делеции и инсерции составляют не более 5%.
Нефробластома – генетически гетерогенное заболевание, но около трети случаев – результат инактивации гена-супрессора WT1.
Гемизиготные делеции хромосомного района 11p13 вызывают синдром WAGR, при котором кроме опухоли Вильмса обнаруживаются аномалии развития мочеполового тракта, аниридия и задержка физического и умственного развития.
Точковые мутации в 7-10 экзонах, кодирующих мотив цинковых пальцев, определяются у больных с синдромом Дениса-Драша, для которого характерна прогрессирующая нефропатия, псевдогермафродитизм, нефро- и гонадобластомы. Соматические точковые и структурные мутации гена выявлены в мезотелиомах, десмопластических мелко-кругло-клеточных опухолях и при острых лейкозах.
К хранителям клеточного цикла относятся оба гена NF1 и NF2, вызывающие нейрофиброматоз I и II типов, но выполняющие в геноме различную функцию. Первый обеспечивает негативную регуляцию онкогена Ras, второй стабилизирует клеточные контакты. Мутации и структурные перестройки первого гена вызывают шванномы периферической нервной системы, а второго – шванномы ЦНС и менингиомы. Изменений этих генов не обнаружено в опухолях другого происхождения.
Синдромы множественной эндокринной неоплазии I и II типов (МЭН 1, МЭН 2) возникают в результате мутаций в гене-супрессоре MEN1 и протоонкогене RET соответственно. В случае синдрома МЭН 1 могут развиваться карциноидные опухоли, ангиофибромы лица, липомы, коллагеномы, гастриномы и инсулиномы.
Герминальные миссенс-мутации RET выявлены у пациентов с синдромом МЭН 2A (медуллярный рак щитовидной железы, феохромоцитома, гиперплазия паращитовидных желез, ганглионейромы слизистой ротовой полости) и семейной формой медуллярного РЩЖ, причем мутации в основном затрагивают пять цистеиновых кодонов в 10 и 11 экзонах и на них приходится 97% и 85% всех мутаций в этих экзонах соответственно.
Спорадические мутации в тирозинкиназном домене гена более характерны для синдрома МЭН 2В, отличающегося от типа 2А более агрессивным течением и отсутствием гиперплазии паращитовидных желез и ганглионейром, а точковая мутация в экзоне 16 (T918C) была идентифицирована в 93% случаев.
Синдром фон Хиппель-Линдау наследуется аутосомно-доминантно с частотой 1 на 36 тыс. Среди злокачественных новообразований, являющихся диагностическим признаком и позволяющих выделить 4 типа заболевания, наиболее часто встречаются гемангиобластомы ЦНС, карциномы почки, феохромоцитомы и опухоли поджелудочной железы. При заболевании обнаруживаются мутации в гене VHL, клонированном в коротком плече хромосомы 3p25.5. Мутации представлены большими делециями, мутациями сдвига рамки считывания и миссенс-мутациями, повреждающими преимущественно 1 и 3 экзоны. Соматические мутации, повреждающие 1 и 2 экзоны гена, выявлены только в светлоклеточном раке почки. Почти в 20% опухолей обнаружено аномальное метилирование промоторной области гена.
Семейный аденоматозный полипоз желудочно-кишечного тракта – аутосомно-доминантное заболевание, вызываемое мутациями в гене APC. Вариантом заболевания является синдром Гарднера, при котором преимущественно поражается толстый кишечник. При заболевании достаточно часто выявляются гепатоцеллюлярный рак, опухоли щитовидной железы, медуллобластомы, фибромы и другие опухоли. Основным типом мутаций являются делеции и нонсенс-мутации, приводящие к преждевременной терминации синтеза белка. Герминальные мутации преимущественно повреждают первую половину гена, причем обнаружено два «горячих» кодона – 1061 и 1309. Мутации последнего кодона приводят к раннему появлению полипов и быстрой малигнизации. Соматические мутации в основном локализуются в районе кодонов 1286-1513, с преимущественным повреждением кодонов 1309 и 1450.
Синдром Горлина или базально-клеточных невусов – редкое аутосомно-доминантное заболевание (частота 1:57000), возникает в результате мутаций в гене-супрессоре PTC1, представленных в 86% делециями и инсерциями, равномерно распределенными по длине гена. Проявляется множественными аномалиями развития, невусами и различными новообразованиями, наиболее частыми среди которых являются базально-клеточные рак кожи, медуллобластомы, астроцитомы, фибромы и аденокарциномы яичников. Во всех опухолях у синдромальных пациентов и в спорадических базально-клеточных карциномах обнаруживается потеря гетерозиготности по маркерам района хромосомы 9q22.3.
Целая группа синдромов – Ковдена, Банаяна-Зонана, Банаяна-Райли-Рувалкаба и часть случаев ювенильного полипоза характеризуется сходными фенотипическими аномалиями и предрасположенностью к злокачественным новообразованиям. Причиной всех заболеваний является патология гена-супрессора PTEN. У больных наиболее часто встречаются рак молочной железы, предстательной железы, яичников, эндометрия, щитовидной железы. Мутации равномерно повреждают весь ген и представляют полный спектр. Только при синдроме Ковдена установлено, что 43% мутаций повреждают 5 экзон, а в экзонах 1, 4 и 9 мутаций не описано. Соматические делеции и мутации PTEN обнаружены в глиобластомах, раках простаты, молочной железы и неходжкинских лимфомах.
Герминальные миссенс-мутации гена-супрессора p53, участвующего в задержке клеточного цикла необходимой для репарации ДНК, приводят к довольно редкому синдрому Ли-Фраумени, при котором типичными являются опухоли мозга, саркомы костей и мягких тканей, лейкозы и РМЖ. Выявлена кластеризация мутаций в районе 14 кодонов (245-258). В то же время различные повреждения гена выявляют практически во всех типах опухолей. Частота миссенс-мутаций гена составляет 74%, сдвига рамки считывания – 11%, нонсенс-мутаций – 7%, мутаций сайта сплайсинга – 4%. Определено 7 «горячих» точек, повреждаемых мутациями: кодоны 130-142, 151-164, 171-181, 193-200, 213-223, 234-258 и 270-286, причем все они локализованы в эволюционно консервативном ДНК-связывающем домене, кодируемом экзонами 5-8. Мутации кодонов 157 и 179 наиболее часто выявляются при раке легкого, кодона 175 – при раке толстой кишки, кодона 248 – при плоскоклеточном раке головы и шеи, кодона 249 – при гепатоцеллюлярном раке и кодона 278 – при опухолях кожи.
Атаксия-телангиоэктазия – аутосомно-рецессивное заболевание, сопровождающееся ломкостью хромосом, причем ионизирующее излучение повышает нестабильность кариотипа. При заболевании наиболее часто обнаруживаются Т-клеточные лейкозы, В-лимфомы, медуллобластомы и глиомы. Причиной являются гомозиготные мутации или делеции гена ATM, регулирующего клеточный цикл, причем делеции составляют основную массу патологии. У гетерозиготных носителей герминальных мутаций часто возникают карциномы молочной железы. Основным типом являются мутации, приводящие к преждевременной терминации синтеза белка.
Недавно клонирован еще один ген, участвующий в репарации двухцепочечных разрывов ДНК. Герминальные мутации гена NBS1 приводят к синдрому Ниджмиген – очень редкому аутосомно-рецессивному заболеванию, основными признаками которого являются аномалии фенотипа, иммунодефицит, предрасположенность к возникновению опухолей (лимфома, лимфолейкоз, нейробластома) и нестабильность хромосом, чувствительных к ионизирующему излучению. Наиболее характерными мутациями гена являются делеции и инсерции, преимущественно повреждающие 6 и 7 экзоны, а мутация 675del5 выявлена у 90% пациентов.
Клонирование генов, вовлеченных в развитие наследственных онкологических заболеваний, практически полностью изменило тактику медико-генетического консультирования, определение конкретной мутации конкретного гена в семье позволяет эффективно проводить пренатальную диагностику.
Читайте также:
- Ядерное сканирование пищеварительного тракта
- Патологическая анатомия мастоидита. Альтеративно-пролиферативная стадия мастоидита
- Факультативные анаэробные грамотрицательные ферментирующие бактерии. Энтеробактерии. Бактерии семейства энтеробактерий. Свойства энтеробактерий.
- Erythema elevatum diutinum. Кольцевидная гранулема
- Синдром Розенберга—Хаториана: атрофия зрительных нервов, полинейропатия и глухота