Клиника хронического идиопатического миелофиброза (ХИМФ) - спленомегалия, портальная гипертензия, анемия, асцит
Добавил пользователь Валентин П. Обновлено: 30.01.2026
Хронический идиопатический миелофиброз (ХИМФ) - определение, эпидемиология, патогенез
Хронический идиопатический миелофиброз (ХИМФ) относится к группе клоновых хронических миелопролиферативных заболеваний (ХМПЗ). Уникальной особенностью ХИМФ является раннее и значительное развитие фиброза костного мозга и миелоидной метаплазии селезенки (ММС).
Существует много синонимов для обозначения данного заболевания: сублейкемический миелоз, алейкемический миелоз, остеомиелопоэтическая дисплазия, агногенная или просто миелоидная метаплазия, миелосклероз с миелоидной метаплазией, хронический гранулоцитарно-мегакариоцитарный миелоз и многие другие. В классификации ВОЗ 2001 г. данное заболевание обозначено как «хронический идиопатический миелофиброз» и отнесено в группу хронических миелопролиферативных заболеваний.
Хронический идиопатический миелофиброз (ХИМФ) — сравнительно редкая патология. Заболеваемость составляет 0,3—1,5 на 100 000 населения в год, а средний возраст на момент заболевания — 60 лет. По данным французских авторов, в год регистрируется от 0,3 до 0,7 новых случаев заболевания.
Мужчины болеют несколько чаще женщин. В 90 % заболевание диагностируется после 40 лет, однако известны случаи заболевания хроническим идиопатическим миелофиброзом в молодом и даже детском возрасте. Отмечается более доброкачественное течение заболевания у детей.

Патогенез хронического идиопатического миелофиброза (ХИМФ)
Хронический идиопатический миелофиброз является клоновым миелопролиферативным заболеванием с первичным поражением стволовой кроветворной клетки, что доказывается обнаружением одного типа изоэнзимов глюкозо-6-фосфатдегидрогеназы в лейкоцитах, эритроцитах и мегакариоцитах больных миелофиброзом женщин-мулаток, а также анализом рестрикции Х-хромосомы у этих пациенток.
Имеются отдельные данные о том, что к патологическому клону при хроническом идиопатическом миелофиброзе относятся также В- и Т-лимфоциты, что является лишним подтверждением стволового уровня поражения гемопоэза при данном заболевании.
Патогенез хронического идиопатического миелофиброза (ХИМФ) нельзя считать полностью расшифрованным. Заболевание характеризуется пролиферацией гранулоцитов и мегакариоцитов с образованием экстрамедуллярных очагов кроветворения, главным образом в селезенке, т. е. развитием миелоидной метаплазии и фиброза костного мозга.
Характер и степень миелофиброза настолько отличает данное заболевание от других хронических миелопролиферативных заболеваний (ХМПЗ), что составляет основу его нозологического обозначения как хронический идиопатический миелофиброз. Между тем миелофиброз при этом заболевании вторичен, реактивен. Реактивный характер миелофиброза доказывается наличием двух типов изоэнзимов глюкозо-6-фосфатдегидрогеназы в фибробластах костного мозга у больных с одним типом фермента в кроветворных клетках: поликлональность фибробластов у больных хроническим идиопатическим миелофиброзом (ХИМФ) свидетельствует о реактивном характере миелофиброза.
Раннее образование экстрамедуллярных очагов гемопоэза, возможно, обусловлено повышенной при этом заболевании циркуляцией в крови полипотентных CD34-позитивных и линейно-рестриктированных предшественников гемопоэза — CFU-GEMM, BFU-E, CFU-GM, GFU-MK. В периферической крови больных ХИМФ количество циркулирующих гемопоэтических предшественников (CD34-позитивных клеток) почти в 2000 раз превышает норму. Высокая экспрессия стволовыми клетками при ХИМФ рецептора фактора стволовых клеток (c-Kit), по-видимому, обусловливает преимущества в пролиферации патологического клона.

Специфические хромосомные аберрации при хроническом идиопатическом миелофиброзе (ХИМФ) не описаны, но те же изменения, которые характерны для других заболеваний этой группы, встречаются у 45—60 % больных. В 90 % обнаруженные изменения представлены 13q-, 20q-, +8, +9, 12р-, +lq. В 46 % обнаруживаются только 13q- и 20q-. В одной из работ цитогетический анализ 14 больных выявил 13q- у 3 больных, 20q- у 1, у остальных десяти больных изменений не было. Эти и многие другие исследования подтверждают факт высокой частоты 13q- аберрации при хроническом идиопатическом миелофиброзе, что позволяет некоторым авторам высказать предположение о патогенетической связи между развитием миелофиброза и делецией 13q-.
Механизм этой связи может заключаться в том, что делегированный участок приходится на район хромосомы 13, в котором локализован какой-то пока не идентифицированный ген, кодирующий один из факторов, ингибирующих развитие опухоли. Другие авторы, однако, не считают, что 13q- и 20q- могут быть связаны с инактивацией какого-то гена. Данные, полученные авторами, которые использовали метод геномной гибридизации, позволил им предположить, что к патогенезу заболевания, скорее всего, имеет отношение изменение хромосомы 9.
В исследовании популяции CD34-позитивных клеток частота цитогенетических аномалий составила 80 %, причем в подавляющем большинстве обнаруживалась 13q-делеция. Когда исследовались зрелые лейкоциты, такие же нарушения кариотипа обнаруживались только в 34,8 % случаев. Часть CD34+-клеток характеризовалась также экспрессией CD38, антигена, экспрессированного на самых ранних гемопоэтических клетках, что подтверждает происхождение хронического идиопатического миелофиброза из самых примитивных гемопоэтических предшественников.
Об их пролиферативном преимуществе свидетельствовало нарастание числа этих клеток в культуре на 7-й и 14-й дни. Ни селезеночные фибробласты, ни В-лимфоциты не имели цитогенетических нарушений.
Несмотря на отсутствие диагностических хромосомных аберраций при хроническом идиопатическом миелофиброзе, значение цитогенетических исследований в прогнозе заболевания несомненно.

Анализ течения заболевания у 165 больных показал, что наличие 20q- и 13q- не влияет на прогноз, в то время как +8 и 12р- часто сочетаются с короткой продолжительностью заболевания и повышенной частотой развития острого лейкоза.
При хроническом идиопатическом миелофиброзе повышено содержание эритропоэти-на в крови, в то же время возможен спонтанный рост эритроидных колоний, с высокой частотой обнаруживается носительство гена PRV-1, повышено содержание тромбопоэтина, и степень этого повышения коррелирует с выраженностью миело-фиброза.
Установлено повышение чувствительности клеток-предшественниц при хроническом идиопатическом миелофиброзе к воздействию различных цитокинов и увеличение экспрессии рецепторов к соответствующим цитокинам: ИЛ-3, GM-CSF, эритропоэтину и стволовому фактору роста (SCF).
В то же время уровень экспрессии тромбопоэтинового рецептора Mpl на мегакариоцитах и тромбоцитах снижен, как это наблюдается при эссенциальной тромбоцитемии (ЭТ) и истинной полицитемии (ИП), из чего следует, что механизм гиперплазии мегакариоцитов и их повышенного колониеобразования в культуре не связан непосредственно с тромбопоэтином. В этом отношении представляет интерес установление повышенной экспрессии фактора транскрипции — GATA-1 — в мегакариоцитах и CD34-позитивных клетках по сравнению со здоровыми лицами. Этот фактор участвует в генерации из стволовой клетки мегакариоцитарных предшественников и бипотенциальных эритроидно-мегакариоцитарных колоний.
Обнаружено, что фибробласты селезенки при хроническом идиопатическом миелофиброзе экспрессируют определенный набор молекул адгезии, отличный от такового у здоровых, и что рост CD34-позитивных предшественников при этом заболевании зависит от их взаимодействия с фибробластами селезенки, что снижает их зависимость от внешних воздействий и способствует экспансии патологического клона в органе и прогрессированию ММС.
Развитие миелофиброза происходит под влиянием фиброгенных цитокинов с участием ряда других факторов и белков.
Многочисленными исследованиями показано, что при хроническом идиопатическом миелофиброзе (ХИМФ) увеличена концентрация как в крови, так и внутри клеток многих цитокинов, участвующих в формировании фиброза и в неоангиогенезе — ИЛ-1, ростового фактора, выделяемого тромбоцитами (platelet-derived growth factor — PDGF, трансформирующего ростового фактора b — TGF-b), основного фактора роста фибробластов (basic fibroblast growth factor- bFGF), фактора роста эндотелия сосудов (vascular endothelial growth factor — VEGF). TGF-b усиливает синтез коллагена и фибронектина, способствует деградации компонентов экстрамедуллярного матрикса, участвующего в образовании фиброзной ткани. Высвобождение фиброгенных цитокинов происходит непосредственно в костном мозге.
Уровень кальмодулина — белка, связывающего ионы кальция, участвующего в их транспорте и тем самым вносящего вклад в формирование фиброза, в моче больных хроническим идиопатическим миелофиброзом (ХИМФ) превышает уровень кальмодулина у здоровых в 3 раза.
Цитокины, повышение уровня которых обнаружено при хроническом идиопатическом миелофиброзе, продуцируются мегакариоцитами, тромбоцитами и моноцитами.
Основные цитокины, участвующие в формировании фиброза (PDGF, bFGF и TGF-b), содержатся в а-гранулах мегакариоцитов и высвобождаются под действием ИЛ-1.
Установлено, что пептид, носящий название субстанции Р и участвующий в передаче сигнала нейронами, содержится в повышенном количестве в крови больных хроническим идиопатическим миелофиброзом. Этот пептид индуцирует продукцию ИЛ-1. Другим источником повышенного уровня ИЛ-1 являются моноциты. При хроническом идиопатическом миелофиброзе они активированы, содержат увеличенное по сравнению с нормой количество TGF-b и даже в отсутствие стимуляции продуцируют ИЛ-1.
С помощью моноклональных антител получены дополнительные доказательства локализации TGF-b в мононуклеарах периферической крови, хотя этот фактор долго считался маркером мегакариоцитов.
Одновременно с фиброзирующим костный мозг действием TGF-b совместно с гранулоцитарно-макрофагальным и макрофагальным факторами роста (GM-CSF и M-SCF) стимулирует рост гранулоцитарно-макрофагальных предшественников, что способствует увеличенной при хроническом идиопатическом миелофиброзе миелопролиферации. Таким образом, при данном заболевании существует аутокринный механизм стимуляции как миелопролиферации, так и миелофиброза. Активированные стромальные клетки также синтезируют цитокины и компоненты экстрацеллюлярного матрикса. Эти факторы поддерживают клональную гемопоэтическую пролиферацию, ответ которой на регуляторные факторы роста при этом заболевании нарушен.
Неоангиогенез поддерживает как фиброзирование костного мозга, так и миелопролиферацию. Экспансия гемопоэтических клеток происходит не только вследствие стимуляции их роста, но и ослабления отрицательных регуляторных сигналов.
Между степенью развития миелофиброза и экспансией гемопоэтических предшественников в периферическую кровь и селезенку имеется прямая связь, однако ее нельзя объяснить простым механическим вытеснением гемопоэтических предшественников фиброзированным костным мозгом. Эта возможность выглядела бы вполне логично, если бы не было нарушений в рецепторном аппарате гемопоэтических клеток по отношению к цитокинам и экспансии кроветворения уже на клеточно-пролиферативной, дофиброзной стадии заболевания. Вероятны более сложные клеточно-клеточные взаимоотношения между гемопоэтическими предшественниками и фибробластами на уровне костного мозга и селезенки.
В заключение следует осветить еще одну сторону этой проблемы. Роль тромбопоэтина и его рецептора в патогенезе миелофиброза в эксперименте на мышах выглядит в высшей степени убедительно: мыши, инфицированные протоонкогеном V-Mpl, дают развитие синдрома миелопролиферации и миелофиброза, аналогичного человеческому. В развитии тромбопоэтининдуцированного миелофиброза у мышей ведущая роль принадлежит TGF-b.
У мышей, подвергшихся чрезмерному воздействию тромбопоэтина, а также у мышей — носителей мутантного гена фактора транскрипции GATA-1, в связи с чем у них имеется редуцированная экспрессия этого фактора транскрипции, принимающего участие в конечной дифференцировке мегакариоцитов и эритроцитов, а также при трансплантации мыши костно-мозговых клеток, которые генно-инженерным путем изменены так, что они гиперэкспрессируют ген тромбопоэтина, также развивается синдром, полностью идентичный хронический идиопатический миелофиброз (ХИМФ), — гиперпролиферация мегакариоцитов, миелофиброз и экстрамедуллярный гемопоэз с постепенным увеличением селезенки.
После трансплантации нормального костного мозга наступает реверсия миелофиброза, без трансплантации болезнь прогрессирует и часто заканчивается острым лейкозом. В то же время показано, что значительная часть мегакариоцитов больных хроническим идиопатическим миелофиброзом плохо или совсем не реагирует на антитела против GATA-1, что совместно с обнаруженным нарушением транскрипционных факторов Scl и FOG-1 в CD34+-клетках больных хроническим идиопатическим миелофиброзом свидетельствует, скорее всего, о множественных молекулярных нарушениях, имеющих значение в патогенезе данного заболевания у человека.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Портальная гипертензия
Портальная гипертензия – синдром, развивающийся вследствие нарушения кровотока и повышения кровяного давления в бассейне воротной вены. Портальная гипертензия характеризуется явлениями диспепсии, варикозным расширением вен пищевода и желудка, спленомегалией, асцитом, желудочно-кишечными кровотечениями. В диагностике портальной гипертензии ведущее место занимают рентгеновские методы (рентгенография пищевода и желудка, кавография, портография, мезентерикография, спленопортография, целиакография), чрескожная спленоманометрия, ЭГДС, УЗИ и др. Радикальное лечение портальной гипертензии – оперативное (наложение портокавального анастомоза, селективного спленоренального анастомоза, мезентерико-кавального анастомоза).
МКБ-10
Общие сведения
Под портальной гипертензией (портальной гипертонией) понимается патологический симптомокомплекс, обусловленный повышением гидростатического давления в русле воротной вены и связанный с нарушением венозного кровотока различной этиологии и локализации (на уровне капилляров или крупных вен портального бассейна, печеночных вен, нижней полой вены). Портальная гипертензия может осложнять течение многих заболеваний в гастроэнтерологии, сосудистой хирургии, кардиологии, гематологии.
Причины
Этиологические факторы, приводящие к развитию портальной гипертензии, многообразны. Ведущей причиной выступает массивное повреждение печеночной паренхимы вследствие заболеваний печени:
- острых и хронических гепатитов
- опухолей печени, холедоха
- паразитарных инфекций (шистосоматоза),
- интраоперационном повреждении или перевязке желчных протоков.
Определенную роль играет токсическое поражение печени при отравлениях гепатотропными ядами (лекарствами, грибами и др.). К развитию портальной гипертензии может приводить заболевания сосудистого генеза:
- тромбоз, врожденная атрезия, опухолевое сдавление или стеноз портальной вены
- тромбоз печеночных вен при синдроме Бадда-Киари
- повышение давления в правых отделах сердца при рестриктивной кардиомиопатии, констриктивном перикардите.
В некоторых случаях развитие портальной гипертензии может быть связано с критическими состояниями при операциях, травмах, обширных ожогах, ДВС-синдроме, сепсисе. Непосредственными разрешающими факторами, дающими толчок к развитию клинической картины портальной гипертензии, нередко выступают инфекции, желудочно-кишечные кровотечения, массивная терапия транквилизаторами, диуретиками, злоупотребление алкоголем, избыток животных белков в пище, операции.
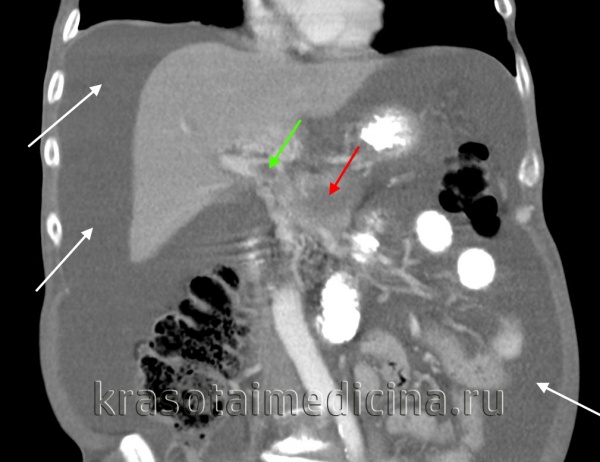
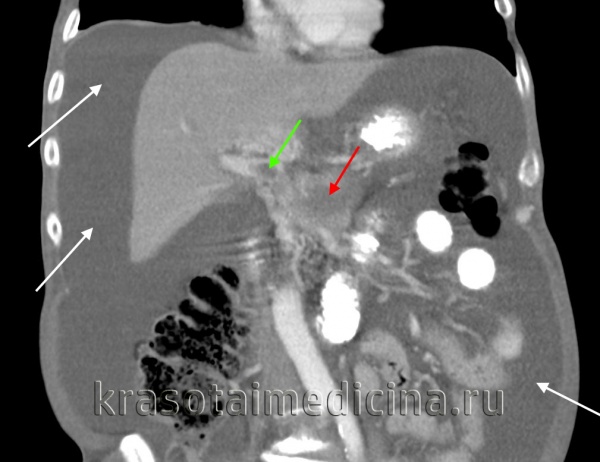
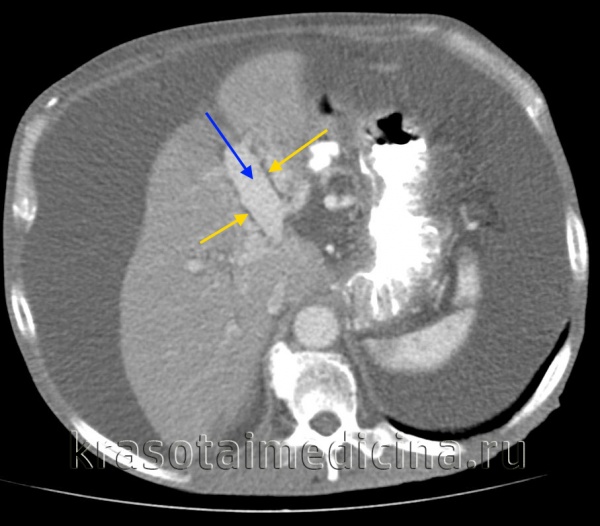
КТ ОБП. Портальная гипертензия на фоне обструкции воротной вены опухолью поджелудочной железы (красная стрелка). Опухолевый тромб (зеленая стрелка) в воротной вене. Асцит (белая стрелка).
Патогенез
Основными патогенетическими механизмами портальной гипертензии выступают наличие препятствия для оттока портальной крови, увеличение объема портального кровотока, повышенное сопротивление ветвей воротной и печеночных вен, отток портальной крови через систему коллатералей (потртокавальных анастомозов) в центральные вены.
В клиническом течении портальной гипертензии может быть выделено 4 стадии:
- начальная (функциональная)
- умеренная (компенсированная) – умеренная спленомегалия, незначительное расширение вен пищевода, асцит отсутствует
- выраженная (декомпенсированная) – выраженные геморрагический, отечно-асцитический синдромы, спленомегалия
- портальная гипертензия, осложненная кровотечением из варикозно-расширенных вен пищевода, желудка, прямой кишки, спонтанным перитонитом, печеночной недостаточностью.
Классификация
В зависимости от распространенности зоны повышенного кровяного давления в портальном русле различают тотальную (охватывающую всю сосудистую сеть портальной системы) и сегментарную портальную гипертензию (ограниченную с нарушением кровотока по селезеночной вене с сохранением нормального кровотока и давления в воротной и брыжеечных венах).
По локализации венозного блока выделяют предпеченочную, внутрипеченочную, постпеченочную и смешанную портальную гипертензию. Различные формы портальной гипертензии имеют свои причины возникновения.
- Развитие предпеченочной портальной гипертензии (3-4 %) связано с нарушением кровотока в портальной и селезеночных венах вследствие их тромбоза, стеноза, сдавления и т. д.
- В структуре внутрипеченочной портальной гипертензии (85-90 %) различают пресинусоидальный, синусоидальный и постсинусоидальный блок. В первом случае препятствие на пути внутрипеченочного кровотока возникает перед капиллярами-синусоидами (встречается при саркоидозе, шистосомозе, альвеококкозе, циррозе, поликистозе, опухолях, узелковой трансформации печени); во втором – в самих печеночных синусоидах (причины - опухоли, гепатиты, цирроз печени); в третьем – за пределами печеночных синусоидов (развивается при алкогольной болезни печени, фиброзе, циррозе, веноокклюзионной болезни печени).
- Постпеченочная портальная гипертензия (10-12%) бывает обусловлена синдромом Бадда-Киари, констриктивным перикардитом, тромбозом и сдавлением нижней полой вены и др. причинами. При смешанной форме портальной гипертензии имеет место нарушение кровотока, как во внепеченочных венах, так и в самой печени, например, при циррозе печени и тромбозе воротной вены.
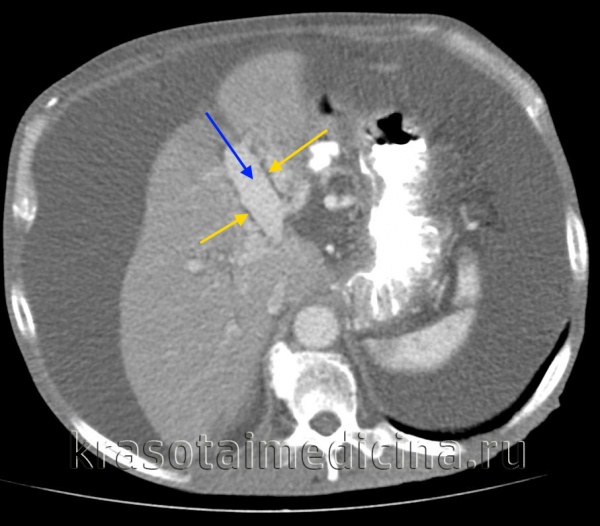
КТ ОБП. Этот же пациент, расширенный левый долевой ствол воротной вены (синяя стрелка) с периваскулярным отеком (желтая стрелка).
Симптомы портальной гипертензии
Наиболее ранними клиническими проявлениями портальной гипертензии служат диспепсические симптомы: метеоризм, неустойчивый стул, чувство переполнения желудка, тошнота, ухудшение аппетита, боли в эпигастрии, правом подреберье, подвздошных областях. Отмечается появление слабости и быстрой утомляемости, похудание, развитие желтухи.
Иногда первым признаком портальной гипертензии становится спленомегалия, выраженность которой зависит от уровня обструкции и величины давления в портальной системе. При этом размеры селезенки становятся меньше после желудочно-кишечных кровотечений и снижения давления в бассейне портальной вены. Спленомегалия может сочетаться с гиперспленизмом – синдромом, характеризующимся анемией, тромбоцитопенией, лейкопенией и развивающимся в результате повышенного разрушения и частичного депонирования в селезенке форменных элементов крови.
Асцит при портальной гипертензии отличается упорным течением и резистентностью к проводимой терапии. При этом отмечается увеличение объемов живота, отеки лодыжек, при осмотре живота видна сеть расширенных вен передней брюшной стенке в виде «головы медузы».
Осложнения
Характерными и опасными проявлениями портальной гипертензии являются кровотечения из варикозно измененных вен пищевода, желудка, прямой кишки. Желудочно-кишечные кровотечения развиваются внезапно, имеют обильный характер, склонны к рецидивам, быстро приводят к развитию постгеморрагической анемии. При кровотечении из пищевода и желудка появляется кровавая рвота, мелена; при геморроидальном кровотечении – выделение алой крови из прямой кишки. Кровотечения при портальной гипертензии могут провоцироваться ранениями слизистой, увеличением внутрибрюшного давления, снижением свертываемости крови и т. д.
Диагностика
Выявить портальную гипертензию позволяет тщательное изучение анамнеза и клинической картины, а также проведение совокупности инструментальных исследований. При осмотре больного обращают внимание на наличие признаков коллатерального кровообращения: расширения вен брюшной стенки, наличия извитых сосудов около пупка, асцита, геморроя, околопупочной грыжи и др.
- Лабораторный комплекс. Объем лабораторной диагностики при портальной гипертензии включает исследование клинического анализа крови и мочи, коагулограммы, биохимических показателей, АТ к вирусам гепатита, сывороточных иммуноглобулинов (IgA , IgM , IgG).
- Рентгенография. В комплексе рентгеновской диагностики используется кавография, портография, ангиография мезентериальных сосудов, спленопортография, целиакография. Данные исследования позволяют выявить уровень блокировки портального кровотока, оценить возможности наложения сосудистых анастомозов. Состояние печеночного кровотока может быть оценено в ходе статической сцинтиграфии печени.
- Сонография. УЗИ брюшной полости необходимо для выявления спленомегалии, гепатомегалии, асцита. С помощью допплерометрии сосудов печени производится оценка размеров воротной, селезеночной и верхней брыжеечной вен, расширение которых позволяет судить о наличии портальной гипертензии.
- Функциональные исследования. С целью регистрации давления в портальной системе прибегают к проведению чрескожной спленоманометрии. При портальной гипертензии давление в селезеночной вене может достигать 500 мм вод. ст., тогда как в норме оно составляет не более 120 мм вод. ст.
Обследование пациентов с портальной гипертензией предусматривает обязательное проведение эзофагоскопии, ФГДС, ректороманоскопии, позволяющих обнаружить варикозное расширение вен ЖКТ. Иногда вместо эндоскопии проводится рентгенография пищевода и желудка. К биопсии печени и диагностической лапароскопии прибегают в случае необходимости получения морфологических результатов, подтверждающих заболевание, приведшее к портальной гипертензии.
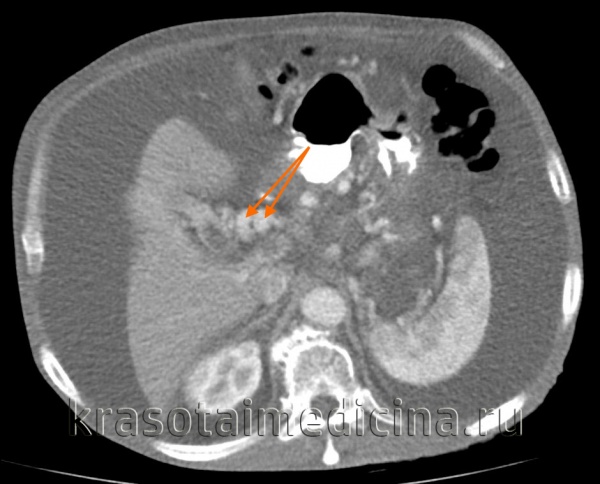
КТ ОБП. Этот же пациент, варикозно расширенные вены как проявление синдрома портальной гипертензии (коричневая стрелка).
Лечение портальной гипертензии
Терапевтические методы лечения портальной гипертензии могут быть применимы только на стадии функциональных изменений внутрипеченочной гемодинамики. В терапии портальной гипертензии используется:
- Фармакотерапия: нитраты (нитроглицерин, изосорбид), β-адреноблокаторы (атенолол, пропранолол), ингибиторы АПФ (эналаприл, фозиноприл), гликозаминогликаны (сулодексид) и др.
- Эндоскопические манипуляции. При остро развившихся кровотечениях из варикозно-расширенных вен пищевода или желудка прибегают к их эндоскопическому лигированию или склерозированию. При неэффективности консервативных вмешательств показано прошивание варикозно-измененных вен через слизистую оболочку.
- Оперативное лечение. Основными показаниями к хирургическому лечению портальной гипертензии служат желудочно-кишечные кровотечения, асцит, гиперспленизм. Операция заключается в наложении сосудистого портокавального анастомоза, позволяющего создать обходное соустье между воротной веной или ее притоками (верхней брыжеечной, селезеночной венами) и нижней полой веной или почечной веной. В зависимости от формы портальной гипертензии могут быть выполнены операции прямого портокавального шунтирования, мезентерикокавального шунтирования, селективного спленоренального шунтирования, трансъюгулярного внутрипечёночного портосистемного шунтирования, редукции селезеночного артериального кровотока, спленэктомия.
- Паллиативные вмешательства. Паллиативными мерами при декомпенсированной или осложненной портальной гипертензии, могут являться дренирование брюшной полости, лапароцентез.
Прогноз
Прогноз при портальной гипертензии, обусловлен характером и течением основного заболевания. При внутрипеченочной форме портальной гипертензии исход, в большинстве случаев, неблагоприятный: гибель пациентов наступает от массивного желудочно-кишечного кровотечения и печеночной недостаточности. Внепеченочная портальная гипертензия имеет более доброкачественное течение. Наложение сосудистых портокавальных анастомозов может продлить жизнь иногда на 10—15 лет.
Миелофиброз ( Агногенная миелоидная метаплазия , Сублейкемический миелоз )
Миелофиброз – это хроническое гематологическое заболевание, характеризующееся опухолевой пролиферацией гемопоэтических стволовых клеток и фиброзом костного мозга. Основные клинические проявления включают симптомы опухолевой интоксикации и анемического синдрома (прогрессирующую слабость, бледность кожи и слизистых оболочек, потерю веса), а также увеличение селезенки (спленомегалию). Диагноз устанавливается на основании молекулярно-генетических исследований, изучения гистологической картины костного мозга. Лечение проводится с помощью химиотерапевтических препаратов. Хирургические методы лечения подразумевают трансплантацию костного мозга и удаление селезенки.
Миелофиброз (агногенная миелоидная метаплазия, сублейкемический миелоз) – злокачественное заболевание, при котором происходит постепенное замещение костного мозга опухолевыми стволовыми клетками и разрастающейся соединительной тканью. Впервые эту патологию описал немецкий врач Г. Хойк в 1879 году. А в 1951 году американским гематологом Уильямом Дамешеком миелофиброз был выделен в самостоятельную нозологическую единицу. При неблагоприятном течении миелофиброз способен трансформироваться в более тяжелую болезнь ‒ острый лейкоз. Распространенность миелофиброза составляет от 0,3 до 0,7 случаев на 100 тыс. населения. Пик заболеваемости приходится на возраст от 50 до 70 лет, но встречаются и молодые пациенты. Чаще страдают мужчины.
Причины миелофиброза
Существует первичный и вторичный сублейкемический миелоз. Точная причина первичного миелофиброза до сих пор не установлена. Наибольшей популярностью среди специалистов в области гематологии пользуется теория влияния генетической мутации. У большинства пациентов выявляются мутации гена тирозинкиназы (JAK2V617F), кальретикулина (CALR), тромбопоэтина (MPL), регулирующих экспрессию белков JAK-STAT сигнального пути. Гены локализуются в локусе хромосомы del3p24.
В качестве этиологического фактора изучается действие большой дозы радиоактивного излучения. Также рассматривается роль персистирующих вирусных инфекций (вируса простого герпеса, Эпштейна-Барра, цитомегаловируса), длительного приема оральных контрацептивов, миелосупрессивных лекарственных препаратов, контакта с различными органическими и неорганическими соединениями (бензолом, мышьяком). Вторичный миелофиброз развивается как исход других хронических миелопролиферативных заболеваний – истинной полицитемии, эссенциальной тромбоцитемии, хронического миелолейкоза.
В результате повышенной экспрессии сигнальных белков в одной из стволовых костномозговых клеток запускается активная пролиферация (опухолевая трансформация). Этот процесс сопровождается вторичным воспалением с выделением цитокинов и факторов роста. Факторы роста фибробластов и эндотелия сосудов индуцируют выработку стромальными клетками костного мозга большого количества коллагена и разрастание соединительной ткани (собственно фиброз). Постепенно нормальная ткань костного мозга замещается опухолью и соединительной тканью.
При массивном поражении опухолью костного мозга клетки крови, не достигнув стадии полного созревания, попадают в системный кровоток. Это приводит к образованию очагов экстрамедуллярного (внекостномозгового) кроветворения, главным образом в печени и селезенке. Распад опухоли ведет к высвобождению мочевой кислоты, которая откладывается в тканях суставов и почечных канальцах.
Симптомы миелофиброза
Длительное время пациент чувствует себя удовлетворительно. Через несколько лет от начала заболевания постепенно появляется опухолевая интоксикация в виде общей слабости, повышения температуры до субфебрильных цифр, потливости, усиливающейся по ночам. У больного снижается аппетит, он стремительно теряет в весе. Присоединяется анемический синдром (бледность кожных покровов, головокружение, учащение сердцебиения). Характерны носовые, десневые кровотечения, геморрагические высыпания на коже. Возникают боли в суставах, кожный зуд, боли в костях.
Пациент ощущает тяжесть и боли в левом подреберье вследствие выраженного увеличения селезенки. На фоне спленомегалии развивается синдром гиперспленизма, который заключается в массивном разрушении клеток крови (в основном эритроцитов) в синусоидах селезенки. В этом случае встречаются признаки гемолиза (желтушность кожи, слизистых оболочек, потемнение мочи).
Редкие симптомы связаны с необычной локализацией очагов экстрамедуллярного кроветворения – в легких (кашель, затруднение дыхания, кровохарканье), желудочно-кишечном тракте (боли в животе, кровавая диарея). При расположении очагов в центральной и периферической нервной системе наблюдаются эпилептические судороги, нарушения чувствительности, слабость движений в конечностях, вплоть до полного паралича.
При миелофиброзе часто образуются тромбы, которые приводят к острому нарушению мозгового кровообращения, инфаркту миокарда, тромбоэмболии легочной артерии. Стойкое снижение уровня лейкоцитов нередко сопряжено с различными инфекциями, приобретающими тяжелое течение. Наиболее неблагоприятным осложнением считается трансформация миелофиброза в миелолейкоз (бластный криз), трудно поддающийся терапии. К нетипичным осложнениям следует отнести патологические переломы из-за деструкции трубчатых костей и портальную гипертензию, причиной которой служит длительная обструкция микротромбами внутрипеченочных вен.
Курацией пациентов с миелофиброзом занимаются врачи-гематологи. При общем осмотре обращает на себя внимание изменение цвета кожных покровов, слизистых (бледность или желтушность), спленомегалия при пальпации и перкуссии селезенки, иногда достигающей гигантских размеров (до лобкового симфиза). Дополнительные методы диагностики включают:
- Общие лабораторные исследования. В начале заболевания в общем анализе крови выявляется увеличение эритроцитов, тромбоцитов, лейкоцитов, со временем сменяющееся на низкие показатели. Часто в периферической крови присутствуют незрелые формы эритроцитов, лейкоцитов (миелоциты, промиелоциты). В биохимическом анализе крови наблюдаются повышенные концентрации лактатдегидрогеназы (ЛДГ), ионизированного кальция. Отмечаются изменения коагулограммы – ускорение свертывания крови, уменьшение активированного частичного тромбопластинового времени, торможение процессов фибринолиза. В анализе мочи обнаруживаются уробилин, гемоглобин, ураты (соли мочевой кислоты).
- Исследование костного мозга. Образец костного мозга получают с помощью трепанобиопсии. Гистологическая картина зависит от фазы заболевания. Для ранней (префибротической фазы) характерны гиперплазия всех ростков кроветворения (гранулоцитарного, мегакариоцитарного, эритроидного) с незрелостью клеток. В позднюю (фибротическую) фазу определяется большое количество коллагеновых и ретикулярных волокон (фиброз), замещающих гемопоэтическую ткань, выраженная клеточная атипия. Высокий уровень бластных клеток (более 20%) свидетельствует о трансформации миелофиброза в острый лейкоз.
- Молекулярно-генетические тесты. Диагностика мутации генов JAK2V617F, CALR, MPL осуществляется методом FISH. Для идентификации аллельной нагрузки мутации проводится полимеразная цепная реакция real-time. Также выполняется HLA-типирование для решения вопроса о возможности трансплантации костного мозга.
- Цитогенетические и цитохимические анализы. При цитогенетическом исследовании (кариотипировании) клеток костного мозга находят аномалии 1, 3, 6 хромосом (транслокация, трисомия, комплексные нарушения). При анализе химического состава (цитохимии) нейтрофилов активность щелочной фосфатазы оказывается в 3 раза выше нормы.
Для достоверной постановки диагноза гематологическим сообществом разработаны специальные критерии. Большие критерии включают повышенную клеточность костного мозга с ретикулярным и коллагеновым фиброзом, наличие мутаций генов JAK2V617F, MPL, CALR. К малым критериям относятся анемия, спленомегалия, лейкоэритробластоз (присутствие в крови незрелых форм лейкоцитов, эритроцитов), а также повышение лактатдегидрогеназы. Диагноз считается подтвержденным, если имеются 2 больших критерия или 1 большой и 3 малых критерия.
Миелофиброз следует дифференцировать в первую очередь с гематологическими заболеваниями, такими как аутоиммунные гемолитические анемии, гемобластозы (лейкозы, лимфомы). Сочетание спленомегалии с симптомами интоксикации (слабостью, субфебрилитетом, ночной потливостью) требует исключения туберкулеза, подострого инфекционного эндокардита.
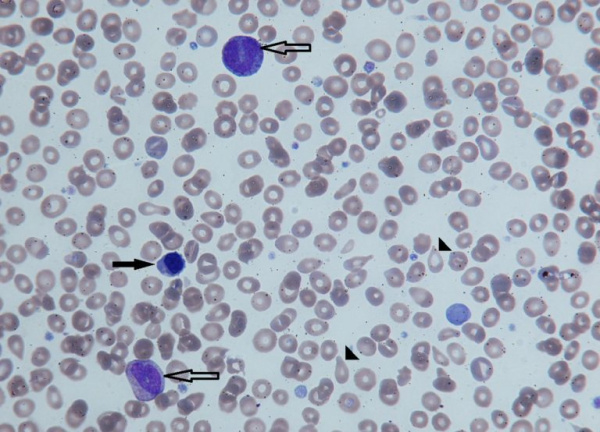
Незрелые формы эритроцитов (черная стрелка) и гранулоцитов (контурная стрелка) в периферической крови
Лечение миелофиброза
После постановки диагноза пациент должен быть госпитализирован в гематологический стационар. Для принятия решения о выборе тактики лечения необходимо определить степень риска, а именно - вероятность бластной трансформации и ориентировочную продолжительность жизни. С этой целью была создана «Международная шкала оценки риска и прогноза» (DIPSS). Она учитывает возраст пациента, количество форменных элементов крови, выраженность симптомов опухолевой интоксикации. Каждый признак соответствует одному баллу. Различают низкий, первый и второй промежуточный, высокий риски, при которых проводится дифференцированная терапия:
Прогноз и профилактика
Миелофиброз – это тяжелое заболевание с неблагоприятным прогнозом. С момента постановки диагноза средняя продолжительность жизни составляет около 5 лет. При манифестации в более молодом возрасте миелофиброз имеет менее агрессивное течение, что сопряжено с лучшим ответом на терапию и большей выживаемостью больных. Эффективных методов профилактики не разработано ввиду неизвестности этиологического фактора. Предупреждение развития вторичного миелофиброза заключается в своевременной диагностике и лечении патологий, на фоне которых он возникает - истинной полицитемии и эссенциальной тромбоцитемии.
2. Патофизиологические основы лечения сублейкемического миелоза. Патофизиология крови. Экстремальные состояния/ Под ред. А.И. Воробьева и Н.А. Горбуновой - 2004.
3. Критерии диагностики и современные методы лечения первичного миелофиброза/ Абдулкадыров К. М., Шуваев В. А., Мартынкевич И. С.// Вестник гематологии. - 2013 - №9(3).
4. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитопения, первичный миелофиброз. - 2014.
Клиника хронического идиопатического миелофиброза (ХИМФ) - спленомегалия, портальная гипертензия, анемия, асцит
Картина крови при хроническом идиопатическом миелофиброзе (ХИМФ) отличается большим разнообразием. Показатели красной крови на момент постановки диагноза чаще нормальны или несколько снижены, но у 15—25 % больных уже имеется анемия. Повышение показателей красной крови, обычно клинически бессимптомное, отмечается у 5—6 % больных.
У большинства больных наблюдается нейтрофильный лейкоцитоз (10—15•10 9 /л), очень характерен выраженный палочкоядерный сдвиг и присутствие единичных мета- и миелоцитов.
У 10—15 % больных наблюдается более выраженный лейкоцитоз (>20•10 9 /л) и большая степень левого сдвига лейкоцитарной формулы крови, циркуляция единичных бластов. Число тромбоцитов нормально у 30—40 % больных и увеличено примерно у 15 %. Отмечается повышение уровня лактатдегидрогеназы, коррелирующее с числом лейкоцитов. Морфологические изменения эритроцитов — частая находка при хроническом идиопатическом миелофиброзе (ХИМФ). Клинические симптомы хронического идиопатического миелофиброза можно разделить на:
1) ассоциированные со значительным увеличением селезенки;
2) обусловленные усиленным клеточным катаболизмом;
3) обусловленные недостаточностью костного мозга.
Под последними имеются в виду анемический и тромбоцитопенический синдромы, хотя в действительности в их развитии участвуют разные патогенетические механизмы.
В зависимости от давности хронического идиопатического миелофиброза (ХИМФ) размеры селезенки широко варьируют. Со временем она достигает гигантских размеров, занимает всю левую и часть правой половины живота, отличается повышенной плотностью и бугристостью. Субъективные расстройства, вызываемые большой селезенкой, — это чувство тяжести, ощущение сдавления (малой вместимости) желудка и кишечника (неустойчивый стул), периодические острые боли, вызываемые инфарктом селезенки и периспленитом.
Классической причиной увеличения селезенки оказывается миелоидная метаплазия селезенки (ММС), но следующей возможной причиной спленомегалии является осложнение портальной гипертонией, а также увеличение депонирующей и секвестрирующей функций селезенки, т. е. рабочая гипертрофия органа.

Гепатомегалия и портальная гипертензия при хроническом идиопатическом миелофиброзе
Более чем у половины больных при установлении диагноза определяют гепатомегалию. Изолированное увеличение печени изредка возможно, как и преобладание гепатомегалии над спленомегалией. Значительное увеличение печени наблюдается обычно у больных, перенесших спленэктомию (СЭ), а частота прогрессирующей гепатомегалии после СЭ составляет 26—22 %.
Функциональные нарушения печени редки, чаще наблюдаются в терминальной стадии заболевания.
Развитие синдрома портальной гипертонии сопровождается значительным увеличением селезенки, не обусловленным ее участием в кроветворении, варикозным расширением вен пищевода, а затем периферическими отеками и асцитом. По данным Silverstein, портальная гипертония осложняет течение хронического идиопатического миелофиброза в 6—8 %, из них в 70 % она является результатом гиперкинетического тока крови и в 30 % — внутрипеченочных блоков. Их причины — миелоидная метаплазия, которая локализуется в синусоидах печени, вызываемый ею реактивный фиброз, а в отдельных случаях — постнекротический цирроз печени, обусловленный перенесенным гепатитом.
Среди вариантов портальной гипертонии, частота которой другими авторами оценивается в 10—15 %, выделяют пресинусоидальный тромботический блок, синусоидальную обструкцию, вызываемую миелоидной метаплазией печени в сочетании с гиперкинетическим током крови, постсинусоидальный, очевидно, тромботический блок, аналогичный синдрому Бадда — Киари.
Считается, что при функциональном внутрипеченочном портальном блоке анатомических изменений в печени нет, но есть и такая точка зрения, что для развития портальной гипертонии одного гиперкинетического тока крови недостаточно и структурные нарушения в печени должны присутствовать.
Уровень блока току крови в портальной системе в настоящее время определяется неинвазивным методом — ультразвуковой допплерографией.
При подпеченочной портальной гипертензии (ПГ) (тромбоз селезеночной или воротной вены) имеются большие размеры селезенки и отсутствие или незначительность увеличения печени. При пункции селезенки кровь поступает в шприц под большим давлением, в пунктате селезенки элементов миелоидной метаплазии обычно мало, за исключением случаев с большой продолжительностью заболевания и смешанным генезом спленомегалии.
При осложнении тромбозом надпеченочных вен клиническая картина выглядит значительно более драматично: болевой синдром имеет различную выраженность, но может и отсутствовать; формируется массивный, резистентный к лечению асцит, нередко желтуха, признаки печеночной недостаточности (клинические и лабораторные) тяжелое общее истощение; возможны периодические кровотечения из расширенных вен пищевода и желудка (кровавая рвота и мелена). Печень обычно значительно увеличена, тогда как селезенка — умеренно. Течение синдрома Бадда — Киари может быть острым, подострым и хроническим.
Неизвестно почему, но у всех шести наблюдаемых нами больных это осложнение возникло на раннем этапе, до постановки гематологического диагноза, и у женщин молодого возраста. Изменения в анализах крови были не столь однозначными, чтобы на их основании поставить гематологический диагноз и уточнить, какой именно. С подобной ситуацией сталкиваются и другие авторы. Расширение возможностей углубленного обследования больных с помощью мегакариоцитарной и эритроидной культур и цитогенетического анализа позволило прийти к заключению, что у 2/3 больных этот синдром является осложнением ХМПЗ.
Склонность к тромбозам в системе воротной, надпеченочных и чревных вен в целом отмечена преимущественно у больных эссенциальной тромбоцитемией (ЭТ) с моноклональным ге-мопоэзом, у носителей гена PRV-1, у больных со спонтанным ростом эритроидной культуры. Пока неизвестно, в какой степени эти патогенетические особенности распространяются на больных хроническим идиопатическим миелофиброзом (ХИМФ).
В клиническом отношении значение проблемы портальной гипертензии у больных хроническим идиопатическим миелофиброзом весьма существенно. Ее своевременная диагностика позволяет принять решение в пользу назначения спленэк-томии у больных с под- и внутрипеченочной портальной гипертензией и назначить адекватное консервативное или хирургическое (наложение шунтов) лечение при осложнении тромбозом надпеченочных вен.
Чаще всего большие размеры селезенки врачи относят за счет основного заболевания и проводят довольно агрессивную терапию с целью ее сокращения, что в случаях осложнения портальной гипертензией обречено на неудачу и может привести к серьезным проблемам.
В одном нашем наблюдении больную лечили по месту жительства миелосаном в течение 6 мес с целью сокращения размеров селезенки. В анализах крови наблюдались только небольшой тромбоцитоз и нейтрофилез. Заболевание расценивалось как сублейкемический миелоз. Селезенка была увеличена до уровня пупка. К концу этого лечения развился тяжелый геморрагический тромбоцитопенический синдром, в течение 3 мес больная находилась в критическом состоянии. Еще через 3 мес она была подвергнута спленэктомии. На операции выявлен цирроз печени без миелоидной метаплазии селезенки (ММС). Диагноз пересмотрен в пользу эссенциальной тромбоцитемии (ЭТ).
В течение последующих 5 лет больная находилась в хорошем состоянии и принимала гидроксимочевину в небольшой дозе, которая контролировала тромбоцитоз. Затем внезапно развился тромбоз в системе мезентериальных сосудов, распознанный с опозданием. Во время операции удалена значительная часть тонкого кишечника.
Случай демонстрирует часто имеющую место неточность гематологического диагноза в группе ХМПЗ, нераспознанную внутрипеченочную портальную гипертензию, в данном случае, видимо, обусловленную сопутствующим заболеванием, неадекватную цитостатическую терапию, осложнившуюся гипоплазией кроветворения и геморрагическим синдромом, а также развитие тромбоза мезентериальных сосудов при контролируемом тромбоцитозе.
Причиной развития асцита может оказаться не только портальная гипертензия, но и имплантация очагов кроветворения на брюшине и сальнике. В таких случаях в асцитической жидкости обнаруживают мегакариоциты и гранулоциты. Плевральный и абдоминальный выпот часто носит геморрагический характер. Эта и другие атипичные локализации миелоидной метаплазии: в лимфатических узлах со сдавлением спинного мозга, тонком кишечнике, средостении, почках, легких, других висцеральных органах — относятся к числу раритетов. Имеются данные об увеличении периферических лимфатических узлов у 32 % больных, но, по нашим наблюдениям, это значительно более редкий феномен.
К симптомам, обусловленным клеточным гиперкатаболизмом, относятся потеря массы тела и повышение температуры тела, гиперурикемия. Она может быть бессимптомной или протекать с признаками подагрической полиартралгии, подагры, мочекаменной болезни, осложняться хроническим пиелонефритом, обтурацией мочеточников, хронической почечной недостаточностью. У отдельных больных интенсивность камнеобразования в почках необычайно велика. Развитию урикемии способствует проведение массивной цитостатической терапии .
Хотя повышение температуры тела может быть результатом клеточного гиперкатаболизма, это справедливо по отношению к умеренному субфебрилитету, а значительные подъемы температуры тела обычно обусловлены инфекцией, особенно мочевыводящих путей, или латентно протекающим МДС-синдромом, который может проявиться как типичный, развернутый острый лейкоз через ряд месяцев и даже лет.

В случаях, протекающих с количественной и качественной патологией тромбоцитов, возможны сосудистые осложнения: тромботические микроциркуляторные расстройства, тромбозы артерий и вен, геморрагический синдром, ДВС-синдром.
Внутренние кровотечения обычно обусловлены разрывом вен пищевода при осложнении портальной гипертонией. Присущая этому заболеванию, как и другим ХМПЗ, качественная дефектность тромбоцитов объясняет появление экхимозов на коже при сравнительно умеренной тромбоцитопении. Несостоятельность гемостаза особенно четко проявляет себя при спленэктомии.
Анемический синдром нередко выходит на передний план, особенно в поздних стадиях заболевания. Его причины разнообразны. Среди них могут быть:
• недостаточное образование эритроцитов;
• гиперволемия;
• усиление депонирования и секвестрации клеток крови в увеличенной селезенке (гиперспле-низм);
• аутоиммунный гемолиз эритроцитов;
• ускоренный гемолиз эритроцитов в результате синдрома пароксизмальной ночной гемоглобинурии или ферментных дефектов (дефицит Г-6-ФДГ и др.);
• дефицит железа и фолиевой кислоты.
Количественная недостаточность эритропоэза определяется замещением кроветворного костного мозга миелофиброзом и остеомиелосклерозом с возможным присутствием жировой ткани. Компенсаторный эритропоэз в трубчатых костях со временем также редуцируется, а компенсаторные возможности селезеночного эритропоэза ограничены его частой неэффективностью и одновременным усилением депонирования и деструкции клеток крови в большой селезенке.
Гемодилюционная анемия является результатом гиперволемии, обусловленной спленомегалией. Она хорошо переносится больными и является по существу только лабораторным феноменом.
Дефекты мембраны эритроцитов, сходные с наблюдаемыми при пароксизмальной ночной гемоглобинурии (ПНГ), описаны при хроническом идиопатическом миелофиброзе многими авторами. Их последствием является синдром гемолитической анемии. Повышенному гемолизу эритроцитов способствует и усиление перекисного окисления липидов клеточных мембран эритроцитов.
Дефицит фолиевой кислоты, приводящий к появлению макроцитарной анемии с кольцами Кебота, базофильной пунктацией эритроцитов, тельцами Жолли, наблюдается в поздней стадии заболевания. Его объясняют повышенным расходом фолиевой кислоты при усиленном гемопоэзе.
К количественной недостаточности эритропоэза приводит и его подавление при прогрессирующей гиперплазии лейкоцитарного ростка, хронической и острой. Возможно развитие сидеробластной анемии без- и с малопроцентной бластемией, которая является предстадией острого лейкоза. Описаны случаи парциальной красноклеточной аплазии, завершившиеся острым лейкозом. Отметим, что обычно к анемии приводит сочетание нескольких причинных факторов. Удельный вес каждого из них подлежит уточнению, что особенно необходимо при решении вопроса о назначении спленэктомии. Это же относится и к тромбоцитопеническому синдрому. Причинами его развития являются:
• усиление депонирования и деструкции тромбоцитов в увеличенной селезенке (и печени);
• вторичный аутоиммунный гемолиз тромбоцитов;
• нарушение образования тромбоцитов в результате редукции числа мегакариоцитов или их качественной дефектности;
• сочетание этих процессов;
• ДВС-синдром (тромбоцитопения потребления).
Среди известных клинических проявлений заболевания могут иметь место и аутоимунные симптомы, такие как дерматиты и кожные васкулиты, опосредованные активацией Т-лимфоцитов.
При рентгенографическом исследовании костного скелета обнаруживаются признаки уплотнения структуры плоских костей, особенно позвонков, иногда эбурнеация трубчатых костей с сужением их просвета, иногда определяют очаговый остеолиз.
Эволюция хронического идиопатического миелофиброза характеризуется постепенным нарастанием лейкоцитоза при исходно различном числе лейкоцитов: нормальном, сниженном и повышенном. Развитие острого лейкоза наблюдается как в случаях прогрессирующего лейкоцитоза (более 30•109/л), так и лейкопении (3•109/л). Большинство больных не доживает до развития типичного острого лейкоза. Картины рефрактерной анемии, тромбоцитопении, панцитопении или, наоборот, нарастающего лейкоцитоза и левого сдвига лейкоцитарной формулы, выход из-под контроля размеров селезенки, появление упорной асептической лихорадки являются показателями терминальной фазы заболевания.
Терминальное состояние больных может определяться и висцеральными осложнениями: сердечной, печеночной и почечной недостаточностью, к которым имеются соответствующие патофизиологические предпосылки. Жизнь больных может оборвать острое кровотечение из расширенных вен пищевода при осложнении портальной гипертонией. Гематологические и соматические терминальные состояния нередко сочетаются с тяжелой общей дистрофией и компрессионными осложнениями.
Течение заболевания обычно хроническое, медленно прогрессирующее. Длительное время оно может протекать почти полностью бессимптомно, и только случайное исследование анализа крови выявляет небольшие отклонения или осмотр позволяет обнаружить спленомегалию. При доброкачественном, медленно протекающем варианте хронического идиопатического миелофиброза (ХИМФ) наблюдаются «мягкая» степень развития миелофиброза или только клеточная пролиферация, отсутствие анемии, нормальное или незначительно повышенное число лейкоцитов и тромбоцитов, отсутствие циркулирующих бластов и незначительное число CD334-позитивных клеток в периферической крови. У подобных больных прогрессия заболевания может длительное время отсутствовать.
Возможны варианты более злокачественного течения, которые терминологически обозначаются как варианты с ускоренным развитием терминальной фазы. Выделяют и вариант с подострым течением, при котором морфологические изменения костного мозга не отличаются от обычного хронического идиопатического миелофиброза, но при этом нет значительной спленомегалии, и довольно быстро развиваются анемия и другие проявления недостаточности кроветворения.
К атипичным хронически протекающим вариантам хронического идиопатического миелофиброза можно отнести и так называемые гибридные формы, имеющие признаки двух ХМПЗ:
• истинной полицитемии и хронического идиопатического миелофиброза — упорная плетора, но раннее и значительное увеличение селезенки за счет миелоидной метаплазии, лейкоцитозные формы, устойчивость к цитостатической терапии, эволюция в острый лейкоз через длительный период зрелоклеточного лейкоцитоза, частые цитогенетические аномалии;
• хронического идиопатического миелофиброза и эссенциальная тромбоцитемия. С первым заболеванием их сближает выраженный миелофиброз, со вторым — небольшая величина селезенки, значительный тромбоцитоз, отсутствие лейкоэритробластической картины периферической крови, характерной для иМФ.
Атипичными в определенном смысле являются и случаи хронического идиопатического миелофиброза с существенным увеличением печени, а не селезенки, а также осложненные портальной гипертонией, синдромом Бадда — Киари, при которых проявления гематологического заболевания по анализу крови могут быть минимальными, и только культуральные и цитогенетические исследования выявляют их принадлежность к ХМПЗ.
Читайте также: