Клиника лейкоэнцефалита. Лейкоэнцефалит Ван-Богарта
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Лейкоэнцефалит (leukoencephalitis; греч. leukos белый + enkephalos головной мозг + -itis) — воспалительно-дистрофическое поражение белого вещества головного мозга. Лейкоэнцефалит относятся к демиелинизирующим заболеваниям (см.).
Впервые заболевание из группы Лейкоэнцефалитов описал Дансон (J. Danson) в 1933 г. под названием «подостро» форма летаргического энцефалита». В 4939 г. Петте и Деринг (H. Pette, G. Doring) сообщили об энцефалите с хроническим прогрессирующим течением, несколько отличающимся по клиническим и патоморфологическим проявлениям, назвав его узелковым панэнцефалитом. В 1945 г. это же заболевание описано Ван-Богартом (L. Van Bogaert) как «подострый склерозирующий лейкоэнцефалит». В дальнейшем Ван-Богарт тщательно изучил клинику и морфологию этой хронической прогрессирующей формы энцефалита. С группой Лейкоэнцефалита также сходны описанный в 1912 г. Шильдером (Р. F. Schilder) диффузный периаксиальный энцефалит и геморрагический Лейкоэнцефалит, о к-ром сообщил Херст (E. W. Hurst) в 1941 г.
Содержание
Этиология и патогенез
Предполагается, что Лейкоэнцефалит являются заболеваниями инфекционно-аллергической природы. Дискутируется роль миксовирусов, вирусов кори, бешенства и Herpes zoster как пусковых факторов гиперергического аутоиммунного процесса.
Классификация
Выделяют следующие клинико-морфол, формы Л.: подострый склерозирующий лейкоэнцефалит Ван-Богарта, периаксиальный лейкоэнцефалит Шильдера, острый геморрагический Л. При Л. демиелинизирующий процесс обычно сочетается с поражением нейронов в той или иной степени, поэтому для некоторых его форм употребляется также термин «панэнцефалит».
Патологическая анатомия
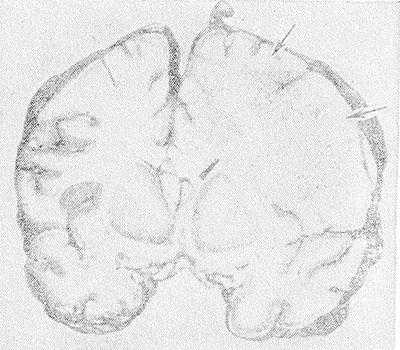
Рис. 1. Фронтальный срез головного мозга больного, умершего от геморрагического лейкоэнцефалита: белое вещество справа более отечное, усеяно мелкими петехиальными кровоизлияниями (указаны стрелками), прилежащие участки коры нечетко очерчены.
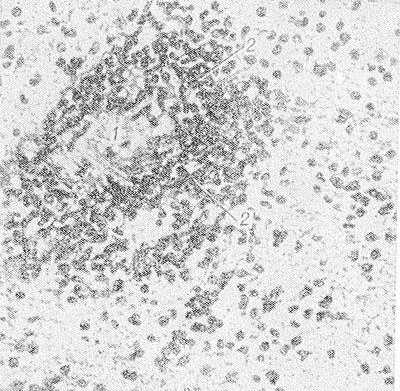
Рис. 2. Микропрепарат ткани белого вещества мозга больного, умершего от геморрагического лейкоэнцефалита: стенка кровеносного сосуда (1) инфильтрирована и окружающая нервная ткань пропитана полиморфно-ядерными лейкоцитами (2).
Макроскопическое исследование мозга при Л. выявляет расширение борозд и атрофию извилин. На срезе полушарий определяются различного размера участки деструкции и демиелинизации во всех отделах мозга, преимущественно в белом веществе, но захватывающие различные участки серого вещества коры (рис. 1). В наиболее пораженных отделах мозг имеет губчатую консистенцию, желудочки мозга умеренно расширены.
Гистол, картина характеризуется диффузной подострой воспалительной реакцией с периваскулярной инфильтрацией лимфоцитами и плазмоцитами и очаговой демиелинизацией (рис. 2). Воспалительные изменения преимущественно локализованы в белом веществе мозга, иногда в коре, подкорковых ганглиях, мозговых оболочках. Разрушается нормально сформированный миелин (миелинокластический тип поражения). Степень демиелинизации и деструкции нервной ткани варьирует в различных очагах. Отдельные мелкие очаги могут сливаться. У краев очага демиелинизации олигодендроциты увеличены, содержат амфофильные включения, в более пораженных участках они полностью исчезают. Кроме того, встречается много больших причудливой формы астроцитов с гиперхроматическими многодольчатыми или несколькими ядрами. Аксоны остаются относительно сохранными на ранних стадиях процесса, позднее в них могут быть дистрофические изменения. Нейроны коры полушарий большого мозга могут содержать включения двух типов: сферические частицы диам. 30—40 мкм и продолговатые, или тубулярные, структуры несколько меньшего диаметра. Включения чаще встречаются при небольшой длительности заболевания. Гистохимические исследования обнаруживают во включениях большое количество белка. В большинстве случаев находят пролиферативную реакцию глии. Глиоз может быть мелкоузелковый или в виде крупных очагов (псевдоопухоль). Диффузное разрастание волокнистой глии приводит иногда к уплотнению мозгового вещества, так что мозг на разрезе имеет хрящевидную консистенцию. Стенки артерий и вен утолщены, с избытком ретикулярных волокон в адвентиции.
Клиническая картина
Нервно-психические нарушения являются наиболее ранним проявлением заболевания. Вначале отмечаются жалобы на повышенную утомляемость, вялость, раздражительность, неустойчивость настроения. Постепенно круг нервно-психических расстройств расширяется. Появляется злобность, эффективность, жадность, эгоистичность, жестокость, недисциплинированность, инертность мышления. Больные часто совершают немотивированные поступки, теряют навыки опрятности.
На фоне психических нарушений постепенно в течение нескольких недель или месяцев прогрессирует очаговая неврологическая симптоматика: апрактические расстройства, приводящие к потере навыков самообслуживания (см. Апраксия); гностические нарушения (см. Агнозия); возникают расстройства чтения, письма, счета. У некоторых больных наблюдаются зрительные, слуховые галлюцинации. Двигательные нарушения вначале представлены преимущественно экстрапирамидными расстройствами: выявляется ригидность, феномен «зубчатого колеса» (см. Дрожательный паралич). Наблюдаются полиморфные гиперкинезы мышц лица, конечностей, туловища — тремор (см. Дрожание), торсионный спазм (см. Торсионная дистония), гемибаллизм (см. Гиперкинезы), миоклонии (см.). Пирамидные нарушения в типичных случаях развиваются на более поздних стадиях в виде моно-, геми- или тетрапарезов и параличей (см. Параличи, парезы). К часто встречающимся симптомам очагового поражения относятся статическая и локомоторная атаксия (см.) мозжечкового или лобного типа. Двустороннее поражение корково-ядерных путей приводит к нарушениям фонации, глотания. Бульбарный паралич развивается довольно редко.
Постоянным признаком заболевания являются судороги (см.). Они могут появляться на разных стадиях болезни. Наиболее характерны малые и абортивные судорожные припадки, реже генерализованные большие припадки. В поздней стадии заболевания развиваются трофические и вегетативные расстройства: кахексия, пролежни, нарушения терморегуляции, профузный пот и т. д. В терминальной стадии больные обездвижены, иногда наблюдается децеребрационная ригидность (см.).
Течение Лейкоэнцефалита может быть неуклонно прогрессирующим или ремиттирующим. В последнем случае клин, картина может напоминать рассеянный склероз (см.).
При электроэнцефалографии регистрируют периодическую пароксизмальную активность с интервалом 5 — 15 сек. одновременно в большинстве отведений в виде медленных (1—2 в 1 сек.) высоковольтажных волн.
В крови определяется лейкоцитоз, повышение фракции гамма-глобулина, обычно повышен титр антител к коревому вирусу или к миксовирусам (вирус jc, sv-40).
В цереброспинальной жидкости в большинстве случаев не наблюдается цитоза и увеличения содержания белка. Однако при электрофоретическом исследовании белков обнаруживают, что гамма-глобулин составляет до 40 и более процентов от общего количества белка, а фракция альбумина снижена. Коллоидные реакции дают максимальную флоккуляцию в первых пробирках (паралитический тип реакции Ланге).
Лечение
Лечение должно быть комплексным. Показана гормональная и симптоматическая терапия. Положительный эффект получают при назначении кортикостероидов. Лечение глюкокортикоидами (преднизолоном) следует начинать в ранней стадии патологического процесса с учетом ритма гормональной деятельности надпочечников. Гормональная терапия дополняется противоаллергическими (димедрол, пипольфен, супрастин, диазолин) и противосудорожными препаратами. Показаны препараты, снижающие мышечный тонус (мидокалм, амедин, мидантан, циклодол и др.), витамины группы В и другие симптоматические средства. Применение активной терапии может задерживать течение болезни и способствовать ремиссиям на несколько лет.
Прогноз
При неуклонно прогрессирующем течении больные погибают через 2—12 мес. после появления первых симптомов. При ремиттирующем течении заболевание длится до 3 лет и более, а ремиссии могут продолжаться от нескольких месяцев до нескольких лет, в течение которых симптомы заболевания почти или полностью отсутствуют.
Особенности отдельных форм лейкоэнцефалита
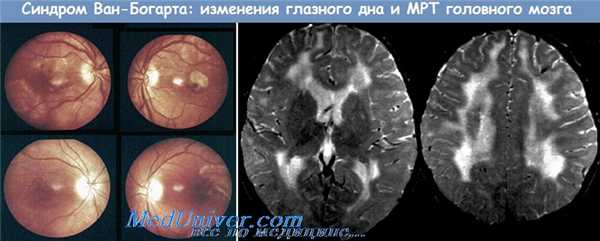
Подострый склерозирующий лейкоэнцефалит Ван-Богарта. При патоморфол, исследовании мозга больных с этой формой Л., как правило, обнаруживаются внутриклеточные включения. Степень поражения уменьшается в направлении от коры к филогенетически более древним образованиям, но чаще, чем при других формах, поражается ствол и спинной мозг.
Клинической особенностью этой формы Лейкоэнцефалита является раннее проявление и преобладание экстрапирамидных нарушений (Гиперкинетическая форма), к к-рым лишь на поздних этапах присоединяются пирамидные симптомы. Эпилептические припадки не характерны.
Периаксиальный диффузный лейкоэнцефалит Шильдера. Патоморфол, особенностью по сравнению с другими Л. и рассеянным склерозом является относительно ранняя дистрофия аксонов. Эта форма отличается от предыдущей преобладанием пирамидных симптомов и частыми эпилептическими припадками. Обычно наблюдаются большие припадки. Характерно развитие ретробульбарного неврита зрительных нервов или центральной формы слепоты, связанной с демиелинизацией затылочных долей (см. Шильдера болезнь).
Острый геморрагический лейкоэнцефалит. По клин, и патоморфол, признакам эта форма Л. сходна с вирусными и поствакцинальными энцефалитами. При патологоанатомическом исследовании выявляют отек мозга, на срезах в веществе мозга — большие очаги мягкой розовато-серой или желтоватой окраски с множественными точечными кровоизлияниями. Гистол. картина характеризуется фибринозным некрозом стенок мелких сосудов, в основном венул, окруженных экссудатом фибрина, воспалительными клетками и кольцевидными геморрагическими зонами. В этих же периваскулярных зонах— демиелинизация с умеренной или выраженной деструкцией аксонов. На самых ранних стадиях периваскулярные инфильтраты представлены гл. обр. нейтрофилами, однако в более старых очагах находят много лимфоцитов и плазмоцитов.
Клиника острого геморрагического Лейкоэнцефалита характеризуется чрезвычайно острым началом, молниеносным нарастанием тяжести симптомов поражения мозга. Заболевают лица обоего пола в возрасте от 20 до 40 лет. Длительность течения от 2 дней до 2 нед. Развернутой клин, картине предшествуют катаральные явления в зеве, лихорадка с лейкоцитозом в периферической крови. Через 2—4 дня появляется головная боль, ригидность мышц шеи, нарушается сознание, иногда развивается кома. Характерны фокальные или генерализованные судороги, двигательные нарушения в виде геми- или тетраплегии, псевдобульбарный паралич. На глазном дне — отек диска (соска) зрительного нерва. Редко наблюдаются подострые и хронические формы. С помощью ЭЭГ и артериографии могут быть обнаружены фокальные изменения. В цереброспинальной жидкости — выраженный плеоцитоз за счет полиморфно-ядерных лейкоцитов, встречаются также лимфоциты; содержание белка повышено до 1 г/л и более; часто выявляется ксантохромия цереброспинальной жидкости, микроскопически можно обнаружить единичные эритроциты.
Исход обычно летальный.
Библиография: Маркова Е. Д. и др. Клинико-анатомические и вирусологические данные в случае подострого склерозирующего панэнцефалита, Журн, невропат. и психиат., т. 77, № 7, с. 100 7, 1977, библиогр.; Цукер М. Б. Клиническая невропатология детского возраста, М., 1978, библиогр.; Чумаков М. Вирусологические аспекты изучения этиологии некоторых хронических заболеваний нервной системы (подострый склерозирующий панэнцефалит, вилюйский энцефаломиелит, множественный склероз), в кн.: Демиелинизирующие заболевания нервн. системы в Эксперим, и клин., под ред. А. И. Булыгина, с. 79, Минск, 1975; Balakоva H., Kvicalа V. Radiologicke a scintigraficke nalezy u akutnich zanetlivech procesii CNS, Cs. Neurol. Neurochir., sv. 40, s. 350, 1977; Gilroy J. a. Meyer J. S. Medical neurology, N. Y., 1975; Lhermitte F. Les leucoencephalites, P., 1950, bibliogr.; Pette H. u. Doring G. Uber einheimische Panencephalomyelitis vom charakter der Encephalitis japonica, Dtsch. Z. Nervenheilk., Bd 149, S. 7, 1939.
Клиника лейкоэнцефалита. Лейкоэнцефалит Ван-Богарта
Синдром Ван-Богарта (Van-Bogaert) - синонимы, авторы, клиника
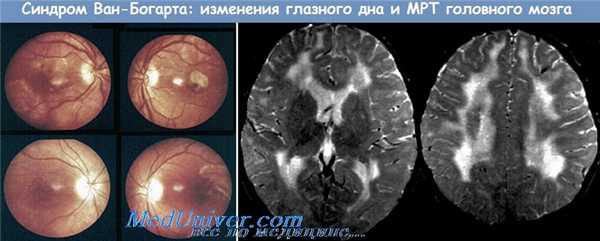
Синонимы синдрома Ван-Богарта. Подострый склерозирующий лейкоэнцефалит. Диффузный воспалительный склероз белого вещества мозговых полушарий.
Определение синдрома Ван-Богарта. Своеобразная, этиологически не ясная форма подострого энцефалита с преимущественной локализацией в подкорковых отделах.
Автор. Van Bogaert Ludo — современный бельгийский невропатолог, Антверпен. Впервые синдром описали van Bogaert и Busscher (одно наблюдение) под названием «диффузный воспалительный склероз белого вещества мозговых полушарий». В последующей работе, опубликованной в 1945 г., van Bogaert привел подробное описание синдрома, предложив его новое название—«подострый склерозирующий лейкоэнцефалит».
Симптоматология синдрома Ван-Богарта:
1. Заболевание обычно начинается в школьном возрасте и характеризуется подострым прогрессирующим фазообразным течением с продолжительностью от нескольких месяцев до нескольких лет.
2. Течение:
I стадия. Постепенно развивающиеся изменения в поведении и снижение интеллекта (медлительность, снижение успеваемости).
II стадия. К явлениям деперсонализации присоединяются ритмические экстрапирамидные гиперкинезы, внезапно возникающие ритмические судороги типа хореи, атетоза и бализма или миоклонические судороги джексоновского типа отдельных конечностей или всего тела. Эти судороги развиваются приступообразно, приводят к значительному снижению мышечного тонуса, что нередко является первым обнаруживавмым проявлением заболевания. Часто отмечают также приступы немотивированной плаксивости и крикливости.
III стадия. Постепенно полностью прекращается интеллектуальная активность, усиливается экстрапирамидный тонус вплоть до полной децеребрации; нарушаются центральные вегетативные регулирующие механизмы (потливость, гипертермия центрального происхождения), наступает кахексия.
3. Спинномозговая жидкость: нормальный цитоз, незначительное увеличение белка; глубокий, широкий зубец в кривой реакции Ланге.
4. На пневмоэнцефалограмме существенных изменений нет.
5. ЭЭГ: приступы ритмичных экстрапирамидных моторных нарушений соответствуют периодически возникающим колебаниям потенциалов с большой амплитудой.
Этиология и патогенез синдрома Ван-Богарта. Возможно, заболевание идентично или близко к так называемому панэнцефалиту (японский энцефалит), описанному Pette и Doring, а также энцефалиту, при котором обнаруживают клеточные включения (Dawson). Во всяком случае, клинические и гистопатологические проявления этих трех заболеваний весьма близки друг к другу. Этиологический агент этих трех заболеваний не выявлен.
Дифференциальный диагноз синдрома Ван-Богарта. Сифилис головного мозга. Опухоли основания мозга и мозгового ствола. Рассеянный склероз. S. Тау — Sachs. S. Dollinger — Bielschowsky. Все формы синдрома диффузного склероза головного мозга. S. Gaucher. S. Niemann — Pick. S. West.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Энцефалит после прививок АКДС у детей. Лейкоэнцефалиты у детей
Поражения нервной системы после введения АКДС-вакцины наблюдается сравнительно часто. Это связано с высокой реактогенностью вакцины, которая гораздо выше, чем реактогенность каждого из компонентов в отдельности. И. С. Левенбук с соавторами (1971) считают, что возникновение неврологических осложнений при АКДС-вакцинации обусловлено коклюшным компонентом вакцины. Вакцина оказывает выраженное аллергизирующее действие на организм ребенка. У отдельных детей после АКДС-вакцинации наблюдаются уртикарная сыпь, синдром ложного крупа, астматический и геморрагический синдромы, в редких случаях — анафилактический шок.
Вакцинальный энцефалит (энцефаломиелит) возникает у детей с различными аллергическими проявлениями на фоне или сразу после перенесенных инфекционных заболеваний, при нарушении сроков вакцинации. Он может развиться после любого введения вакцины, независимо от кратности.
Симптомы энцефалита появляются через несколько суток после вакцинации внезапно, остро. Поднимается температура, появляются рвота, судороги, адинамия, нарушается сознание, возможно развитие комы. На фоне общемозговых расстройств могут развиться центральные и периферические двигательные нарушения различной степени, вплоть до параличей, расстройство функции сфинктеров. Однако несмотря на тяжесть острого периода в дальнейшем наступает сравнительно быстрое выздоровление. Нормализуется температура, ребенок приходит в сознание, прекращаются судороги. В течение 1—2 нед исчезает очаговая неврологическая симптоматика.
В течение 2—3 мес после энцефалита держится астенический синдром. Выраженная очаговая симптоматика после энцефалита наблюдается при тяжелых формах, протекающих с длительной потерей сознания, дыхательными и сердечно-сосудистыми нарушениями.
Поражение нервной системы при вакцинации АКДС может быть также в виде преходящих, различной тяжести энцефалитических реакций, главным образом в виде судорожного синдрома. Судороги могут быть однократные или повторные, генерализованные или малые — «кивки», сааламовы судороги, вздрагивания, «обмякания», подергивания лицевых мышц. Судороги обычно сочетаются с беспокойством, расстройством сна или вялостью. Эти нарушения держатся обычно всего в течение 3—5 дней, но сопровождаются последующей длительной астенией.
При появлении неврологических нарушений назначается комплексная терапия с включением гормонов и других десенсибилизирующих средств, дегидратационной терапии. Показано применение витаминов группы В, аскорбиновой кислоты, никотиновой кислоты и др. При появлении бактериальных осложнений назначаются антибиотики.
Поскольку поражения нервной системы, безусловно, чаще развиваются у детей с измененной реактивностью, при определении показаний к вакцинации необходимо тщательное изучение анамнеза, внимательное отношение к жалобам матери, строгое соблюдение правил применения вакцины.
Лейкоэнцефалиты
Это группа демиелинизирующих заболеваний воспалительно-дегенеративного характера. Хотя при лейкоэнцефалитах поражается преимущественно белое вещество мозга, обычно в той или иной степени страдают и нейроны. Поэтому четкой грани между лейкоэнцефалитами и панэнцефалитами провести нельзя. Вопрос об этиологических факторах лейкоэнцефалитов и этиологической однородности отдельных форм в настоящее время не решен однозначно. Предполагается, что лейкоэнцефалиты относятся к заболеваниям инфекционно-аллергической природы. Остается спорным вопрос о роли миксовирусов, вирусов кори, бешенства и Herpes zoster как пусковых факторов гиперергического аутоиммунного процесса.
При исследовании мозга больных, умерших от лейкоэнцефалита, находят диффузные воспалительно-дегенеративные изменения, очаги демиелинизации и деструкции в белом веществе мозга, иногда в коре, подкорковых ганглиях и мозговых оболочках. Отдельные очаги могут сливаться, выраженность дегенеративных изменений в них варьирует. Деструкции подвергаются нормально сформированный миелин (миелинокластический тип поражения). В большинстве случаев выражен глиоз, иногда столь значительно, что мозг на разрезе имеет хрящевидную консистенцию.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Симптомы появляются исподволь и неуклонно прогрессируют или ремиттируют. К самым ранним относятся психические нарушения, которые у маленьких детей проявляются апатией, вялостью или раздражительностью, капризностью, непослушанием, немотивированным агрессивным поведением. Дети теряют навыки опрятности, речь становится более скудной, исчезает логика мышления. На фоне психических нарушений постепенно, в течение нескольких недель или месяцев, развивается очаговая неврологическая симптоматика (апраксия, агнозия, зрительные и слуховые галлюцинации, полиморфные гиперкинезы, повышение мышечного тонуса по пластическому типу равномерно в сгибательных и разгибательных группах мышц, что приводит к скованности движений). Позднее развиваются спастические моно-, геми- или тетрапарезы и параличи. Наблюдаются также нарушения координации движений и неустойчивая походка (мозжечкового или лобного происхождения). Расстройства фонации и глотания обычно сочетаются с высокими нёбным и глоточным рефлексами. Бульбарный паралич бывает редко.
Постоянным симптомом лейкоэнцефалитов являются судороги, чаще в виде малых или абортивных припадков, перемежающихся с редкими развернутыми приступами. В поздней стадии заболевания присоединяются вегетативные и трофические расстройства: кахексия, профузный пот, икота, нарушения терморегуляции, пролежни и т. д. В терминальной стадии больные обездвижены, могут возникать состояния децеребрационной ригидности.
На ЭЭГ регистрируется периодическая пароксизмальная активность с интервалом 5—15 с одновременно в большинстве отведений в виде медленных (1—2 с) высоковольтажных волн. По мере нрогрессирования заболевания эти изменения становятся более выраженными.
В крови может быть лейкоцитоз, фракция альфа-глобулина повышена. У больных обычно повышен титр антител к коревому вирусу или к миксовирусам — вирусу JC или SV-40.
В спинномозговой жидкости в большинстве случаев не наблюдается цитоза и увеличения содержания белка. При электрофо-ретическом исследовании белков, однако, обнаруживается, что Y-глобулин составляет до 40 и более процентов от общего количества белка, а фракция альбумина снижена. Коллоидные реакции дают максимальную флоккуляцшо в первых пробирках (паралитический тип реакции Ланге).
При неуклонно прогрессирующем течении больные умирают через 2—12 мес после появления первых симптомов. При длительном течении продолжительность жизни может быть 3 года и более, а ремиссии продолжаются от нескольких месяцев до нескольких лет, в течение которых симптомы заболевания могут уменьшаться.
Существует несколько клшшко-морфологических вариантов лейкоэнцефалитов, из которых у маленьких детей описаны лишь подострый склерозирующий лейкоэицефалит Ван-Богарта и Даусона и острый геморрагический лейкоэицефалит.
Лейкоэнцефалит Ван-Богарта выделен из группы лейкоэнцефалитов на основе регулярного обнаружения в клетках нервной системы характерных включений наряду с типичными для лейкоэнцефалитов изменениями. Выраженность поражения мозга уменьшается в направлении от коры к филогенетически более древним образованиям, но чаще, чем при других формах заболевания, поражены ствол и спинной мозг.
Клиническая особенность этой формы лейкоэнцефалита — раннее проявление и преобладание экстрапирамидных нарушений (гиперкинетическая форма), к которым лишь на поздних этапах присоединяются пирамидные симптомы. Для нее не характерны эпилептические припадки.
Лейкоэнцефалит Шильдера
Лейкоэнцефалит Шильдера — дегенеративно-демиелинизирующее поражение головного мозга, сопровождающееся образованием крупных или сливных зон демиелинизации. Имеет неуклонно прогрессирующее течение с неспецифичной и полиморфной клинической картиной, которая может включать психические нарушения, пирамидный и экстрапирамидный синдромы, когнитивный дефицит, поражение черепно-мозговых нервов, эписиндром. Диагностируется лейкоэнцефалит Шильдера по клиническим критериям и результатам МРТ после исключения другой патологии с подобными проявлениями. Терапия осуществляется глюкокортикостероидами, антиконвульсантами, миорелаксантами и психотропными средствами. Однако лечение малоэффективно.
МКБ-10
Общие сведения
Лейкоэнцефалит Шильдера впервые был рассмотрен в качестве самостоятельной нозологии в 1912 г. психоневрологом, имя которого прочно закрепилось в названии заболевания, хотя сам автор обозначил описанную им патологию термином «периаксиальный диффузный лейкоэнцефалит». Позже различными исследователями были представлены описания других клинических форм лейкоэнцефалита: в 1941 г. - геморрагического лейкоэнцефалита, в 1945 г. - подострого склерозирующего лейкоэнцефалита. Поскольку основной патоморфологический субстрат болезни составляют диффузные зоны демиелинизации белого вещества, лейкоэнцефалит Шильдера входит в группу демиелинизирующих заболеваний.
Преимущественный возраст манифестации болезни Шильдера до сих пор остается спорным вопросом. Зарубежные специалисты в области неврологии считают характерным дебют в возрастном периоде от 7 до 12 лет, а отдельные авторы предлагают относить заболевание к детской форме рассеянного склероза. Наблюдения отечественных неврологов, напротив, свидетельствуют о равной степени поражения лиц различной возрастной категории.
Причины лейкоэнцефалита Шильдера
Этиопатогенез болезни Шильдера находится в стадии изучения. Из названия заболевания видно, что первоначально подразумевалась воспалительная этиология церебрального поражения, т. е. энцефалит. Предполагается вирусная теория заболевания по типу медленных инфекций. Среди возможных инфекционных агентов дискутируется роль кори, герпетической инфекции, миксовирусов, которые, возможно, запускают процесс аутоиммунного церебрального воспаления. Однако безуспешные попытки выделения возбудителя привели к возникновению иной этиопатогенетической теории. Последняя предполагает связь лейкоэнцефалита Шильдера с дисфункцией регуляторных механизмов липидного обмена, что сближает заболевание с наследственными лейкодистрофиями.
Морфологические изменения заключаются в образовании в белом церебральном веществе полушарий значительных зон демиелинизации, имеющих четкие заостренные очертания и зачастую асимметрично расположенных. В ряде случаев подобные очаги формируются в мозжечке и мозговом стволе. У пациентов, заболевших в пубертатном периоде и во взрослом возрасте, описаны случаи, когда наряду с зонами обширной демиелинизации наблюдаются округлые бляшковидные очаги, напоминающие бляшки рассеянного склероза.
Симптомы лейкоэнцефалита Шильдера
Заболевание отличается наличием неспецифичной и полиморфной симптоматики. Может манифестировать исподволь развивающимися психическими расстройствами: лабильностью настроения, апатией, нарушением поведения, эпизодами возбуждения с галлюцинаторным синдромом. Интеллектуальное снижение прогрессирует вплоть до деменции. Наблюдаются аграфия, акалькулия, алексия, агнозия, апраксия. Вследствие демиелинизации черепных нервов возникает неврит зрительного нерва, офтальмоплегия, тугоухость, снижение зрения, бульбарные расстройства. При поражении мозжечка появляется мозжечковая атаксия, скандированная речь, интенционный тремор. Поражение зрительной зоны коры проводит к гемианопсии, корковому амаврозу. Возможны экстрапирамидные нарушения в виде гиперкинезов, торсионной дистонии и т. п. Пирамидные расстройства обычно наблюдаются на поздних этапах лейкоэнцефалита в виде моно-, геми- и тетрапарезов. Зачастую присутствует судорожный синдром (по типу джексоновкой эпилепсии или с генерализованными эпиприступами), характеризующийся отсутствием специфической ЭЭГ-картины.
Вариативность сочетаний различных симптомокомплексов настолько выражена, что не позволяет выделить типичный вариант течения болезни Шильдера. В ряде случаев клиника сходна с прогредиентным вариантом рассеянного склероза, в других - имеет псевдотуморозный характер, в третьих — напоминает психиатрическую патологию. В последнем случае пациенты могут проходить лечение у психиатра вплоть до развития явной неврологической симптоматики.
Диагностика лейкоэнцефалита Шильдера
Прижизненно диагностировать лейкоэнцефалит Шильдера весьма затруднительно. Эта задача требует от невролога тщательного сопоставления анамнестических, клинических и томографических данных, внимательного проведения дифдиагностики со схожими заболеваниями. С целью обследования зрительного и слухового анализаторов к консультациям могут привлекаться офтальмолог и отоларинголог.
Электроэнцефалография выявляет признаки диффузного церебрального поражения: снижение альфа-активности и дезорганизацию ритма; зачастую определяется эпилептиформная активность. При исследовании цереброспинальной жидкости обнаруживается повышение уровня гамма-глобулина на фоне снижения удельного веса альбуминовой фракции. Наиболее информативным способом инструментальной диагностики выступает МРТ головного мозга. Болезнь Шильдера подтверждает наличие как минимум одного большого или пары сливных очагов демиелинизации в белом церебральном веществе.
Для установления окончательного диагноза многие неврологи руководствуются критериями C.M. Poser 1985 г.: наличие по данным МРТ 1-2 округлых зон демиелинизации величиной не менее 2х3 см; отсутствие патологии надпочечников; исключение любой иной церебральной патологии (внутримозговой опухоли, рассеянного энцефаломиелита, инсульта и пр.); соответствие норме уровня жирных кислот в сыворотке крови; выявление на аутопсии зон диффузного хронического склероза. В некоторых случаях отличить лейкоэнцефалит Шильдера от лейкодистрофии позволяют лишь гистологические исследования церебральных тканей пораженной зоны.
Лечение и прогноз лейкоэнцефалита Шильдера
Отсутствие ясных представлений об этиопатогенезе болезни Шильдера пока не позволило разработать более или менее эффективные методы ее лечения. Отмечен некоторый эффект глюкокортикостероидной терапии, в связи с чем многим пациентам назначают метилпреднизолон, вначале парентерально в ударной дозе, а затем внутрь с постепенным снижением дозы. Параллельно проводится курс нейропротекторной, антиоксидантной и сосудистой терапии, при необходимости назначаются антиконвульсантное лечение (карбамазепин, диазепам), миорелаксанты (амантадин, толперизон, амидин), противоотечные мероприятия (фуросемид, ацетазоламид, магния сульфат), психотропные фармпрепараты.
Своевременно начатое лечение способно лишь несколько задержать прогрессирование патологии. Однако, не смотря на его проведение, все пациенты погибают. Время наступления летального исхода варьирует от нескольких месяцев до 3 лет с момента дебюта лейкоэнцефалита.
Читайте также: