Хромосомные перестройки. Делеции и дупликации
Добавил пользователь Валентин П. Обновлено: 29.01.2026
Хромосомные перестройки. Делеции и дупликации
В пределах одной хромосомы могут происходить разрывы И слияния, в результате этого появляется возможность инверсии генетического материала. У людей с инверсией хромосом видимые изменения фенотипа часто отсутствуют, однако, поскольку при такой перестройке может нарушаться конъюгация хромосом во время мейоза, у потомков могут произойти более серьезные аберрации.
Межхромосомные перестройки, различимые с помощью светового микроскопа и не сопровождаемые видимой утратой сегмента хромосомы, называют сбалансированными транслокациями. У 6-10% людей с явно сбалансированными хромосомными транелокациями выявляют клинические аномалии; это позволяет предположить разрыв одного или, возможно, двух тепов в месте транслокации. И таких ситуациях для выяснения причины следует провести анализ родительских хромосом.
Если у одного из родителей обнаружат такие же хромосомные перестройки при нормальном фенотипе, у ребенка следует искать другие причины заболевания.
Делеции и дупликации. Эти хромосомные аберрации представляют собой утрату или увеличение хромосомного материала. Многие клинические синдромы связывают с аберрациями в специфических участках хромосом. Минимальная делеция, видимая в световом микроскопе, связана с потерей значительного участка ДНК, длиной порядка 1 млн пар нуклеотидов, иными словами, потенциально утрачивается или разрушается не один ген.
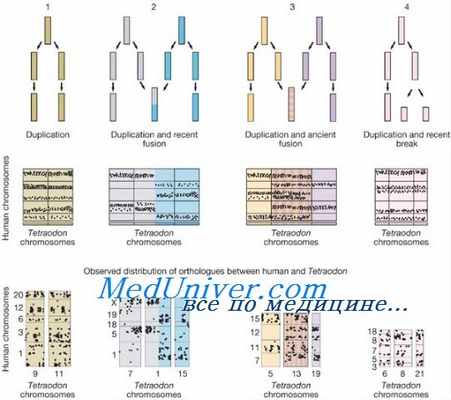
Целый ряд патологических состояний, которые ранее связывали с мутацией в одном локусе, обусловлен небольшими интерстициальными аберрациями, затрагивающими генные кластеры. Скорее всего, эти состояния определяются отсутствием нескольких, возможно, многих локусов на одной хромосоме, а не плейотропизмом проявления одной мутации, и поэтому их лучше считать геномными нарушениями. В прошлом их называли смежными генными дефектами.
В тех участках генома, где произошли такие делеции, наличие периодически повторяющихся нуклеотидных последовательностей предрасполагает к аберрантной рекомбинации, чем и объясняется удивительно высокая частота их возникновения. Такие дефекты могут передаваться по наследству, а их проявление в семье наследуется по законам Менделя как доминантное. Некоторые интерстициальные делеции встречаются довольно часто и являются основными причинами врожденных пороков сердца. Например, делеция участка 22q 11.2 служит причиной синдрома DiGeorge, а также отвечает более чем за 50% случаев разрывов дуги аорты типа В и 5% случаев кажущейся изолированной тетрады Fallot.
Применение сравнительной геномной гибридизации, осуществляемой с помощью молекулярных биочипов, повысило качество поиска делений и дупликаций. Чувствительность метода опережает возможность определения связи изменений генома с фенотипом.
Если у пациента с помощью молекулярного биочипа обнаружена аномалия, то, как и при хромосомных аберрациях, следует обследовать его родителей для выяснения связи этой аномалии с фенотипом.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
ХРОМОСО́МНЫЕ ПЕРЕСТРО́ЙКИ
ХРОМОСО́МНЫЕ ПЕРЕСТРО́ЙКИ (хромосомные аберрации), тип мутаций, характеризующихся изменением структуры хромосом. В основе Х. п. лежат спонтанные или индуцированные мутагенами разрывы хромосом с дальнейшим нетождественным воссоединением образовавшихся хромосомных фрагментов. Возможность существования такой негомологичной рекомбинации впервые предположил А. С. Серебровский (1929). Ныне механизм репарации ДНК с двунитевыми разрывами общепринят. При внутрихромосомных перестройках – делеции, дефишенси (концевые нехватки хромосом), дупликации, инверсии, транспозиции – происходят разрывы, затрагивающие одну хромосому или две гомологичные. Такие аберрации могут приводить к изменению количества генетич. материала или изменять только локализацию генов, не изменяя их баланс. При межхромосомных перестройках разрывы затрагивают две и более негомологичных хромосомы. Обмен фрагментами между ними приводит к изменению локализации генов, образованию новых групп сцепления. Особые типы Х. п. представляют собой «слияние» негомологичных хромосом (т. н. робертсоновские транслокации), образование кольцевых хромосом из нормальных «палочковидных» и др. Многие Х. п. могут вызывать изменение морфологич. признаков организма, оказывать на него неблагоприятное воздействие (см. Хромосомные болезни ). Однако некоторые Х. п. могут оказаться выгодными для популяции или вида и приобрести адаптивное и эволюционное значение. Так, дупликации отд. генов обеспечивают высокий уровень синтеза важных для организма молекул, создавая «запас прочности» при реализации функций этих молекул. Напр., у эукариот множественно дуплицированными являются гены, контролирующие структуру рибосомных и транспортных РНК, гистонов, тубулинов, актина и др. Дупликации генов могут приводить к появлению новых генов и псевдогенов : существуют гены, контролирующие структуру изоферментов , и т. н. семейства генов, кодирующих разные белки со сходной аминокислотной последовательностью (субъединицы гемоглобинов, вариабельные участки субъединиц иммуноглобулинов, константные участки тяжёлых цепей разных классов иммуноглобулинов, разл. типов гистонов). Следствием робертсоновских транслокаций являются различия кариотипов (разного числа хромосом) при сходстве генетич. материала близких видов животных: коз, бизонов и коров ( n= 30), овец и красных буйволов ( n= 27), овцебыков ( n= 24). С разл. целями Х. п. используют в генетич. анализе (напр., для картирования мутантных аллелей нормальных генов). Анализ частоты Х. п. в культуре клеток при действии изучаемого фактора позволяет быстро оценить его гено-токсикологич. эффект (мутагенность). Как правило, Х. п. выявляют и анализируют цитологически, существуют генетич. методы их исследования. См. также ст. Мутации и лит. при ней.
ПГТ хромосомных перестроек
Преимплантационное генетическое тестирование хромосомных перестроек — это одно из разновидностей преимплантационного генетического скрининга эмбрионов на наличие/отсутствие хромосомного дисбаланса в их геноме.
Помимо численных аномалий (трисомии, моносомии, полисомии, триплоидии, др.) хромосомный дисбаланс у эмбриона может быть представлен различными структурными аномалиями в самих хромосомах – хромосомные аберрации.
В основе таких хромосомных аномалий лежит первоначальное нарушение целостности хромосомы — разрывы. Повреждение ДНК в виде двухцепочечных разрывов может быть обусловлено внешними факторами (химиотерапией, ионизирующим излучением), структурные перестройки могут наследоваться от отца или матери.
В результате таких разрывов может произойти либо утрата генетического материала (делеция), либо удвоение участка хромосомы – дупликация, либо различные внутрихромосомные и межхромосомные перестройки.
Патогенное влияние структурных дефектов хромосом у эмбриона на исход беременности и на здоровье потомства зависит от типа хромосомной перестройки, её локализации на хромосоме, дозы генов, попавших в участок дефекта/аберрации. Урон, наносимый такими структурными хромосомными деффектами, выразится либо в прекращении эмбриогенеза (развития эмбриона), либо в рождении ребенка с патологическим синдромом.
Структурные хромосомные аномалии в виде делеций, дупликаций и транслокаций встречаются наиболее часто среди иных возможных структурных дефектов и признаны наиболее клинически значимыми.
Делеции – это потеря участка хромосом, потеря генов.
Дупликации – это, напротив, увеличение количества хромосомного материала.
Эти дефекты могут быть разных размеров - от крупных, доступных современным микроскопам, до микроделеций и микродупликаций, которые могут проявляться с достаточной частотой, но которые можно обнаружить только современными молекулярными методами с высоким разрешением (способностью их увидеть). Примеры наиболее частых врожденных синдромов после таких «поломок»: Ди Джорджи, Прадера-Вилли, Ангельмана, «кошачьего крика». Все они сопровождаются не только физическими расстройствами, но и умственной отсталостью.
Транслокации – ещё один из не менее клинически значимых дефектов хромосом. Это межхромосомная перестройка, когда происходит перенос участка из одной хромосомы на другую, негомологичную. Носительство транслокаций встречается практически у каждого из 500 человек, но лишь в 5,5% случаев носительства сбалансированных хромосомных перестроек, по данным наблюдений специалистов, не влияет на реализацию репродуктивной функции.
Особое опасение перед переносом эмбриона в полость матки вызывает риск формирования несбалансированной транслокации у эмбриона, когда родители являются скрытыми носителями сбалансированных перестроек. Сбалансированные перестройки у будущих родителей могут никак не проявлять себя. Проблема всплывает при зачатии, когда сбалансированная перестройка может явиться причиной формирования несбалансированной у эмбриона. Беременность эмбрионом с такой поломкой может закончиться самопроизвольным абортом или привести к появлению ребенка с врожденными дефектами и/или умственной отсталостью.
Обнаружить за одно исследование максимально все возможные такие «поломки» в хромосомном материале эмбрионов могут полногеномные тесты на основе двух современных технологий последнего поколения - aCGH и NGS, которые мы и используем в нашей лаборатории.
XI Международная студенческая научная конференция Студенческий научный форум - 2019

Типы хромосомных аберраций в эукариотической клетке.
Текст работы размещён без изображений и формул.
Полная версия работы доступна во вкладке "Файлы работы" в формате PDF
Хромосомные аберрации (хромосомные мутации, хромосомные перестройки) — изменения структуры хромосом. Классифицируют делеции (удаление участка хромосомы), инверсии (изменение порядка генов участка хромосомы на обратный), дупликации (повторение участка хромосомы), транслокации (перенос участка хромосомы на другую). Хромосомные перестройки носят, как правило, патологический характер и нередко приводят к гибели организма. Показано значение хромосомных перестроек в видообразовании и эволюции.
ВОЗНИКНОВЕНИЕ ХРОМОСОМНЫХ АББЕРАЦИЙ
В ходе кроссинговера образуются разрывы хромосом, которые затем репарируются. Нарушения процесса репарации могут привести к появлению хромосомных перестроек. Разрывы хромосом и, как следствие, образование перестроек происходят под действием различных мутагенных факторов: физической (ионизирующее излучение), химической или биологической (транспозоны, вирусы) природы. Также некоторые хромосомные перестройки (делеции) характерны для носителей специфических сайтов ломкости.
Различают терминальные (утрата концевого участка хромосомы) и интеркалярные (утрата участка на внутреннем участке хромосомы) делеции. Если после образования делеции хромосома сохранила центромеру, она аналогично другим хромосомам передается при делении, участки же без центромеры как правило утрачиваются. При конъюгации гомологов во время кроссинговера у нормальной хромосомы на месте делеции в мутировавшей хромосоме образуется т. н. делеционная петля, которая компенсирует отсутствие делетированного участка.
Исследованные делеции редко захватывает протяженные участки хромосом, обычно такие аберрации летальны. Самым хорошо изученным заболеванием, обусловленным делецией, является синдром кошачьего крика, описанный в 1963 году Джеромом Леженом. В его основе лежит делеция небольшого участка короткого плеча 5 хромосомы. Для больных характерен ряд отклонений от нормы: нарушение функций сердечно-сосудистой, пищеварительной систем, недоразвитие гортани (с характерным криком, напоминающим кошачье мяуканье), общее отставание развития, умственная отсталость, лунообразное лицо с широко расставленными глазами. Синдром встречается у 1 новорожденного из 50000.
Другой интересной делецией является делеция в гене, кодирующем рецептор CCR5. Этот рецептор используется вирусом иммунодефицита человека (ВИЧ) для распознавания своей цели — Т-лимфоцитов. Продукта гена с делецией получил название CCR5-Δ32, этот вариант CCR5 не узнается ВИЧ, и носители такой мутации к ВИЧ невосприимчивы (это порядка 10 % европейцев).
Дупликации появляются в результате неравного кроссинговера (в этом случае второй гомолог несет делецию) или в результате ошибки в ходе репликации. При конъюгации хромосомы с дупликацией и нормальной хромосомы как и при делеции формируется компенсационная петля.
Практически у всех организмов в норме наблюдается множественность генов, кодирующих рРНК (рибосомальную РНК). Это явление назвали избыточностью генов. Так у E. coli на рДНК (ДНК, кодирующее рРНК) приходится 0,4 % всего генома, что соответствует 5-10 копиям рибасомальных генов.
Другой пример дупликации — мутация Bar у Drosophila, обнаруженная в 20-х годах XX века Т. Морганом и А. Стертевантом. Мутация обусловлена дупликацией локуса 57.0 X-хромосомы. У нормальных самок (B+/B+) глаз имеет 800 фасеток, у гетерозиготных самок (B+/B) глаз имеет 350 фасеток, у гомозигот по мутации (B/B) — всего 70 фасеток. Обнаружены также самки с трижды повторенным геном — double Bar (BD/B+).
В 1970 году Сусумо Оно в монографии «Эволюция путем дупликации генов» разработал гипотезу об эволюционной роли дупликаций, поставляющих новые гены, не затрагивая при этом функций исходных генов. В пользу этой идеи говорит близость ряда генов по нуклеотидному составу, кодирующих разные продукты. Это трипсин и хемотрипсин, гемоглобин и миоглобин и ряд других белков.
Различают парацентрические (инвертированный фрагмент лежит по одну сторону от центромеры) и перицентрические (инвертированный фрагмент лежит по разные стороны от центромеры) инверсии. При инверсиях не происходит потери генетического материала, потому как таковые инверсии как правило не влияют на фенотип, но если в инверсионной гетерозиготе (то есть организме, несущем как нормальную хромосому, так и хромосому с инверсией) происходит кроссинговер, то существует вероятность формирования аномальных хроматид. В случае парацентрической инверсии образуется одна нормальная и одна инвертированная (фенотипически нормальная) хроматиды, дицентрическая хроматида с дупликацией и делецией (при расхождении хроматид она обычно разрывается на две) и ацентрическая хроматида с дупликацией и делецией (обычно утрачивается). В случае перицентрической инверсии образуется одна нормальная и одна инвертированная хроматиды, а также две хроматиды с дупликацией и делецией. Гаметы, несущие дефектные хромосомы, обычно не развиваются или погибают на ранних этапах эмбриогенеза. Но гаметы с инвертированной хромосомой развиваются в организмы, 50 % гамет которых нежизнеспособны. Т.о. мутация сохраняется в популяции.
У человека наиболее распространенной является инверсия в 9 хромосоме, не вредящая носителю, хотя существуют данные, что у женщин с этой мутацией существует 30 % вероятность выкидыша.
Существует несколько форм транслокации:
• собственно транслокация (перенос участка с одной негомологичной хромосомы на другую);
• реципрокная транслокация (две негомологичные хромосомы обмениваются участками);
• робертсоновская транслокация (две негомологичные хромосомы объединяются в одну);
• транспозиция (перенос участка хромосомы на другое место на той же хромосоме).
Транслокация, реципрокная транслокация и транспозиция, которые не сопровождаются утратой генетического материала (т. н. сбалансированные транслокации), часто не проявляются фенотипически. Однако, как и в случае с инверсиями, в процессе гаметогенеза часть сформированных гамет несет летальные аберрации. К примеру, в случае реципрокной транслокации обычно выживает не более 50 % зигот.
Примером транслокации может служить т. н. семейный синдром Дауна. При этом заболевании у одного из родителей обнаруживается фенотипически непроявляющаяся транслокация 21 хромосомы на 14. У такого человека с вероятностью в 1/4 образуются гаметы с двумя 21 хромосомами (одна свободная и одна траслоцированная). При слиянии такой гаметы с нормальной образуется трисомик по 21 хромосоме.
Другой пример — транслокация типа Philadelphia, транслокация между 9 и 22 хромосомами. В 95 % случаев эта мутация является причиной одной из форм хронической лейкемии (chronic myelogenous leukemia).
Робертсоновские транслокации, возможно, являются причиной различий между числом хромосом у близкородственных видов. Существуют данные, что два плеча 2-й хромосомы человека соответствуют 12 и 13 хромосомам шимпанзе. Возможно, 2-я хромосома образовалась в результате робертсоновской транслокации двух хромосом обезьяноподобного предка человека. Таким же образом объясняют тот факт, что различные виды дрозофилы имеют от 3 до 6 хромосом.
Робертсоновские транслокации привели к появлению в Европе нескольких видов-двойников (хромосомные расы) у мышей группы видов Mus musculus, которые, как правило, географически изолированы друг от друга. Набор и, как правило. экспрессия генов при робертсоновских транслокациях не изменяются, поэтому виды практически неотличимы внешне. Однако они имеют разные кариотипы, а плодовитость при межвидовых скрещиваниях резко понижена.
ВСЕ ХРОМОСОМНЫЕ ПЕРЕСТРОЙКИ МОГУТ БЫТЬ ПОДРАЗДЕЛЕНЫ НА:
При сбалансированных перестройках изменяется порядок сегментов (локусов, генов) на хромосоме, но не происходит количественных нарушений генетического материала. (Например, инверсия и взаимные транслокации).
При возникновении несбалансированных перестроек всегда имеет место нарушение «дозы» определенных сегментов хромосомы. Это сопровождается изменением баланса генов и манифестацией той или иной формы хромосомной болезни.
СИНДРОМ ВОЛЬФА-ХИРШХОРНА (делеция короткого плеча хромосомы 4)
• Популяционная частота - 1:100000.
• обусловлен делецией сегмента короткого плеча хромосомы 4.
• Клинически характеризуется мВПР (микроцефалия, клювовидный нос, гипертелоризм, эпикант, аномальные ушные раковины, расщелины верхней губы и нёба, аномалии глазных яблок, антимонголоидный разрез глаз, маленький рот, пороки внутренних органов) с последующей резкой задержкой физического и психомоторного развития.
• Жизнеспособность детей резко снижена. Большинство умирают в возрасте до 1 года.
СИНДРОМ КОШАЧЬЕГО КРИКА (делеция короткого плеча хромосомы 5)
• Популяционная частот 1:45000 – 1:50000.
• Синдром обусловлен делецией короткого плеча 5 хромосомы.
• Кариотип: 46,ХХ, del 5p или 46,XY,del 5p.
• Специфический плач, напоминающий кошачье мяуканье или крик. Он обусловлен изменением гортани (сужение, мягкость хрящей, уменьшение надгортанника, необычная складчатость слизистой оболочки). С возрастом этот симптом исчезает.
СИНДРОМ ЧАСТИЧНОЙ ТРИСОМИИ ПО КОРОТКОМУ ПЛЕЧУ ХРОМОСОМЫ 9
• Клиническая картина многообразна и включает внутриутробные и постнатальные нарушения развития: задержку роста, умственную отсталость, микробрахицефалию, антимонголоидный разрез глаз, энофтальм (глубоко посаженные глаза), гипертелоризм, округлый кончик носа, опущенные углы рта, низко расположенные оттопыренные ушные раковины с уплощенным рисунком, гипоплазию (иногда дисплазию) ногтей. Врожденные пороки сердца обнаружены у 25% больных.
СИНДРОМ ПРАДЕРА-ВИЛЛИ И АНГЕЛЬМАНА
• у 70% больных наблюдается частичная делеция длинного плеча 15-й хромосомы (отцовская аллель), у 5% заболевание связано с другими перестройками хромосомы 15.
• Характерные внешние признаки: череп со сдавленной с боков лобной частью, миндалевидный разрез глаз, опущенные углы рта, маленькие стопы и кисти)
Наблюдается отставание умственного развития, поведенческие нарушения, задержка физического развития, низкорослость, гипотония, гипогонадизм.
Синдром Ангельмана – генетическое заболевание, для которого характерны психическая задержка развития, судорожные припадки, нарушения сна, хаотичные движении рук, частый смех и практически постоянная улыбка.
• Встречаемость 1:20 000
• Кариотип больных с синдромом Ангельмана — 46 XX или XY, 15р−.
• Продолжительность жизни 20-50 лет
1. Курчанов, Н. А. Генетика человека с основами общей генетики / Н.А. Курчанов. - М.: СпецЛит, 2009.
2. Гнатик, Е. Н. Генетика человека. Былое и грядущее / Е.Н. Гнатик. - М.: ЛКИ, 2010
3. Баранов, В. С. Цитогенетика эмбрионального развития человека / В. С. Баранов, Т. В. Кузнецова. -Спб. : Изд-во Н-Л, 2007. - 640 с.
4. Клаг У., Каммингс М. Основы генетики — М.: Мир, 2007.
5. Биология. Книга 1. Под ред. акад. РАМН Ярыгина В. Н. — М.: Высшая школа, 2003.
6. Грин Н. и др., Биология — М.: Мир, 1990. Т. 1-3.
7. Жимулев И. Ф. Общая и молекулярная генетика. — Новосибирск: Изд-во НГУ, 2003.
8. Р.В. Тузова, Н.А. Ковалев. Молекулярно-генетические механизмы эволюции органического мира. Генетическая и клеточная инженерия. – Минск: Беларусь, 2010
Хромосомные перестройки
Хромосомные аберрации (хромосомные мутации, хромосомные перестройки) — тип мутаций, которые изменяют структуру хромосом. Классифицируют делеции (утрата участка хромосомы), инверсии (изменение порядка генов участка хромосомы на обратный), дупликации (повторение участка хромосомы), транслокации (перенос участка хромосомы на другую), а также дицентрические и кольцевые хромосомы. Известны также изохромосомы, несущие два одинаковых плеча. Если перестройка изменяет структуру одной хромосомы, то такую перестройку называют внутрихромосомной (инверсии, делеции, дупликации, кольцевые хромосомы), если же двух разных, то межхромосомной (дупликации, транслокации, дицентрические хромосомы). Хромосомные перестройки подразделяют также на сбалансированные и несбалансированные. Сбалансированные перестройки (инверсии, реципрокные транслокации) не приводят к потере или добавлению генетического материала при формировании, поэтому их носители, как правило, фенотипически нормальны. Несбалансированные перестройки (делеции и дупликации) меняют дозовое соотношение генов, и, как правило, их носительство сопряжено с клиническими отклонениями от нормы.
Хромосомные перестройки играют определенную роль в эволюционном процессе и видообразовании, в нарушении фертильности, в онкологических и врождённых наследственных заболеваниях человека.
Хромосомные перестройки были открыты у дрозофил при помощи генетического анализа. В некоторых скрещиваниях соотношение числа потомков в разных классах сильно отличалось от ожидаемого, и это объяснили наличием перестроек в хромосомах родителей. Делеции, дупликации и транслокации обнаружил К. Бриджес в 1916, 1919 и 1923 годах, соответственно. Первую инверсию описал А. Стёртевант в 1921 году, сравнивая порядок генов в хромосоме 3 у D.melanogaster и D.simulans. Первые наблюдения хромосомных перестроек были сделаны на политенных хромосомах слюнных желез. Лишь спустя некоторое время существование перестроек было доказано цитологически на митотических хромосомах. Однако проще всего перестройки можно увидеть в политенных хромосомах у гетерозиготных особей, благодаря образованию петель и крестообразных структур. Также перестройки можно увидеть в профазе мейоза при образовании синаптонемных комплексов, где, благодаря синапсису гомологичных хромосом, также образуются петли и крестообразные структуры. [1] :1
Содержание
Возникновение хромосомных аберраций
Основной предпосылкой для возникновения хромосомных перестроек является появление в клетке двунитевых разрывов ДНК, то есть разрывов обоих нитей спирали ДНК в пределах нескольких п.о. Двунитевые разрывы ДНК возникают в клетке спонтанно или под действием различных мутагенных факторов: физической (ионизирующее излучение), химической или биологической (транспозоны, вирусы) природы. Двунитевые разрывы ДНК возникают запрограммированно во время профазы I мейоза, а также при созревании Т- и B-лимфоцитов во время специфической соматической (V(D)J рекомбинации. Нарушения и ошибки процесса воссоединения двунитевых разрывов ДНК приводят к появлению хромосомных перестроек.
Классификация
Делеции
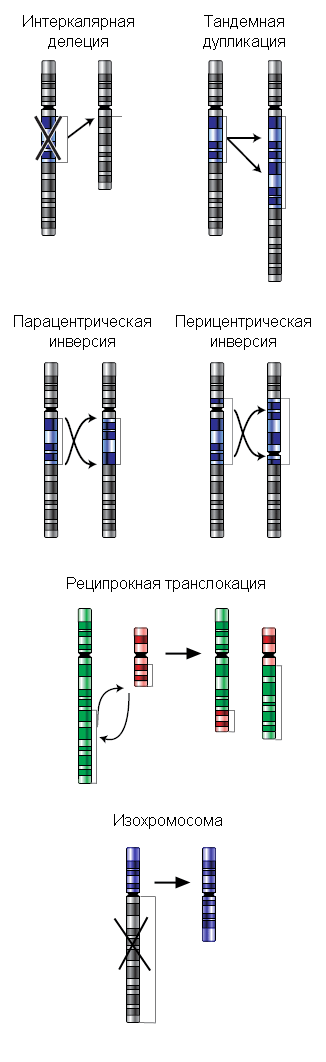
Различают терминальные (утрата концевого участка хромосомы) и интеркалярные (утрата участка на внутреннем участке хромосомы) делеции. Если после образования делеции хромосома сохранила центромеру, она аналогично другим хромосомам передается при митозе, участки же без центромеры, как правило, утрачиваются. При конъюгации гомологичных хромосом во время мейоза у нормальной хромосомы на месте, соответствующем интеркалярной делеции у дефектной хромосомы, образуется делеционная петля, которая компенсирует отсутствие делетированного участка.
Врождённые делеции у человека редко захватывает протяженные участки хромосом, обычно такие аберрации приводят к гибели эмбриона на ранних этапах развития. Самым хорошо изученным заболеванием, обусловленным достаточно крупной делецией, является синдром кошачьего крика, описанный в 1963 году Жеромом Леженом. В его основе лежит делеция участка короткого плеча 5 хромосомы. Для больных характерен ряд отклонений от нормы: нарушение функций сердечно-сосудистой, пищеварительной систем, недоразвитие гортани (с характерным криком, напоминающим кошачье мяуканье), общее отставание развития, умственная отсталость, лунообразное лицо с широко расставленными глазами. Синдром встречается у 1 новорожденного из 50000.
Современные методы выявления хромосомных нарушений, прежде всего флуоресцентная гибридизация in situ, позволили установить связь между микроделециями хромосом и рядом врождённых синдромов. Микроделециями, в частности, обусловлены давно описанные синдром Прадера-Вилли и синдром Вильямса.
Дупликации
Дупликации представляют собой класс перестроек, который объединяет как внутри- , так и межхромосомные перестройки. Вообще, любая дупликация — это появление дополнительной копии участка хромосомы, которая может располагаться сразу за тем районом, который дуплицирован, тогда это тандемная дупликация, либо в новом месте или в другой хромосоме. Новая копия может образовать отдельную маленькую хромосому со своими собственными теломерами и центромерой, тогда это свободная дупликация [1] :2 . Тандемные дупликации появляются в половых клетках при мейозе в результате неравного кроссинговера (в этом случае второй гомолог несет делецию) или в соматических клетках в результате неаллельной гомологичной рекомбинации при репарации двунитевого разрыва ДНК. В процессе кроссинговера у гетерозиготы при конъюгации хромосомы с тандемной дупликацией и нормальной хромосомы, как и при делеции, формируется компенсационная петля.
Практически у всех организмов в норме наблюдается множественность генов, кодирующих рРНК (рибосомальную РНК). Это явление назвали избыточностью генов. Так у E. coli на рДНК (ДНК, кодирующее рРНК) приходится 0,4 % всего генома, что соответствует 5-10 копиям рибосомальных генов.
Другой пример дупликации — мутация Bar у Drosophila, обнаруженная в 20-х годах XX века Т. Морганом и А. Стёртевантом. Мутация обусловлена дупликацией локуса 57.0 X-хромосомы. У нормальных самок (B + /B + ) глаз имеет 800 фасеток, у гетерозиготных самок (B + /B) глаз имеет 350 фасеток, у гомозигот по мутации (B/B) — всего 70 фасеток. Обнаружены также самки с трижды повторенным геном — double Bar (B D /B + ).
В 1970 году Сусумо Оно в монографии «Эволюция путем дупликации генов» разработал гипотезу об эволюционной роли дупликаций, поставляющих новые гены, не затрагивая при этом функций исходных генов. В пользу этой идеи говорит близость ряда генов по нуклеотидному составу, кодирующих разные продукты. Это трипсин и химотрипсин, гемоглобин и миоглобин и ряд других белков.
Инверсии
Инверсией называют поворот участка хромосомы на 180°. Различают парацентрические (инвертированный фрагмент лежит по одну сторону от центромеры) и перицентрические (инвертированный фрагмент лежит по разные стороны от центромеры) инверсии. При инверсиях не происходит потери генетического материала, поэтому инверсии, как правило, не влияют на фенотип носителя. Однако, если у гетерозигот по инверсиям (то есть у организма, несущего как нормальную хромосому, так и хромосому с инверсией) в процессе гаметогенеза при мейозе происходит кроссинговер в пределах инвертированного участка, то существует вероятность формирования аномальных хромосом, что в свою очередь может привести к частичной элиминации половых клеток, а также формировании гамет с несбалансированным генетическим материалом.
Более 1% человеческой популяции являются носителями перицентрической инверсии в 9 хромосоме, которую считают вариантом нормы [2]
Транслокации
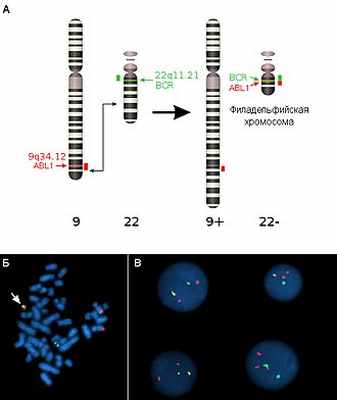
Транслокации представляют собой межхромосомную перестройку, при которой происходит перенос участка одной хромосомы на другую. Отдельно выделяют реципрокные транслокации (когда две негомологичные хромосомы обмениваются участками) и Робертсоновские транслокации, или центрические слияния (при этом две негомологичные акроцентрические хромосомы объединяются в одну с утратой материала коротких плеч). Первым центрические слияния описал американец У.Робертсон (W.R.B.Robertson) в 1916 г., сравнивая кариотипы близких видов саранчовых.
Реципрокные транслокации не сопровождаются утратой генетического материала, их также называют сбалансированными транслокациями, они, как правило, не проявляются фенотипически. Однако, у носителей реципрокных транслокаций половина гамет несёт несбалансированный генетический материал, что приводит к снижению фертильности, повышенной вероятности спонтанных выкидышей и рождения детей с врождёнными аномалиями. Частота гетерозигот по реципрокным транслокациям оценивается как 1 на 600 супружеских пар. Реальный риск рождения детей с несбалансированным кариотипом определяется характером реципрокной транслокации (спецификой хромосом, вовлеченных в перестройку, размерами транслоцированных сегментов) и может достигать 40 %.
Примером реципрокной транслокации может служить транслокация типа «филадельфийская хромосома» (Ph) между хромосомами 9 и 22. В 95 % случаев именно эта мутация в гемопоэтических клетках-предшественниках является причиной хронического миелобластного лейкоза. Эту перестройку описали П.Новелл (P.Nowell) и Д.Хангерфорд (D.Hungerford) в 1960 г. и назвали в честь города в США, где оба работали. В результате этой транслокации ген ABL1 из хромосомы 9 объединяется с геном BCR хромосомы 22. Активность нового химерного белка приводит к нечувствительности клетки к воздействию факторов роста и вызывает её безудержное деление.
Робертсоновские транслокации являются одним из наиболее распространенных типов врождённых хромосомных аномалий у человека. По некоторым данным, их частота составляет 1:1000 новорожденных. Их носители фенотипически нормальны, однако у них существует риск самопроизвольных выкидышей и рождения детей с несбалансированным кариотипом, который существенно варьирует в зависимости от хромосом, вовлеченных в слияние, а также от пола носителя. Большинство Робертсоновских транслокаций (74 %) затрагивают хромосомы 13 и 14. В структуре обращаемости на пренатальную диагностику лидерами оказываются носители der(13;14) и der(14;21) [3] :1 . Последний случай, а именно, Робертсоновская транслокация с участием хромосомы 21 приводит к так называемому «семейному» (наследуемому) синдрому Дауна.
Робертсоновские транслокации, возможно, являются причиной различий между числом хромосом у близкородственных видов. Показано, что два плеча 2-й хромосомы человека соответствуют 12 и 13 хромосомам шимпанзе. Возможно, 2-я хромосома образовалась в результате робертсоновской транслокации двух хромосом обезьяноподобного предка человека. Таким же образом объясняют тот факт, что различные виды дрозофилы имеют от 3 до 6 хромосом. Робертсоновские транслокации привели к появлению в Европе нескольких видов-двойников (хромосомные расы) у мышей группы видов Mus musculus, которые, как правило, географически изолированы друг от друга. Набор и, как правило, экспрессия генов при робертсоновских транслокациях не изменяются, поэтому виды практически неотличимы внешне. Однако они имеют разные кариотипы, а плодовитость при межвидовых скрещиваниях резко понижена.
Изохромосомы
Изохромосомы состоят из двух копий одного плеча хромосомы, соединенных центромерой таким образом, что плечи образовавшейся хромосомы представляют собой зеркальные «отражения» друг друга. В определенном смысле изохромосома представляет собой гигантскую инвертированную дупликацию размером с целое плечо и делецию другого плеча. Пациенты с 46 хромосомами, из которых одна представляет собой изохромосому, являются моносомиками по генам утраченного хромосомного плеча и трисомиками по генам, присутствующим в изохромосоме. Если изохромосома является добавочной, то данный пациент является тетрасомиком по генам, представленным в изохромосоме. В целом, чем меньше изохромосома, тем меньше генетический дисбаланс, и тем более вероятно выживание плода или ребенка с такой перестройкой. Следовательно, не удивительно, что наиболее частые из описанных случаев аутосомных изохромосом вовлекают хромосомы с маленькими плечами. Некоторые из наиболее частых участников формирования изохромосом — это короткие плечи хромосом 5, 8, 12, 18 [4] .
Для объяснения возникновения изохромосом можно предположить два механизма: (1) вследствие аномального поперечного разделения центромеры при делении клетки или (2) в результате неправильного слияния концов изохроматидного разрыва, образовавшегося в прицентромерной области [3] :2 .
Сайты ломкости
В 70-х годах XX века было обнаружено явление повышенной ломкости хромосом в определённых сайтах — при окраске метафазных хромосом некоторых индивидов одни и те же участки хромосом в части клеток имели разрыв или пробел в окрашивании. В дальнейшем было показано, что разрывы и пробелы появляются в определённых хромосомных сайтах при умеренном репликативном стрессе в клетках у всех людей. Первый тип ломких сайтов получил название редкие, или наследуемые, фрагильные сайты, а второй — обычные, или конститутивные, фрагильные сайты [5] .
Наследуемые фрагильные сайты находят у пациентов, у которых экспансия микро- или минисателлитных повторов ведёт к «хрупкости» хромосомы в месте такого повтора. Большинство наследуемых фрагильных сайтов не связано с какой-либо клинически значимой патологией, кроме наследуемого сайта ломкости FRAXA, который наблюдается у больных синдромом хрупкой Х-хромосомы (Синдром Мартина — Белл). Наследуемый фрагильный сайт FRAXA локализуется в 5'-нетранслирумой области гена FMR1 и содержит повтор из триплетов ЦЦГ. Аномальная длина этого повтора вызывает гиперметилирование промотора гена FMR1 и, как следствие, нарушение экспрессии гена [6] .
Конститутивные фрагильные сайты вызывают особенный интерес, потому что они являются «горячими точками» для хромосомных перестроек в различных раковых заболеваниях. Природа явления ломкости хромосом до конца не изучена. Известно, что конститутивные фрагильные сайты, как правило, ассоциированы с позднореплицирующимся хроматином [7] . Они нередко находятся в пределах очень длинных генов (около 1 млн п.о. и более), таких как FHIT и WWOX [8] . Многие обычные сайты ломкости являются тканеспецифичными. Последние исследования связывают ломкость хромосом с дефицитом сайтов инициации репликации в этих районах. Недостаточность сайтов инициации в конце S-фазы клеточного цикла может приводить к локальной незавершенности процесса репликации при репликативном стрессе и формировании в некоторых случаях двунитевого разрыва ДНК [9] .
Хромосомные аберрации и мутагенные воздействия
Мутагенные воздействия, вызывающие двунитевые разрывы ДНК, приводят к появлению хромосомных перестроек в клетках. Самым хорошо охарактеризованным мутагеном, индуцирующим хромосомные аберрации, является ионизирующее излучение. Родоначальником радиационной цитогенетики считается Карл Сакс, чья фундаментальная работа "Chromosome Aberrations Induced by X-Rays" была опубликована в 1938 году [10] . Для классификации радиоиндуцированных хромосомных нарушений создана собственная классификация аберраций, которая лишь частично совпадает с классификацией, используемой в медицинской генетике. В этой классификации выделяют аберрации хромосомного и хроматидного типа, которые, в свою очередь, могут быть обменными и простыми, стабильными и нестабильными. Тип хромосомных аберраций в значительной степени обусловлен фазой клеточного цикла, на котором находилась клетка в момент облучения.
При облучении клеток на стадии G0-G1 клеточного цикла в метафазах затем наблюдают аберрации хромосомного типа. Наиболее характерными среди них являются так называемые обменные хромосомные аберрации, а именно: дицентрические и кольцевые хромосомы, образующиеся в результате неправильного воссоединения двунитевых разрывов ДНК. Дицентрические и кольцевые хромосомы, как правило, сопровождаются фрагментом хромосомы, не содержащем центромеры, т. н. хромосомным ацентрическим фрагментом. К обменным аберрациям хромосомного типа относятся и транслокации. Нерепарированные двунитевые разрывы ДНК приводит к делециям хромосом и формированию ацентрических хромосомных фрагментов, которые можно наблюдать в ближайшем митозе. Дицентрики, кольца и ацентрические фрагменты плохо передаются в череде клеточных делений и в делящихся клетках со временем исчезают, поэтому их относят к нестабильным хромосомным перестройкам. Транслокации, не приводящие к потере генетического материала, беспрепятственно передаются дочерним клеткам в митозе, поэтому их классифицируют как стабильные аберрации.
Если облучение вызвало появление двунитевого разрыва ДНК в участке хромосомы, уже прошедшем удвоение в процессе репликации в S-фазе клеточного цикла, то это может привести к образованию аберраций хроматидного типа. Наиболее типичными аберрациями хроматидного типа являются тетрарадиалы (обменные аберрации, возникающие в процессе неправильно соединения двух двунитевых разрывов ДНК, находящихся на хроматидах разных хромосомах) и хроматидные фрагменты (нерепарированный двунитевой разрыв ДНК).
Дицентрики и кольца, а также некоторые обменные аберрации хроматидного типа часто приводят к формированию «мостов» в анафазе митоза, которые можно детектировать при помощи ана-телофазного метода анализа хромосомных аберраций.
Для частоты радиоиндуцированных хромосомных аберраций характерна строгая зависимость от дозы, мощности и характера ионизирующего излучения, что позволило создать цитогенетические методы биологической дозиметрии [11] .
Методы детекции хромосомных перестроек
Хромосомные перестройки выявляют и анализируют при помощи цитогенетических методов, наиболее часто анализ хромосомных перестроек проводят цитологически на стадии метафазы. Самым распространенным и доступным цитогенетическим методом является метод дифференциальной G-окраски хромосом (G-бэндинг). С конца 80-х годов прошлого века для выявления хромосомных перестроек применяют метод флуоресцентной гибридизации in situ с использованием ДНК-проб к отдельным хромосомам или хромосомным локусам. Одним из наиболее точных методов обнаружения небольших дупликаций и делеций в настоящее время является метод сравнительной геномной гибридизации на препаратах метафазных хромосом или ДНК-микрочипах. Дупликации и делеции могут быть выявлены и при полногеномном SNP-генотипировании. Следует отметить, что два последних метода не позволяют выявлять сбалансированные хромосомные перестройки и, в отличие от других цитогенетических методов, не позволяют проводить анализ хромосомных аберраций на уровне отдельной клетки, то есть являются нечувствительными для случаев мозаицизма.
Читайте также: