Классификация аномалий развития мозжечка
Добавил пользователь Дмитрий К. Обновлено: 30.01.2026
Мальформация Денди-Уокера и атрезия мозжечкового отверстия - клиника, диагностика
Главным признаком классической мальформации Денди-Уокера является полная или частичная агенезия червя и крупная кистозная мальформация в задней ямке, соответствующая дивертикулярному расширению крайне увеличенного четвертого желудочка. Гидроцефалия часто отсутствует у новорожденных и детей раннего возраста и иногда до позднего периода полового созревания. Обычно имеется заметное расширение задней ямки и возвышение места слияния синусов и боковых синусов, проходящих по теменным костям вместо чешуи затылочной кости.
Различные определения синдрома Денди-Уокера включают в себя спектр аномалий задней ямки, различных по клиническим и визуализационным особенностям и исходам. В эти аномалии входит вариант синдрома Денди-Уокера (Sarnat и Alcala, 1980), при котором присутствует часть червя мозжечка и задняя ямка не расширена, случаи, связанные с другими мальформациями периферической или центральной нервной системы. Некоторые авторы (Klein et al., 2003; Parisi et al., 2003) указывают более строгие критерии:
1) крупные средние размеры кисты задней ямки, широко сообщающейся с четвертым желудочком;
2) маленький повернутый, приподнятый червь мозжечка с патологическими бороздами;
3) смещенный кверху Тенториум;
4) расширенная задняя ямка;
5) смещенные кпереди и латерально, но предположительно нормальные, полушария;
6) нормальный ствол мозга.
В соотвествии с недавним определением, используемым и в статьях на сайте, сочетанные соматические мальформации не характерны. Сопутствующие пороки развития ЦНС включают патологическую миграцию в нижней оливе, агенезию мозолистого тела у 7-15% пациентов (Hirschetal, 1984, Murray et al., 1985) и затылочные цефалоцелеу 17% (Hirsch et al., 1984). Периферические мальформации присутствуют в широко изменяющейся доле случаев, в зависимости от используемых диагностических критериев и от происхождения случаев. В ряде диагностированных пренатально случаев, частота сопровождающих мальформаций может быть высокой. Сюда входят периферические мальформации, такие как расщелина губы и неба, пороки развития сердца, аномалии мочевыделительного тракта и малый лицевой дисморфизм (Has et al., 2004).
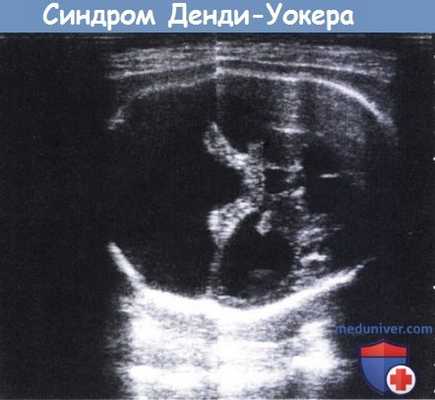
Мальформация Денди-Уокера.
Пренатальная диагностика методом ультразвукового сканирования на 33 неделе беременности.
Хорошо видно отсутствие червя мозжечка и выраженное расширение задней черепной ямки (левая часть рисунка).
Основным клиническим проявлением синдрома Денди-Уокера является гидроцефалия. Поскольку гидроцефалия обычно становится заметной к концу первого года жизни, в 75% случаев диагноз ставится в возрасте после трех месяцев и редко в первый день рождения. Голова может быть вытянутой в переднезаднем направлении с выступающим затылком, что может натолкнуть на мысль о возможном наличии аномалии. Примечательно, что отсутствуют мозжечковые симптомы или нарушения позы. Некоторая степень задержки умственного развития отмечается в 30-50% случаев, что является показанием для проведения нейровизуализации.
Визуализация имеет характерную картину. Во всех случаях присутствуют огромные скопления, которые в половине случаев распространяются через тенториальное отверстие в тригеминальную цистерну и/или вниз через большое затылочное отверстие. Это проявляется значительным расширением четвертого желудочка. Полушария маленькие, но нормальные. Червь мозжечка часто уменьшен и смещен, прижат к намету. Сагиттальные срезы позволяют лучше увидеть остатки червя, которые смещены вверх к намету мозжечка и повернуты кпереди. По мнению Klein et al. (2003), у большинства пациентов имеется только две щели червя и три доли; такие пациенты не имеют сочетающихся мальформаций и их уровень развития почти всегда остается в пределах нормы. В наименьшей группе червь развит неправильно и имеет только одну щель или ни одной. В этих случаях всегда присутствуют сопутствующие аномалии и большая или меньшая задержка умственного развития.
Дифференциальная диагностика мальформации проста в типичных случаях. В мегацистерне большой ретроцеребеллярный карман (соответствующий карману Блейка) сообщается с четвертым желудочком, но червь мозжечка полный. Дифференцировка варианта синдрома Денди-Уокера от увеличенной цистерны магна или ретроцеребеллярной арахноидальной кисты представляет некоторые трудности (Tortori-Donati et al., 1996, Boltshauser et al, 2002). В вариантных формах червь может быть атрофичным, но содержит все дольки и сдавлен стволом. В случаях с мегацистерной червь по сути нормальный, и четвертый желудочек отличается от кисты. В случаях Денди-Уокера червь может быть аномальным, атрофированным, неполным или вообще отсутствовать, четвертый желудочек трудно отличить от кисты и переднее положение сосудистого сплетения смещено к верхней стенке кисты, что является значимым диагностическим признаком (Nelson et al., 2004). Намет мозжечка смещен вверх, его вырезка видна на теменной, а не на затылочной кости, приводя к значительному расширению задней ямки.
Исход для пациентов, страдающих синдромом Денди-Уокера, относительно неблагоприятный при использовании обычных критериев. Уровень смертности в 144 случаях, рассмотренных Hirsch et al. (1984), составил 27% и только около половины выживших имели нормальный IQ. Напротив, исход благоприятный при применении строгих параметров Klein et al. (2003), если оперативное вмешательство по поводу гидроцефалии выполнено рано (Parisi и Dobyns, 2003). Генетический прогноз хороший, с рецидивами в 1-5% случаев. Это контрастирует с более тяжелым прогнозом атипичных и/или ассоциированных форм. Хотя увеличенная цистерна магна обычно рассматривается как доброкачественное изменение, Bodensteiner et al. (1988) обнаружили, что у 62% пациентов с мегацистерной магна была неврологическая патология, полагая, что это может быть частью комплексной мальформации. Случаи Денди-Уокера-подобного синдрома задней ямки, связанные с лицевыми или другими гемангиомами и сердечными и сосудистыми аномалиями, представляют различные формы (PHACE/PHACES синдромы или синдром Паскаля-Кастровьеджо II) (Metry et al., 2001).
Лечение заключается в шунтировании гидроцефалии, а не в открытии кистозного четвертого желудочка. Некоторые хирурги советуют устанавливать шунт в четвертый желудочек, но это мнение не единодушное. После шунтирования боковых желудочков может потребоваться дополнительное шунтирование четвертого желудочка. Хорошие результаты может дать эндоскопическая хирургия (Mohanty et al., 2006).
Механизмом синдрома Денди-Уокера ранее предполагалась атрезия отверстий Мажанди и Люшки. Тем не менее, эта атрезия не является ни постоянным, ни характерным признаком. В настоящее время этот синдром связывают с остановкой развития среднего мозга с сохранением переднего мембранозного участка четвертого желудочка плода. Эта структура в норме исчезает до момента открытия отверстия. Аномалия, по всей видимости, появляется до третьего месяца беременности (Friede, 1989). Полная атрезия отверстия четвертого желудочка редко наблюдается вне комплекса Денди-Уокера (Amacher и Page, 1971). Это вызывает формирование гидроцефалии, которая у одного из моих пациентов началась и была более заметна в четвертом желудочке, чем в остальных частях желудочковой системы. Микроскопическое исследование оболочки, покрывающей четвертый желудочек, выявило глиальную ткань, выстланную по внутренней стороне эпендимой (Friede, 1989). Эта глиальная оболочка позволяет отличить атрезию отверстия от фиброза базальной цистерны с последующей гидроцефалией. Происхождение мембраны неизвестно.
Классификация аномалий развития мозжечка
Аплазия и гипоплазия мозжечка - клиника, диагностика
Основные дефекты развития мозжечка, которые повреждают преимущественно полушария (Robain et al. 1987) или почти в одинаковой степени полушария и червь, составляют гетерогенную группу редких аномалий с различной патологией и клиническими проявлениями (Sarnat и Alcala, 1980; Robain et al, 1987; Altman et al., 1992). Случаи вероятной атрофии и врожденной мосто-мозжечковой атрофии обсуждаются в отдельной статье на сайте.
Диагностика неоднозначна, так как многие очевидные мальформации фактически являются приобретенными заболеваниями в результате деструктивных или метаболических процессов (Sener, 1995; Boltshauser, 2004). Правильная классификация дефектов развития мозжечка затруднена. Friede (1989) утверждает, что классическое разделение на неоцеребеллярную против палеоцеребеллярной гипоплазии является искусственным и во многих случаях целесообразнее говорить об аплазии передней или задней доли мозжечка.
Тотальная аплазия мозжечка является исключением (Glickstein, 2004). Интересно то, что она не всегда проявляется клиническими симптомами. В действительности, остатки мозжечка, вероятно, всегда присутствуют (Gardner, 2001) и определение «гипоплазия» или «атрофия» точнее, чем «агенезия». Сообщалось о мозжечковой агенезии в сочетании с несахарным диабетом (Zafeiriou et al., 2004). Гипоплазия менее характерна и может быть преимущественно односторонней.
В отдельных случаях она ограничена специфическими структурами, такими как долька полушария мозжечка. Часто сопутствуют аномалии глубоких мозжечковых и стволовых ядер и дисплазия нижней оливы. В некоторых случаях имеется гидроцефалия (Friede, 1989), возможны и другие патологические изменения, например, агенезия мозолистого тела или аринэнцефалия.
Односторонняя аплазия (Boltshauser et al., 1996; Kilickesmez et al., 2004) встречается реже и часто бессимптомная. Может сопровождаться ипсилатеральной лицевой ангиомой и различными аномалиями мозговых и/или периферических сосудов при синдроме PHACES (Pascual-Castroviejo et al., 2003, Bhattacharya et al„ 2004).
Клинические проявления крайне неустойчивы. Они могут совсем отсутствовать или проявляться в виде атаксии, нарушения равновесия и других мозжечковых симптомов. Характерна задержка умственного развития в различной степени. Некоторые случаи представлены атаксическим церебральным параличом. В большинстве случаев преобладали двигательные расстройства, но у некоторых пациентов отмечались признаки аутизма или микроцефалии.
Диагноз мозжечковой гипоплазии обычно основан на радиологических методах исследования. Диффузную гипоплазию бывает трудно отличить от варианта Данди-Уолкера по данным КТ, и мегацистерну часто принимают за мозжечковую гипоплазию. Дифференциальный диагноз включает визуализацию серпа мозжечка, что характеризует картину цистерн, и полное наличие червя мозжечка на сагиттальных изображениях МРТ.
Другие необычные пороки развития мозжечка:
1. Текто-мозжечковая дизрафия (Mori et al., 1994, Dehdashti et al., 2004) является сложной аномалией развития, которая включает в себя инвертированный мозжечок, развивающийся внутри четвертого желудочка, гипоплазию червя и затылочное цефалоцеле.
2. Ромбэнцефалосинапсис (Toelle et al., 2002) означает врожденное слияние мозжечковых полушарий с объединением зубчатых ядер по средней линии и атрезией водопровода. Пациенты с умеренными симптомами живут долго.
Комплексные мальформации мозжечка в связи с цефалоцеле могут возникнуть в результате ишемической деструкции прежде нормально сформированного мозжечка, вследствие артериального сдавления дивертикулом четвертого желудочка.
3. Дентато-оливарная дисплазия отвечает за неонатальные тонические судороги, напоминающие синдром Отахара, обнаруженный у нескольких пациентов (Harding и Boyd, 1991, Robain и Dulac, 1992). Предполагают генетическое происхождение патологии (Harding и Сорр, 1997).
Аномалия Киари ( Синдром Арнольда-Киари )
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
МКБ-10
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
- Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
- Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
- Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
- Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Аномалии развития головного мозга ( Пороки развития головного мозга )
Аномалии развития головного мозга — это результат происходящих во внутриутробном периоде нарушений формирования отдельных церебральных структур или головного мозга в целом. Зачастую имеют неспецифическую клиническую симптоматику: преимущественно эпилептический синдром, задержку психического и умственного развития. Тяжесть клиники напрямую коррелирует со степенью поражения головного мозга. Диагностируются антенатально при проведении акушерского УЗИ, после рождения — при помощи ЭЭГ, нейросонографии и МРТ головного мозга. Лечение симптоматическое: противоэпилептическое, дегидратационное, метаболическое, психокоррегирующее.
Наиболее весомой причиной сбоев внутриутробного развития является влияние на организм беременной и на плод, различных вредоносных факторов, обладающих тератогенным действием. Возникновение аномалии в результате моногенного наследования встречается лишь в 1% случаев. Наиболее влиятельной причиной пороков головного мозга считается экзогенный фактор. Тератогенным эффектом обладают многие активные химические соединения, радиоактивное загрязнение, отдельные биологические факторы. Немаловажное значение здесь имеет проблема загрязнения среды обитания людей, обуславливающая поступление в организм беременной токсических химических веществ.
Различные эмбриотоксические воздействия могут быть связаны с образом жизни самой беременной: например, с курением, алкоголизмом, наркоманией. Дисметаболические нарушения у беременной, такие как сахарный диабет, гипертиреоз и пр., могут также стать причиной церебральных аномалий плода. Тератогенным действием обладают и многие медикаменты, которые может принимать женщина в ранние сроки беременность, не подозревая о происходящих в ее организме процессах. Мощный тератогенный эффект оказывают инфекции, перенесенные беременной, или внутриутробные инфекции плода. Наиболее опасны цитомегалия, листериоз, краснуха, токсоплазмоз.
Патогенез
Дифференцировка нейробластов (зародышевых нервных клеток) приводит к образованию нейронов, формирующих серое вещество, и глиальных клеток, составляющих белое вещество. Серое вещество отвечает за высшие процессы нервной деятельности. В белом веществе проходят различные проводящие пути, связывающие церебральные структуры в единый функционирующий механизм. Рожденный в срок новорожденный имеет такое же число нейронов, как и взрослый человек. Но развитие его мозга продолжается, особенно интенсивно в первые 3 мес. жизни. Происходит увеличение глиальных клеток, разветвление нейрональных отростков и их миелинизация.
Сбои могут произойти на различных этапах формирования головного мозга. Если они возникают в первые 6 мес. беременности, то способны приводить к снижению числа сформированных нейронов, различным нарушениям в дифференцировке, гипоплазии различных отделов мозга. В более поздние сроки может возникать поражение и гибель нормально сформировавшегося церебрального вещества.
Виды аномалий мозга
Анэнцефалия — отсутствие головного мозга и акрания (отсутствие костей черепа). Место головного мозга занято соединительнотканными разрастаниями и кистозными полостями. Может быть покрыто кожей или обнажено. Патология несовместима с жизнью.
Энцефалоцеле — пролабирование церебральных тканей и оболочек через дефект костей черепа, обусловленный его незаращением. Как правило, формируется по средней линии, но бывает и асимметричным. Небольшое энцефалоцеле может имитировать кефалогематому. В таких случаях определить диагноз помогает рентгенография черепа. Прогноз зависит от размеров и содержимого энцефалоцеле. При небольших размерах выпячивания и наличии в его полости эктопированной нервной ткани эффективно хирургическое удаление энцефалоцеле.
Микроцефалия — уменьшение объема и массы головного мозга, обусловленное задержкой его развития. Встречается с частотой 1 случай на 5 тыс. новорожденных. Сопровождается уменьшенной окружностью головы и диспропорциональным соотношением лицевого/мозгового черепа с преобладанием первого. На долю микроцефалии приходится около 11% всех случаев олигофрении. При выраженной микроцефалии возможна идиотия. Зачастую наблюдается не только ЗПР, но и отставание в физическом развитии.
Макроцефалия — увеличение объема головного мозга и его массы. Гораздо менее распространена, чем микроцефалия. Макроцефалия обычно сочетается с нарушениями архитектоники мозга, очаговой гетеротопией белого вещества. Основное клиническое проявление — умственная отсталость. Может наблюдаться судорожный синдром. Встречается частичная макроцефалия с увеличением лишь одного из полушарий. Как правило, она сопровождается асимметрией мозгового отдела черепа.
Кистозная церебральная дисплазия — характеризуется множественными кистозными полостями головного мозга, обычно соединенными с желудочковой системой. Кисты могут иметь различный размер. Иногда локализуются только в одном полушарии. Множественные кисты головного мозга проявляются эпилепсией, устойчивой к антиконвульсантной терапии. Единичные кисты в зависимости от размера могут иметь субклиническое течение или сопровождаться внутричерепной гипертензией; зачастую отмечается их постепенное рассасывание.
Голопрозэнцефалия — отсутствие разделения полушарий, в результате чего они представлены единой полусферой. Боковые желудочки сформированы в единую полость. Сопровождается грубыми дисплазиями лицевого черепа и соматическими пороками. Отмечается мертворождение или гибель в первые сутки.
Агирия (гладкий мозг, лиссэнцефалия) — отставание развития извилин и тяжелое нарушение архитектоники коры. Клинически проявляется выраженным расстройством психического и моторного развития, парезами и различными формами судорог (в т. ч. синдромом Веста и синдромом Леннокса-Гасто). Обычно заканчивается летальным исходом на первом году жизни.
Пахигирия — укрупнение основных извилин при отсутствии третичных и вторичных. Сопровождается укорочением и выпрямлением борозд, нарушением архитектоники церебральной коры.
Микрополигирия — поверхность коры мозга представлена множеством мелких извилин. Кора имеет до 4-х слоев, тогда как в норме кора насчитывает 6 слоев. Может быть локальной или диффузной. Последняя, полимикрогирия, характеризуется плегией мимических, жевательных и глоточных мышц, эпилепсией с дебютом на 1-ом году жизни, олигофренией.
Гипоплазия/аплазия мозолистого тела. Часто встречается в виде синдрома Айкарди, описанного только у девочек. Характерны миоклонические пароксизмы и сгибательные спазмы, врожденные офтальмические пороки (колобомы, эктазия склеры, микрофтальм), множественные хориоретинальные дистрофические очаги, обнаруживаемые при офтальмоскопии.
Фокальная корковая дисплазия (ФКД) — наличие в коре головного мозга патологических участков с гигантскими нейронами и аномальными астроцитами. Излюбленное расположение — височные и лобные зоны мозга. Отличительной особенностью эпиприступов при ФКД является наличие кратковременных сложных пароксизмов с быстрой генерализацией, сопровождающихся в своей начальной фазе демонстративными двигательными феноменами в виде жестов, топтания на одном месте и т. п.
Гетеротопии — скопления нейронов, на этапе нейронной миграции задержавшихся на пути своего следования к коре. Гетеротопионы могут быть единичными и множественными, иметь узловую и ленточную форму. Их главное отличие от туберозного склероза — отсутствие способности накапливать контраст. Эти аномалии развития головного мозга проявляются эписиндромом и олигофренией, выраженность которых прямо коррелирует с числом и размером гетеротопионов. При одиночной гетеротопии эпиприступы, как правило, дебютируют после 10-летнего возраста.
Тяжелые аномалии развития головного мозга зачастую могут быть диагностированы при визуальном осмотре. В остальных случаях заподозрить церебральную аномалию позволяет ЗПР, гипотония мышц в неонатальном периоде, возникновение судорожного синдрома у детей первого года жизни. Исключить травматический или гипоксический характер поражения головного мозга можно при отсутствии в анамнезе данных о родовой травме новорожденного, гипоксии плода или асфиксии новорожденного. Пренатальная диагностика пороков развития плода осуществляется путем скринингового УЗИ при беременности. УЗИ в I триместре беременности позволяет предупредить рождение ребенка с тяжелой церебральной аномалией.
Одним из методов выявления пороков головного мозга у грудничков является нейросонография через родничок. Намного более точные данные у детей любого возраста и у взрослых получают при помощи МРТ головного мозга. МРТ позволяет определить характер и локализацию аномалии, размеры кист, гетеротопий и других аномальных участков, провести дифференциальную диагностику с гипоксическими, травматическими, опухолевыми, инфекционными поражениями мозга. Диагностика судорожного синдрома и подбор антиконвульсантной терапии осуществляется при помощи ЭЭГ, а также пролонгированного ЭЭГ-видеомониторинга. При наличии семейных случаев церебральных аномалий может быть полезна консультация генетика с проведением генеалогического исследования и ДНК-анализа. С целью выявления сочетанных аномалий проводится обследование соматических органов: УЗИ сердца, УЗИ брюшной полости, рентгенография органов грудной полости, УЗИ почек и пр.
Лечение аномалий мозга
Терапия пороков развития головного мозга преимущественно симптоматическая, осуществляется детским неврологом, неонатологом, педиатром, эпилептологом. При наличии судорожного синдрома проводится антиконвульсантная терапия (карбамазепин, леветирацетам, вальпроаты, нитразепам, ламотриджин и др.). Поскольку эпилепсия у детей, сопровождающая аномалии развития головного мозга, обычно резистентна к противосудорожной монотерапии, назначают комбинацию из 2 препаратов (например, леветирацетам с ламотриджином). При гидроцефалии осуществляют дегидратационную терапию, по показаниям прибегают к шунтирующим операциям. С целью улучшения метаболизма нормально функционирующих мозговых тканей, в какой-то степени компенсирующих имеющийся врожденный дефект, возможно проведение курсового нейрометаболического лечения с назначением глицина, витаминов гр. В и пр. Ноотропные препараты используются в лечении только при отсутствии эписиндрома.
При умеренных и относительно легких церебральных аномалиях рекомендована нейропсихологическая коррекция, занятия ребенка с психологом, комплексное психологическое сопровождение ребенка, детская арт-терапия, обучение детей старшего возраста в специализированных школах. Указанные методики помогают привить навыки самообслуживания, уменьшить степень выраженности олигофрении и по возможности социально адаптировать детей с церебральными пороками.
Прогноз и профилактика
Прогноз во многом определяется тяжестью церебральной аномалии. Неблагоприятным симптомом выступает ранее начало эпилепсии и ее резистентность к осуществляемой терапии. Осложняет прогноз наличие сочетанной врожденной соматической патологии. Эффективной мерой профилактики служит исключение эмбриотоксических и тератогенных влияний на женщину в период беременности. При планировании беременности будущим родителям следует избавиться от вредных привычек, пройти генетическое консультирование, обследование на наличие хронических инфекций.
Синдром Денди-Уокера
Синдром Денди-Уокера – это врожденная патология нервной системы, для которой характерна триада признаков: гидроцефалия, гипоплазия или аплазия мозжечка, кисты задней черепной ямки. Болезнь имеет полиэтиологическую природу, среди провоцирующих факторов выделяют генетические аномалии, тератогенные влияния. Клиническая симптоматика включает классические признаки гидроцефалии, многообразные неврологические нарушения. Диагностика порока требует проведения нейросонографии, МРТ, эхокардиографии, также возможна пренатальная постановка диагноза при УЗИ-скрининге. Лечение заключается в консервативных и хирургических методах коррекции гидроцефалии.
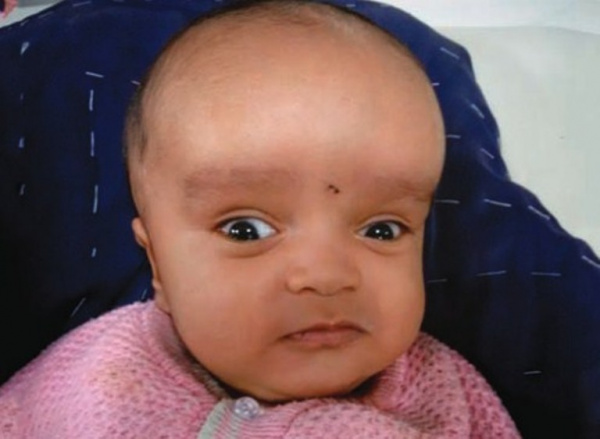
Пороки ЦНС занимают второе место в группе врожденных болезней, уступая только сердечно-сосудистым аномалиям. До 80% всех патологий сопровождаются гидроцефалией, как при синдроме Денди-Уокера. Данное заболевание было описано американским нейрохирургом В. Денди в 1914 году, спустя 28 лет канадско-американский нейрохирург А. Уокер с коллегами предложил оперативный способ лечения порока. Патология встречается с частотой 1:25000-1:35000, девочки болеют чаще. Среди младенцев с врожденной гидроцефалией патология регистрируется в 3,5-12% случаев.
Этиологические факторы синдрома Денди-Уокера до сих пор точно не выяснены. По современным данным, патология имеет мультифакториальную природу, ее развитию способствуют как внутренние причины (хромосомные и генные мутации), так и воздействие экзогенных тератогенов. Провоцирующими факторами заболевания могут выступать:
- вирусные инфекции (цитомегаловирус, краснуха);
- гестационный диабет;
- употребление алкоголя матерью во время беременности.
Новейшие исследования показывают четкую связь изолированной формы аномалии Денди-Уокера с мутациями генов ZIC1 и ZIC4. Риск развития заболевания повышается у младенцев с врожденными нарушениями обмена веществ. Чаще всего синдром ассоциирован с одной из форм 3-метилглутаконовой ацидурии, которая проявляется ретинопатией, почечной дисфункцией, метаболическим ацидозом и повышением активности печеночных ферментов.
Выделяют три основные гипотезы структурных поражений головного мозга при болезни Денди-Уокера. Согласно первой из них, пороки ЦНС вызваны замедлением эмбрионального развития на раннем этапе при закладке ромбовидного мозга. Такая ситуация наблюдается под действием тератогенных факторов либо на фоне генетических мутаций. Вторая гипотеза указывает на заращение выходного отверстия 4 желудочка мозга и позднее открытие апертуры Мажанди.
Новые экспериментальные данные подтверждают вероятность третьей гипотезы: типичные анатомические изменения развиваются при возникновении сосудистого сплетения на тонкой крыше ромбовидного мозга. Патология сопровождается внутриутробной гидроцефалией, вызывает серьезные неврологические нарушения. На фоне этого развивается большая киста в задних отделах черепа, которая препятствует формированию мозолистого тела и червя мозжечка.
Специалисты выделяют открытую и закрытую формы синдрома. В первом случае наблюдается окклюзия отверстий Люшка и Мажанди, что сопровождается серьезными нарушениями ликвородинамики. Второй вариант не имеет таких патологий, поэтому протекает в более благоприятной форме. В нейрохирургии важное значение имеет анатомическая классификация порока, согласно которой выделяют 2 формы синдрома Денди-Уокера:
- Полная. Характеризуется отсутствием червя мозжечка, наличием связи между полостью четвертого желудочка и мозжечково-мозговой цистерной.
- Неполная. Проявляется частичным недоразвитием мозжечкового червя в его нижней части, за счет чего коммуникация с большой цистерной реализуется не на всем протяжении.
В 1989 г. американским педиатром и нейрорадиологом Джеймсом Барковичем была предложена альтернативная классификация, базирующаяся на результатах МР-диагностики. Выделена группа заболеваний, названная «комплексом Денди-Уокера», который включает 4 клинических формы: классическую аномалию (полная форма), вариант Денди-Уокера (менее грубые нарушения), кисту кармана Блейка и mega cisterna magna.
Симптомы
Основное проявление синдрома Денди-Уокера – гидроцефалия, возникающая в первые месяцы жизни ребенка. Степень выраженности симптоматики зависит от варианта синдрома, скорости прогрессирования нарушений, общего состояния новорожденного. К ранним признакам относят беспокойное поведение, монотонный крик, нарушение питания и частые срыгивания. Внешне обнаруживается выбухание родничков, расширение черепных швов, быстрое увеличение окружности головы.
Характерный признак гидроцефалии при синдроме Денди-Уокера – симптом заходящего солнца: при взгляде вниз радужка частично скрывается нижним веком, над ней появляется белая полоска склеры. Неврологические симптомы также представлены нистагмом, экзофтальмом, косоглазием. Нередко у новорожденных отмечается судорожный синдром, парезы и параличи конечностей. При прогрессировании болезни характерно отставание в психомоторном развитии.
Осложнения
В 65-68% случаев синдром Денди-Уокера сопровождается другими структурными неврологическими аномалиями. Чаще всего диагностируется агенезия мозолистого тела, стеноз Сильвиева водопровода, гетеротопия извилин коры мозжечка. При тяжелых формах заболевания определяется недоразвитие ствола головного мозга, что сопряжено с глубоким угнетением витальных функций и высоким риском младенческой летальности.
До 55% детей имеют сопутствующие врожденные синдромы: Уокера-Варбурга, PHACE, Ritscher-Schinzel. Характерны различные формы хромосомных аберраций: делеции, дупликации, трисомии и триплоидии. Поражение кардиоваскулярной системы проявляется септальными дефектами и нарушениями постнатальной гемодинамики. Реже определяются офтальмологические пороки: ретинальная дисплазия, микрофтальмия.
Первичное обследование ребенка с неврологическими нарушениями проводится врачом-педиатром или неонатологом. При гидроцефалии и подозрении на синдром Денди-Уокера к диагностике подключается детский невролог, нейрохирург. Физикальный осмотр выявляет неспецифические признаки внутричерепной гипертензии, нарушения моторного развития, сопутствующие аномалии. Для постановки окончательного диагноза проводится:
- Нейросонография. При ультразвуковом сканировании головного мозга определяется крупное кистозное образование в задней части черепной коробки. На УЗИ мозжечковый червь не визуализируется, недоразвитые полушария мозжечка раздвинуты, желудочковая система мозга расширена и деформирована.
- МРТ головного мозга. Магнитно-резонансная томография в сагиттальной и аксиллярной проекции демонстрирует расширение четвертого желудочка, грубые нарушения развития мозжечка, другие структурные аномалии.
- Эхокардиография. УЗИ сердца рекомендовано всем детям с синдромом Денди-Уокера, поскольку он нередко ассоциирован с врожденной кардиальной патологией. По результатам ЭхоКГ можно определить аномалии развития клапанного аппарата, дефекты внутрисердечной перегородки, патологии расположения крупных сосудов.
- Кариотипирование. При комбинированных врожденных пороках развития необходима консультация генетика и тщательное изучение генетического материала ребенка. Углубленное исследование направлено на диагностику хромосомных аберраций.
- Пренатальная диагностика. При проведении УЗИ плода удается предположить аномалию на сроке 15-16 недель, более четкая визуализация структур IV желудочка возможна после 22 недели гестации. При сочетании патологии с расщелинами лица эхосонография может быть недостаточно информативна.
Дифференциальная диагностика
При постановке диагноза необходимо дифференцировать синдром с гипоплазией мозжечка другой этиологии, ретроцеребральными кистами и расширениями большой мозговой цистерны. Патогномоничным признаком синдрома Денди-Уокера считается дефект червя мозжечка, который не возникает при других вариантах пороков ЦНС. Проводится дифференциальная диагностика с арахноидальными кистами, которая требует дополнительных инструментальных методов исследования.
Лечение синдрома Денди-Уокера
Консервативная терапия
Наибольшую опасность для жизни и здоровья младенца представляет гидроцефалия, для коррекции которой показано лечение в отделении интенсивной терапии. Назначается массивная дегидратационная терапия, которая включает разные типы диуретиков, гипертонические инфузионные растворы. Усилия врачей направлены на уменьшение ликворопродукции и нормализацию внутричерепного давления, чтобы предупредить отек мозга и компрессию церебральных структур.
Хирургическое лечение
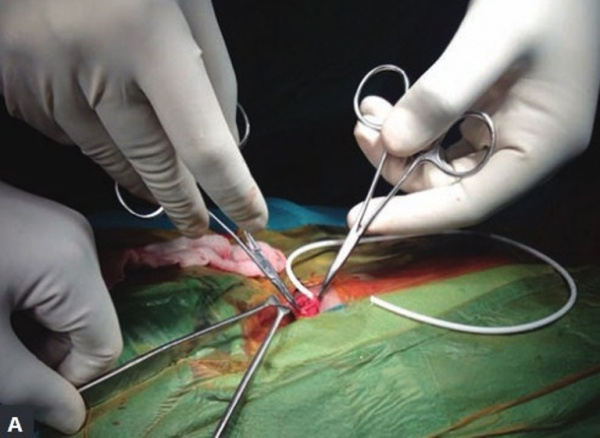
При неэффективности консервативных методов и неуклонном прогрессировании гидроцефалии требуется помощь детских нейрохирургов. Основным методом коррекции при синдроме Денди-Уокера являются шунтирующие операции с имплантацией дренажных трубок или клапанных регулируемых систем. Существует несколько типов шунтирования: вентрикуло-перитонеальное, вентрикуло-атриальное, вентрикулоцистерностомия.
Для коррекции окклюзии ликворных путей применяются методы эндоскопической вентрикулостомии, которые восстанавливают нормальный отток цереброспинальной жидкости, устраняют острую неврологическую симптоматику. При гипертензионно-гидроцефальном кризе показана вентрикулярная разгрузочная пункция, которая относится к вариантам экстренного временного дренирования.
Исход заболевания зависит от глубины поражения ЦНС, скорости нарастания гидроцефалии, наличия сочетанной патологии. Нарастающая гипертензия и грубые неврологические пороки в 90% случаев становятся причиной смерти пациента в первые годы жизни, для выживших детей с неполным вариантом аномалии прогноз более благоприятный. Вследствие недостаточной изученности этиопатогенеза болезни эффективные меры профилактика пока не разработаны.
1. Случая мальформации Денди-Уокера с доношенной беременностью и родами / Е.В. Беляева, Л.В. Лапшина, Е.В. Шапошникова, А.А. Молгачев // Журнал неврологии и психиатрии. – 2018. – №2.
2. Аномалия Денди-Уокера – редкая причина сирингомиелии у взрослых /Г.Ю. Евзиков// Неврология, нейропсихиатрия, психосоматика. – 2017. – №9.
3. Синдром Денди-Уокера у новорожденных/ Л.А. Петрова, А.В. Розанов, Ю.И. Барашнев, В.О. Панов// Российский вестник перинатологии и педиатрии. – 2010. – №1.
Читайте также: