Кости при болезни Эрдгейма-Честера - лучевая диагностика
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
Ключевые слова
Об авторах
ФГБУ «Национальный медицинский исследовательский центр онкологии им. Н.Н. Блохина» Минздрава России
Россия
Александр Сергеевич Крылов
115478 Москва, Каширское шоссе, 24
Список литературы
1. Estrada-Veras J.I., O’Brien K.J., Boyd L.C. et al. The clinical spectrum of Erdheim–Chester disease: an observational cohort study. Blood Adv 2017;1(6):357–66. DOI: 10.1182/bloodadvances.2016001784.
2. Emile J.F., Abla O., Fraitag S. et al. Revised classification of histiocytoses and neoplasms of the macrophage-dendritic cell lineages. Blood 2016;127(22):2672–81. DOI: 10.1182/blood-2016-01-690636.
3. Arnaud L., Gorochov G., Charlotte F. et al. Systemic perturbation of cytokine and chemokine networks in Erdheim–Chester disease: a single-center series of 37 patients. Blood 2011;117(10):2783–90. DOI: 10.1182/blood-2010-10-313510.
4. Tran T.A., Fabre M., Pariente D. et al. Erdheim–Chester disease in childhood: a challenging diagnosis and treatment. J Pediatr Hematol Oncol 2009;31(10):782–6. DOI: 10.1097/MPH.0b013e3181b76827.
6. Campo E., Harris N.L., Jaffe E.S. et al. WHO Classification of tumours of haematopoietic and lymphoid tissues. International Agency for Research on Cancer 2017. P. 586.
7. Emile J.F., Charlotte F., Amoura Z., Haroche J. BRAF mutations in Erdheim– Chester disease. J Clin Oncol 2013;31(3):398. DOI: 10.1200/JCO.2012.46.9676.
8. Cangi M.G., Biavasco R., Cavalli G. et al. BRAFV600E-mutation is invariably present and associated to oncogene-induced senescence in Erdheim–Chester disease. Ann Rheum Dis 2015;74(8):1596–602. DOI: 10.1136/annrheumdis-2013-204924.
9. Emile J.F., Diamond E.L., Hélias-Rodzewicz Z. et al. Recurrent RAS and PIK3CA mutations in Erdheim–Chester disease. Blood 2014;124(19):3016–9. DOI: 10.1182/blood-2014-04-570937.
10. Diamond E.L., Abdel-Wahab O., Pentsova E. et al. Detection of an NRAS mutation in Erdheim–Chester disease. Blood 2013;122(6):1089–91. DOI: 10.1182/blood-2013-02-482984.
11. Chester W. Uber lipoid granulomatose. Virchows Arch (Pathol Anat Phys) 1930;279:561–602.
13. Haroche J., Arnaud L., Amoura Z. Erdheim–Chester disease. Curr Opin Rheumatol 2012;24(1):53–9. DOI: 10.1097/BOR.0b013e32834d861d.
16. Scolaro J.C., Peiris A.N. The Hairy Kidney of Erdheim–Chester Disease. Mayo Clin Proc 2018;93(5):671. DOI: 10.1016/j.mayocp.2018.03.003.
17. Kraniotis P., Daoussis D. Periaortitis, hairy kidneys and bone lesions. Rheumatology (Oxford) 2016;55(12):2118. DOI: 10.1093/rheumatology/kew331.
18. Lee H.J., Lee K.Y., Shin D.Y. et al. A case of Erdheim–Chester disease with asymptomatic renal involvement. Cancer Res Treat 2012;44(2):146–50. DOI: 10.4143/crt.2012.44.2.146.
19. Serratrice J., Granel B., De Roux C. et al. “Coated aorta”: a new sign of Erdheim– Chester disease. J Rheumatol 2000;27(6):1550–3.
21. Suzuki H., Wanibuchi M., Komatsu K. et al. Erdheim–Chester disease involving the central nervous system with the unique appearance of a coated vertebral artery. NMC Case Rep J 2016;3(4):125–8. DOI: 10.2176/nmccrj.cr.2015-0331.
22. Diamond E.L., Hatzoglou V., Patel S. et al. Diffuse reduction of cerebral grey matter volumes in Erdheim–Chester disease. Orphanet J Rare Dis 2016;11(1): 109. DOI: 10.1186/s13023-016-0490-3.
23. Lachenal F., Cotton F., Desmurs-Clavel H. et al. Neurological manifestations and neuroradiological presentation of Erdheim–Chester disease: report of 6 cases and systematic review of the literature. J Neurol 2006;253(10):1267–77. DOI: 10.1007/s00415-006-0160-9.
24. Nicolazzi M.A., Carnicelli A., Fuorlo M. et al. Cardiovascular involvement in Erdheim–Chester disease: a case report and review of the literature. Medicine (Baltimore) 2015t;94(43):e1365. DOI: 10.1097/MD.0000000000001365.
25. Lee K., Kim H.R., Roh J. et al. Erdheim– Chester disease presenting as an anterior mediastinal tumor without skeletal involvement. Korean J Thorac Cardiovasc Surg 2018;51(3):223–6. DOI: 10.5090/kjtcs.2018.51.3.223.
26. Razanamahery J., Jacquier F., Humbert S. A rare case of chylous ascites. Gastroenterology 2017;153(4):903–5. DOI: 10.1053/j.gastro.2017.04.008.
27. Balasubramanian G., Modiri A., Affi M. et al.A fatal case of Erdheim–Chester disease with hepatic involvement. ACG Case Rep J 2017;4:e95. DOI: 10.14309/crj.2017.95.
28. Ambrosini V., Savelli F., Merli E. et al. F-18 FDG PET/CT detects muscle involvement in Erdheim–Chester disease. Clin Nucl Med 2012;37(2):196–7. DOI: 10.1097/RLU.0b013e31823e9d54.
29. Martineau P., Pelletier-Galarneau M., Zeng W. The imaging findings of Erdheim–Chester disease: a multimodality approach to diagnosis and staging. World J Nucl Med 2017;16(1):71–4. DOI: 10.4103/1450-1147.181149.
30. Sabino D., do Vale R.H.B., Duarte P.S. et al. Complementary findings on 18F-FDG PET/CT and 18F-NaF PET/CT in a patient with Erdheim–Chester disease. Radiol Bras 2017;50(3):202–3. DOI: 10.1590/0100-3984.2015.0172.
33. Boissel N., Wechsler B., Leblond V. Treatment of refractory Erdheim–Chester disease with double autologous hematopoietic stem-cell transplantation. Ann Intern Med 2001;135(9):844–5.
34. Gaspar N., Boudou P., Haroche J. et al. Highdose chemotherapy followed by autologous hematopoietic stem cell transplantation for adult histiocytic disorders with central nervous system involvement. Haematologica 2006;91(8):1121–5.
35. Braiteh F., Boxrud C., Esmaeli B., Kurzrock R. Successful treatment of Erdheim–Chester disease, a non-Langerhans-cell histiocytosis, with interferonalpha. Blood 2005;106(9):2992–4. DOI: 10.1182/blood-2005-06-2238.
36. Haroche J., Amoura Z., Trad S.G. et al. Variability in the efficacy of interferon-alpha in Erdheim–Chester disease by patient and site of involvement: results in eight patients. Arthritis Rheum 2006;54(10):3330–6. DOI: 10.1002/art.22165.
37. Arnaud L., Hervier B., Néel A. et al. CNS involvement and treatment with interferon-α are independent prognostic factors in Erdheim–Chester disease: a multicenter survival analysis of 53 patients. Blood 2011;117(10):2778–82. DOI: 10.1182/blood-2010-06-294108.
38. Aouba A., Georgin-Lavialle S., Pagnoux C. et al. Rationale and efficacy of interleukin-1 targeting in Erdheim–Chester disease. Blood 2010;116(20):4070–6. DOI: 10.1182/blood-2010-04-279240.
39. Aubert O., Aouba A., Deshayes S. et al. Favorable radiological outcome of skeletal Erdheim–Chester disease involvement with anakinra. Joint Bone Spine 2013;80(2):206–7. DOI: 10.1016/j.jbspin.2012.07.005.
40. Goyal G., Shah M.V., Call T.G. et al. Efficacy of biological agents in the treatment of Erdheim–Chester disease. Br J Haematol 2018;183(3):520–4. DOI: 10.1111/bjh.14997.
41. Tomelleri A., Cavalli G., De Luca G. et al. Treating heart inflammation with interleukin-1 blockade in a case of Erdheim–Chester disease. Front Immunol 2018;9:1233. DOI: 10.3389/fimmu.2018.01233
43. Nikonova A., Esfahani K., Chausse G. et al. Erdheim–Chester disease: the importance of information integration. Case Rep Oncol 2017;10(2):613–9. DOI: 10.1159/000477658.
44. Hyman D.M., Puzanov I., Subbiah V. et al. Vemurafenib in multiple nonmelanoma cancers with BRAF V600 mutations. N Engl J Med 2015;373(8):726–36. DOI: 10.1056/NEJMoa1502309.
45. Mirouse A., Savey L., Domont F. et al. Systemic vasculitis associated with vemurafenib treatment: case report and literature review. Medicine (Baltimore) 2016;95(46):e4988. DOI: 10.1097/MD.0000000000004988.
46. Bhatia A., Ulaner G., Rampal R. et al. Single-agent dabrafenib for BRAFV600E-mutated histiocytosis. Haematologica 2018;103(4):e177–80. DOI: 10.3324/haematol.2017.185298.
47. Cohen Aubart F., Emile J.F., Carrat F. et al. Targeted therapies in 54 patients with Erdheim–Chester disease, including follow-up after interruption (the LOVE study). Blood 2017;130(11):1377–80. DOI: 10.1182/blood-2017-03-771873.
50. Dagna L., Corti A., Langheim S. et al. Tumor necrosis factor α as a master regulator of inflammation in Erdheim–Chester disease: rationale for the treatment of patients with infliximab. J Clin Oncol 2012;30(28):e286–90. DOI: 10.1200/JCO.2012.41.9911.
51. Cives M., Simone V., Rizzo F.M. et al. Erdheim–Chester disease: a systematic review. Crit Rev Oncol Hematol 2015;95(1):1–11. DOI: 10.1016/j.critrevonc.2015.02.004.
53. Azadeh N., Tazelaar H.D., Gotway M.B. et al. Erdheim–Chester disease treated successfully with cladribine. Respir Med Case Rep 2016;18:37–40. DOI: 10.1016/j.rmcr.2016.03.008.
54. Бялик Т.Е., Якимович О.Ю., Махонова Л.А. и др. Диссеминированная ювенильная ксантогранулема у взрослых. Клиническое наблюдение. Клиническая онкогематология 2011;4(4):329–33.
55. Poiroux L., Paycha F., Polivka M., Ea H.K. Efficacy of zoledronic acid in Erdheim–Chester disease: a case report. Joint Bone Spine 2016;83(5):573–5. DOI: 10.1016/j.jbspin.2015.10.010
Кости при болезни Эрдгейма-Честера - лучевая диагностика
а) Терминология:
• Гистиоцитоз, характеризующийся инфильтрацией скелета и внутренних органов гистиоцитами, нагруженными липидами:
о Вызывает фиброз и остеосклероз
б) Визуализация:
• Трубчатые кости (98%):
о По сравнению с нижними конечностями верхние поражаются менее часто и менее тяжело
• Двустороннее симметричное поражение
• Пятнистый или диффузный склероз костномозгового канала:
о У большинства пациентов наблюдается неоднородный (65%) или однородный (35%) склероз
о В 1/3 случаев наблюдаются смешанные литические/склеротические очаги
(Слева) Рентгенография в ПЗ проекции: определяются склеротичные огрубевшие трабекулы с литическими областями в диафизах длинных трубчатых костей. Склероз приводит к сужению костномозгового канала и отсутствию четкой границы между каналом и кортикальным веществом. Изменения симметричны, эпифизы не поражены. Волнистость кортикального вещества говорит о поражении надкостницы.
(Справа) Сканирование костей скелета, прямая проекция: у этого же пациента определяется поражение дистального отдела лучевой кости и костей лицевого черепа; осевой скелет не поражен. Картина характерна для болезни Эрдгейма-Честера. (ЭЧ). (Слева) МРТ Т1ВИ, корональный срез: у пациента 79 лет определяются пятнистые области ↓ ИС, замещающие нормальный желтый костный мозг в диафизах, и однородная область ↓ ИС в дистальном метадиафизе. В дистальных эпифизах бедренный костей желтый костный мозг не изменен.
(Справа) МРТ Т2ВИ, режим подавления сигнала от жира, корональный срез: в дистальных метадиафзиах определяется область преимущественно однородного сигнала низкой интенсивности, соответствующего области плотного склероза на Т1 ВИ. В среднем отделе диафизов определяется область неоднородного сигнала высокой интенсивности, что говорит об активном процессе замещения костного мозга. По данным биопсии подтвержден ЭЧ.
• Утолщение кортикального вещества:
о Периостит (66%): волнистый контур кортикального вещества о Утолщение эндоста (94% случаев)
о Размытие границы кортикального вещества и костного мозга
• Относительное щажение эпифизов, как минимум в субхондральной области
• МРТ:
о Неоднородный сигнал низкой интенсивности на Т1 ВИ
о Смешанный неоднородный сигнал на Т2 ВИ/STIR
о Интенсивное контрастирование, однородное
о Периостит визуализируется в виде областей высокой интен сивности сигнала по ходу кортикального вещества
о Могут наблюдаться инфаркты кости
в) Дифференциальная диагностика:
• Лангергансоклеточный гистиоцитоз
• Миелофиброз
• Прогрессирующая диафизарная дисплазия
• Интрамедуллярный остеосклероз
г) Клинические особенности:
• Классическая триада: костная боль, экзофтальм, диабет
• Возраст: 7-84 лет; средний возраст - 43 года
• Большинство пациентов умирает в течение трех лет:
о Осложнения со стороны почек, сердечно-сосудистой системы, легких или центральной нервной системы
Рентгенограмма, МРТ костей при болезни Эрдгейма-Честера
а) Терминология:
1. Аббревиатура:
• Болезнь Эрдгейма-Честера (ЭЧ)
2. Определение:
• Гистиоцитоз, характеризующийся инфильтрацией скелета и внутренних органов гистиоцитами, нагруженными липидами:
о Приводит к развитию фиброза и остеосклероза
б) Визуализация:
• Лучший диагностический критерий:
о Диффузный интрамедуллярный склероз трубчатый костей, двусторонняя симметричность, обычно поражаются нижние конечности
• Локализация:
о Преимущественно длинные крупные кости:
- По сравнению с нижними конечностями верхние поражаются менее часто и менее тяжело
- Диафиз (100%), метафиз (83%)
- Считается, что эпифизы не поражаются; в действительности не поражается субхондральная кость; частичное поражение эпифизов наблюдается в 45% случаев
о Могут поражаться плоские кости
о Редко наблюдается поражение позвоночника
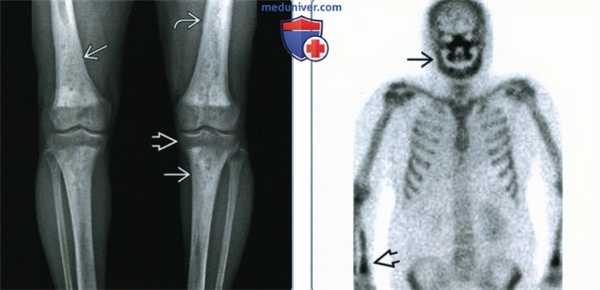
(Слева) Рентгенография в ПЗ проекции: определяются склеротичные огрубевшие трабекулы с литическими областями в диафизах длинных трубчатых костей. Склероз приводит к сужению костномозгового канала и отсутствию четкой границы между каналом и кортикальным веществом. Изменения симметричны, эпифизы не поражены. Волнистость кортикального вещества говорит о поражении надкостницы.
(Справа) Сканирование костей скелета, прямая проекция: у этого же пациента определяется поражение дистального отдела лучевой кости и костей лицевого черепа; осевой скелет не поражен. Картина характерна для болезни Эрдгейма-Честера. (ЭЧ).
1. Рентгенография костей при болезни Эрдгейма-Честера:
• Трубчатые кости (98%):
о Двустороннее симметричное (98%):
о Пятнистый или диффузный склероз костномозгового канала
- В большинстве случаев наблюдается неоднородный (65%) или однородный (35%) склероз
- В 1/3 случаев наблюдаются смешанные литические/склеротические очаги
- Только литическое поражение наблюдается лишь в 5-8% случаев
о Грубые трабекулы
о Утолщение кортикального вещества:
- Периостит (66%); волнистый контур кортикального вещества
- Утолщение эндоста (94% случаев):
Размытость границы между кортикальным веществом и костномозговым каналом
Костномозговой канал может быть облитерирован
о Относительное щажение эпифизов, как минимум субхондральной части:
- Может наблюдаться метаэпифизарная линия просветления
• Позвоночник (редко):
о Литическое поражение со склеротическими краями
о Почти всегда сочетается с поражениями длинных костей
• Очаговое псевдоопухолевое поражение (редко):
о Вариабельная степень лизиса, разрушение кортикального вещества, мягкотканное объемное образование
2. МРТ костей при болезни Эрдгейма-Честера:
• Область однородного сигнала низкой интенсивности на Т1 ВИ
• Область смешанного неоднородного сигнала на Т2 ВИ/STIR
• Интенсивное контрастирование, неоднородность
• Периостит визуализируется в виде области высокой интенсивности сигнала по ходу кортикального вещества
• Могут наблюдаться инфаркты кости
3. Радионуклидная диагностика:
• Аномальное повышение накопления при сканировании костей скелета:
о Двустороннее симметричное в трубчатых костях
• ПЭТ/КТ обладает вариабельной чувствительностью:
о Глазницы (60%), кости (55%), легкие (37%), забрюшинное пространство (7%)
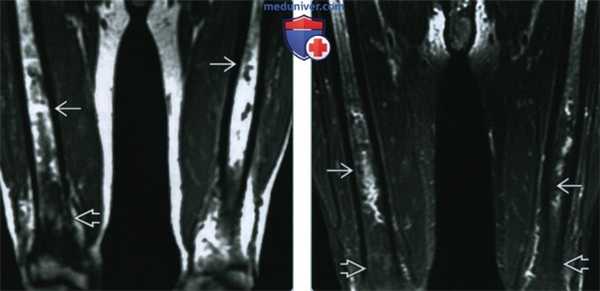
(Слева) МРТ Т1ВИ, корональный срез: у пациента 79 лет определяются пятнистые области ↓ ИС, замещающие нормальный желтый костный мозг в диафизах, и однородная область ↓ ИС в дистальном метадиафизе. В дистальных эпифизах бедренный костей желтый костный мозг не изменен.
(Справа) МРТ Т2ВИ, режим подавления сигнала от жира, корональный срез: в дистальных метадиафзиах определяется область преимущественно однородного сигнала низкой интенсивности, соответствующего области плотного склероза на Т1 ВИ. В среднем отделе диафизов определяется область неоднородного сигнала высокой интенсивности, что говорит об активном процессе замещения костного мозга. По данным биопсии подтвержден ЭЧ.
в) Дифференциальная диагностика изменений костей при болезни Эрдгейма-Честера:
2. Миелофиброз:
• Склероз трубчатых костей с картиной, схожей с ЭЧ
• Поражение осевого скелета отличает от ЭЧ
3. Прогрессирующая диафизарная дисплазия:
• У пациентов более молодого возраста
• Четкое утолщение надкостницы и эндоста; четкая граница между кортикальным веществом и остаточной полостью
4. Интрамедуллярный остеосклероз:
• Ограничен диафизами; возможна облитерация костномозгового канала
• Может быть односторонним или двусторонним и асимметричным
г) Патология. Микроскопия:
• Диффузная инфильтрация костного мозга пенистыми гистиоцитами:
о Ассоциирована с плотным фиброзом, лимфоцитами, плазматическими клетками, гигантскими клетками Тутона
д) Клинические особенности:
1. Проявления:
• Типичные признаки/симптомы:
о Боль в костях, редко с набуханием мягких тканей
• Другие признаки/симптомы:
о Внескелетные проявления > чем в 50% случаев:
- Гипоталамо-гипофизарная ось: несахарный диабет
- Глазницы: экзофтальм, окологлазничные ксантомы
- Забрюшинное пространство: особенно околопочечное
о Классическая триада: костная боль, экзофтальм, диабет
о Липидный профиль сыворотки крови обычно относительно нормальный
2. Демография:
• Возраст:
о Пик встречаемости: 50-70 лет
• Пол: небольшая предрасположенность у мужского пола
3. Течение и прогноз:
• Прогноз ухудшается при поражении внутренних органов
• Большинство пациентов умирает в течение трех лет:
о Осложнения со стороны почек, сердечно-сосудистой системы, легких или центральной нервной системы
4. Лечение:
• Интерферон-α
• Стероидные гормоны
• Бифосфонаты
Болезнь Эрдгейма-Честера глазницы: признаки, гистология, лечение, прогноз
Лангергансоклеточный гистиоцитоз, как правило, является заболеванием детского возраста. Также существует несколько имеющих большое значение ксантогранулематозных заболеваний, развивающихся, в том числе и в глазнице, в основном у взрослых пациентов.
Обычно их относят к идиопатическим, но также они могут являться компонентом специфических синдромов, в том числе болезни Эрдгейма-Честера (Erdheim-Chester), ассоциированной с астмой ксантогранулемы взрослых и некробиотической ксантогранулемы с парапротеинемией.
Болезнь Эрдгейма-Честера — нечасто встречающаяся форма гистиоцитоза неясной этиологии, характеризуемая ксантогранулематозной инфильтрацией различных органов — легких, почек, сердца, костей, забрюшинной клетчатки и, иногда, глазницы (1-13). Может развиваться тяжелое системное поражение, приводящее к смерти вследствие почечной или сердечной недостаточности.
В собственной серии наблюдений авторов из 1264 новообразований глазницы четыре случая болезни Эрдгейма-Честера составили 25% гистиоцитарных новообразований и 1% всех опухолей глазницы (1).
а) Клиническая картина. Поражение глазницы при болезни Эрдгейма-Честера может быть первым очагом поражения или развиваться уже после постановки диагноза системного заболевания. Оно характеризуется экзофтальмом, обычно двусторонним, и смещением глазного яблока.
Нередко наблюдается резко выраженный экзофтальм, вызывающий обнажение роговицы и кератопатию, компрессионную нейрооптикопатию и тяжелые нарушения зрения. Характерным симптомом являются двусторонние атипичные ксантелазмы (плоские ксантомы) кожи периокулярной области (10).
Комбинация двусторонних ксантелазм и экзофтальма должна вызывать подозрения о наличии болезни Эрдгейма-Честера, таким пациентам показано системное обследование с целью диагностики сопутствующих нарушений.
Болезнь Эрдгейма-Честера характеризуется двусторонними ксантелазмами и двусторонним экзофтальмом. Характерная клиническая картина вызывает у врача подозрения о диагнозе. Часто наблюдается массивное поражение глазницы. Ниже кратко проиллюстрированы три случая.
Ксантелазма обоих верхних век и двусторонний экзофтальм у мужчины 78 лет с системными признаками болезни Эрдгейма-Честера, в том числе с фиброзом легких и забрюшинной клетчатки. КТ, аксиальная проекция: пациент, представленный на рисунке выше; определяется очаговая инфильтрация мягких тканей глазницы. Ксантелазма верхнего века левого глаза у престарелой женщины с атипичной односторонней инфильтрацией, вызванной болезнью Эрдгейма-Честера. Двусторонний экзофтальм и атипичные ксантелазмы у пациента 28 лет. У пациента развилось тяжелое нарушение зрения обоих глаз, вызванное массивным поражением глазницы и сдавливанием зрительного нерва. КТ, аксиальная проекция: пациент, представленный на рисунке выше; определяется массивная инфильтрация обеих глазниц. КТ, корональная проекция, срез через среднюю часть глазницы пациента, представленного на рисунке выше; определяется протяженность поражения глазниц.
б) Диагностика. При лучевых исследованиях в глазнице выявляются диффузные мягкотканые образования, иногда заполняющие всю глазницу и вызывающие тяжелый экзофтальм (8). Хотя это заболевание часто поражает длинные кости, в глазнице патологический процесс обычно развивается в мягких тканях и значимое поражение кости отсутствует.
в) Патологическая анатомия. Гистологически болезнь Эрдгейма-Честера характеризуется наличием пластов ксантомных клеток, перемешанных с лимфоцитами и плазматическими клетками, обычно в сочетании с обширным фиброзом. Ксантомные клетки на самом деле являются гистиоцитами, фагоцитировавшими липиды, в основном холестерин. Обычно выявляются отдельные гигантские клетки Touton.
В отличие от заболеваний типа лангергансоклеточного гистиоцитоза, патологическая ткань при болезни Эрдгейма-Честера не прокрашивается красителями на протеин S-100, а при ультраструктурных исследованиях гранулы Birbeck не выявляются. В ходе недавних исследований у некоторых пациентов с болезнью Эрдгейма-Честера была выявлена мутация BRAFV600E.
В одной серии наблюдений семи больных, эта мутация была подтверждена у трех (50%) из шести обследованных пациентов (6). Этими данными подтверждается мнение о болезни Эрдгейма-Честера как о полисистемной клоновой патологии с неопластическими и воспалительными элементами, связанной с нарушением сигнального пути BRAF, а также других ключевых сайтов (6).
г) Лечение. В прошлом большинство методов лечения были практически неэффективны, и заболевание приводило к тяжелым осложнениям со стороны глазницы и слепоте. Иногда эффективен интерферон-альфа. В настоящее время все пациенты обследуются на наличие мутации BRAFV600E и, если результат положительный, им назначается ве-мурафениб и другие препараты — ингибиторы BRAF, что позволяет достичь контроля заболевания (6, 7, 9).
В одном исследовании из восьми пациентов с болезнью Эрдгейма-Честера, имевших мутацию BRAF V600E, у которых другие виды лечения оказались безуспешными, после назначения вемурафениба у всех пациентов эффект развился через шесть месяцев и сохранялся в среднем в течение десяти месяцев (7).
д) Список использованной литературы:
1. Shields JA, Shields CL, Scartozzi R. Survey of 1264 patients with orbital tumors and simulating lesions: The 2002 Montgomery Lecture, part 1. Ophthalmology 2004;111: 997-1008.
2. Shields JA, Bakewell B, Augsburger JJ, et al. Classification and incidence of space-occupying lesions of the orbit. A survey of 645 biopsies. Arch Ophthalmol 1984:102: 1606-1611.
3. Shields JA, Bakewell B, Augsburger JJ, et al. Space-occupying orbital masses in children: A review of 250 consecutive biopsies. Ophthalmology 1986;93:379-384.
4. Shields JA, Shields CL. Clinical spectrum of histiocytic tumors of the orbit. Trans Pa Acad Ophthalmol Otolaryngol 1990;42:931-937.
5. Alper MG, Zimmerman LE, LaPiana FG. Orbital manifestations of Erdheim-Chester disease. Trans Am Ophthalmol Soc 1983;891:64-85.
6. Mazor RD, Manevich-Mazor M, Kesler A, et al. Clinical considerations and key issues in the management of patients with Erdheim-Chester disease: a seven case series. BMC Med 2014; 12:221.
7. Haroche J, Cohen-Aubart F, Emile JF, et al. Reproducible and sustained efficacy of targeted therapy with vemurafenib in patients with BRAFV600E-mutated Erheim-Chester disease. J Clin Oncol 2015;33(5):411-418.
8. De Abreu MR, Chung CB, Biswal S, et al. Erdheim-Chester disease: MR imaging, anatomic, and histopathologic correlation of orbital involvement. AJNR Am J Neuroradiol 2004;25:627-630.
9. Hyman DM, Diamond EL, Vibat CR, et al. Prospective blinded study of BRAFV600E mutation detection cell-free DNA of patients with systemic histiocytic disorders. Cancer Discov 2014;33(5):411—418.
10. Shields JA, Karcioglu Z, Shields CL, et al. Orbital and eyelid involvement with Erdheim-Chester disease. Arch Ophthalmol 1991;109:850-854.
11. Rozenberg I, Wechsler J, Koenig F, et al. Erdheim-Chester disease presenting as malignant exophthalmos. Br J Radiol 1986;59:173-177.
12. Karcioglu ZA, Sharara N, Boles TL, et al. Orbital xanthogranuloma: clinical and morphologic features in eight patients. Ophthal Plast Reconstr Surg 2003;19:372-381.
13. Valmaggia C, Neuweiler J, Fretz C, et al. A case of Erdheim-Chester disease with orbital involvement. Arch Ophthalmol 1997;115:1467-1468.
МСКТ — мультиспиральная КТ
ПЭТ — позитронно-эмиссионная томография
УЗИ — ультразвуковое исследование
IgG4-CЗ — заболевания, связанные с IgG4
IgG4-ССЗ — системное заболевание, связанное с IgG4
Описание клинического случая. Пациент Ш., 65 лет, обратился в НИИР в марте 2015 г. с жалобами на слабость и утомляемость, периодически возникающую фебрильную лихорадку, снижение массы тела до 10 кг за год и выраженную отечность век.
Из анамнеза: перенес холецистэктомию в 2011 г., наблюдался в поликлинике по поводу ишемической болезни сердца, атеросклероза аорты и коронарных артерий, гипертонической болезни I стадии; аллергологический анамнез без особенностей, наследственность не отягощена. Больным себя считает с марта 2014 г., когда по поводу нарастающей слабости, периодического повышения температуры тела и артериального давления (АД) до 170/100 мм рт.ст. обратился в поликлинику по месту жительства. При компьютерной томографии (КТ) органов брюшной полости и забрюшинного пространства и органов малого таза с внутривенным контрастным усилением омнипаком 350—100 мл выявлены признаки каликопиелоэктазии обеих почек (более выражено справа), инфильтративные изменения стенок лоханок в верхней трети обоих мочеточников, выраженные инфильтративные изменения паранефральной клетчатки с обеих сторон, лимфаденопатия брыжейки тонкой кишки с перифокальной инфильтрацией клетчатки. В легких патологических изменений не выявлено. С диагнозом: болезнь Ормонда наблюдался у уролога и получал терапию 20—30 мг преднизолона. При лабораторном обследовании выявлялась умеренная воспалительная активность: л. (11—15)·10 9 /л; СОЭ 36—68 мм/ч, α 1 -глобулины 7,4% (норма 2,3—6,4%), С-реактивный белок 15,1—35,6 г/л; незначительная гипергаммаглобулинемия до 20% (норма 11—18%) в отсутствие в крови ревматоидного фактора (РФ) и онкомаркеров (ПСА, СА-19—9 и РЭА). Заболевание протекало волнообразно, но в январе 2015 г. у больного появился отек век и стал нарастать экзофтальм. Больной направлен на консультацию в НИИР. При осмотре: кожа век пигментирована. На веках ксантелазмы различной величины (см. рис. 1). Лагофтальм 3 мм справа и 4 мм слева. Ограничение подвижности глазных яблок во всех отведениях. Конъюнктива век, переходных и полулунных складок отечны. Складка бульбарной конъюнктивы вдоль всего края нижнего века с двух сторон. Слезный ручей невысокий, ток слезы замедлен. Пальпебральные доли слезных желез увеличены в размерах, отечны, сосуды резко расширены. Другие органы без особенностей. При ультразвуковом исследовании (УЗИ) щитовидной железы выявлены диффузные изменения по типу хронического тиреоидита. Уровни гормонов щитовидной железы в пределах нормы, антитела к тиреоглобулину и тиреоидной пероксидазе не выявлялись. По данным магнитно-резонансной томографии головного мозга и глазниц, картина выраженного двустороннего экзофтальма, который может соответствовать периневральной форме идиопатического псевдотумора глазничной клетчатки с воспалительно-отечными (реактивными?) изменениями слезных желез и мышц глазниц. Исследование с использованием ядерного магнитного резонанса головы выявило единичные мелкие очаги в белом веществе головного мозга, вероятно, сосудистого генеза. КТ брюшной полости и забрюшинного пространства. В полости перикарда определяется незначительное количество жидкости, шириной в области переднего листка до 0,7 см. Печень умеренно увеличена в размерах, структура и плотность ее не изменены. Отмечаются выраженное уплотнение и тяжистость паранефральной клетчатки, обе почечные ножки на этом фоне дифференцируются нечетко. На уровне L II —L IV аорта муфтообразно окружена мягкоткаными структурами, толщина которых достигает 1,2 см. Подобного характера изменения определяются и в области нисходящей аорты на уровне T IX —T X . Кпереди от тел нижнегрудных позвонков визуализируется стелящийся инфильтрат толщиной до 1,3 см. В брыжейке тонкой кишки множество увеличенных до 1,8×1,6 см ЛУ без четких контуров. Компьютерно-томографическая картина не противоречит болезни Ормонда. При сравнении с данными от 2014 г. имеется выраженная отрицательная динамика в виде прогрессирования склеротических изменений и явлений гидронефроза с обеих сторон (больше справа), со значительным замедлением экскреторной функции почек. Для исключения лимфопролиферативного процесса целесообразно дополнительное обследование. При УЗИ глаза и глазницы выявлены признаки выраженного отека век и экзофтальма, значительное увеличение размеров изображения, так называемого стандартного плоскостного ультразвукового среза слезных желез «по глубине и ширине» (OD 2,27×1,52 см; OS 2,23×1,03 см). С обеих сторон имеется неравномерное значительное увеличение толщины прямых глазодвигательных мышц, больше с правой стороны (OD 0,52×0,56 cм; OS 0,48×0,53 см). С правой и левой стороны, между зрительным нервом и прямыми глазодвигательными мышцами, имеются области разряжения, которые особенно хорошо видны с внутренней стороны. При иммунологическом исследовании: высокочувствительный (вч) СРБ 105 мг/л (норма 65 г/л), значительное повышение уровня вчСРБ 181,5 мг/л (норма 5 г/л). Выполнено иммуногистохимическое исследование (ИГХИ) биоптата с использованием антител CD138, IgG, IgG4, panCK, CD20, CD3, CD68 (PGM-1). Полиморфно-клеточный инфильтрат представлен множеством гистиоцитов, СD68 + с примесью лимфоцитов, большая часть которых является Т-лимфоцитами, CD3 + . В-лимфоциты в виде отдельно расположенных небольших групп клеток CD20 + . Плазматические клетки CD138 + в большом количестве, большая часть которых является позитивными по IgG плазмоцитами. При реакции с IgG4 позитивны лишь единичные клетки, составляющие менее 10% позитивных клеток по IgG (что можно объяснить, вероятно, предшествующей терапией стероидами). Заключение: учитывая данные анамнеза и морфологическую картину, следует предполагать заболевание, ассоциированное с IgG4. Исследование, выполненное на проточном цитофлуориметре, выявило увеличение процентного и абсолютного количества Т-клеток (CD3 + 89,3%; 2,2·10 9 /л), Т-цитотоксических клеток (CD3 + CD8 + 44,8%; 1,0·10 9 /л), снижение процентного и абсолютного количества В-клеток (CD19 + CD3 – 0,3%; 0,008·10 9 /л). С учетом неэффективности ранее проводимой терапии глюкокортикостероидами (ГКС) больному начата комбинированная терапия ритуксимабом и эндоксаном, которая является оптимальным лечением при IgG4-CCЗ [29—31]. Внутривенно капельно с премедикацией 500 мг солюмедрола введены 1000 мг ритуксимаба и 1000 мг эндоксана 1 раз в 2 нед (2 вливания на курс) с последующим введением эндоксана 1 раз в 14 дней (4 на курс) и поддерживающей терапии метилпреднизолоном 4 мг. При оценке эффективности терапии через 4 мес объективно сохранялся выраженный экзофтальм, периодически отмечался подъем температуры до фебрильной. При лабораторном исследовании отмечено снижение показателей воспалительной активности (Hb 136 г/л, л. 11,5·10 9 /л, тр. 433·10 9 /л, СОЭ 50 мм/ч по Вестегрену (норма до 20 мм/ч), вчСРБ 15,2 мг/л, снижение уровня IgG4 до 2,1 г/л. Наблюдалось полное истощение B-клеток CD19 + в крови. При мультиспиральной КТ (МСКТ) глазниц после внутривенного введения 100 мл визипака в отсроченную фазу в ретробульбарной клетчатке определяется мягкотканое образование с неровными контурами, полностью выполняющее полости глазниц, гомогенной структуры, размерами справа 3,3×4,3×3,5 см, слева 3,3×3,7×3,6 см. Мышцы глаз, слезные железы на этом фоне нечетко дифференцируются. Зрительные нервы окружены мягкоткаными разрастаниями, истончены. Определяется двусторонний экзофтальм. Костно-деструктивных изменений нет. Выполнена МСКТ органов брюшной полости после контрастирования желудочно-кишечного тракта до и после введения везипака. Заключение: проявление ретроперитонеального фиброза. Умеренная двусторонняя пиелоэктазия. Умеренная гепатомегалия. Состояние после холецистэктомии. Незначительный перикардит. Множественные увеличенные ЛУ брыжейки тонкой кишки. При сравнении с данными КТ от 2015 г. без динамики. С учетом прогрессирования клинических проявлений псевдотумора глазниц (см. рис. 2, рис. 3), отсутствия положительной динамики при МСКТ глазниц и органов брюшной полости на фоне терапии, некоторых необычных клинических (периодические подъемы температуры до фебрильной, наличие множественных ксантелазм век) и морфологических (наличие большого количества гистиоцитов и недостаточного количество плазмоцитов, секретирующих IgG4, при ИГХИ для постановки диагноза IgG4-ССЗ) проявлений решено провести дополнительное обследование больного с пересмотром биоптатов для исключения системного варианта гистиоцитоза. Проведена позитронно-эмиссионная томография (ПЭТ) для исключения генерализованного гистиоцитоза. Заключение: данных о наличии агрессивного лимфопролиферативного заболевания, а также другого неопластического процесса не получено. Псевдотумор глазниц, генерализованный фиброз медиастинальный, внутрибрюшной, внутритазовый и забрюшиной клетчатки с минимальным воспалительным компонентом. Изменения по типу «волосатой почки» и «облицованной аорты». Внутрибрюшная лимфоаденопатия, вероятнее всего, реактивного генеза. Двусторонний пневмофиброз, застойные (?) изменения в базальных отделах легких. Двусторонний гидронефроз. Гепатомегалия. Генерализованная мелкоячеистая перестройка костей скелета (остеопороз?). Накопление препарата в скелете, вероятнее всего, за счет раздражения костного мозга (см. рис. 4). При К.Т. нижних конечностей и костей черепа выявлены множественные остеосклеротические изменения в дистальных отделах бедренных и большеберцовых костей (рис. 5) и плоских костях черепа (рис. 6). Сцинтиграфия костей: выявлены множественные склеротические перестройки костного мозга. Симметричная патологическое избыточное накопление радиофармпрепарата (РФП) в костях лицевого скелета, эпифизах, метафизах и метадиафизах трубчатых костей наиболее выраженное в бедренных, большеберцовых и костях стоп. Повышенное накопление в чашечно-лоханочной системе правой почки. Однофотонная эмиссионная КТ/КТ черепа и коленных суставов: очаги патологического избыточного накопления РФП соответствуют склеротической перестройке костного мозга. Пересмотр гистологических препаратов с ИГХИ: среди полиморфно-клеточного инфильтрата большое количество гистиоцитов, пенистых клеток, скопления плазматических клеток. Гистиоциты СD68 + , S100 – , IgG4 + , единичные плазматические клетки. Субстрат поражения может принадлежать гистиоцитозу, учитывая локализацию следует предполагать БЭЧ (см. рис. 7). Методом полимеразной цепной реакции в реальном времени с анализом кривых плавления (Rotor Gene 6000) выполнено молекулярное исследование биоптатов. В 599—601-м кодонах 15-го экзона гена BRAF мутация не обнаружена. На рис. 8 представлена частота повторяющихся клинико-радиологических находок у больных БЭЧ, согласно данным литературы и в нашем случае заболевания. Таким образом, больному диагностирован генерализованный гистиоцитоз: БЭЧ с конституциональными нарушениями (лихорадка, патологическая утомляемость, снижение массы тела), поражением кожи и области глазниц (ксантелазмы век, экзофтальм, псевдотумор глазниц, дакриоаденит), костей (остеосклеротические поражения бедренных, малоберцовых и плоских костей черепа), поражение сердечно-сосудистой системы (выпотной перикардит, поражение грудной и брюшной аорты по типу «облицованной аорты»), ретроперитонеальных проявлений (гидронефроз, пиелоэктазия за счет инфильтрации околопочечной ткани по типу «волосатой почки») c отрицательной мутацией BRAFV600E.
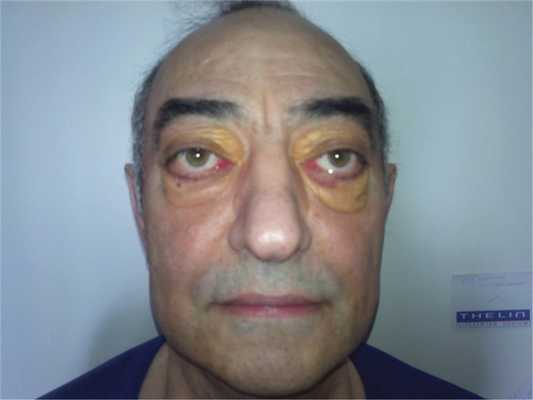
Рис. 1. Экзофтальм, ксантелазмы век.
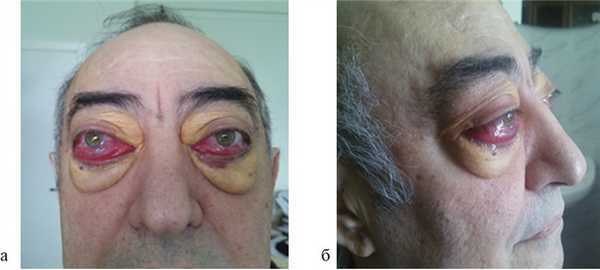
Рис. 2. Прогрессирование псевдотумора глазниц (а, б).
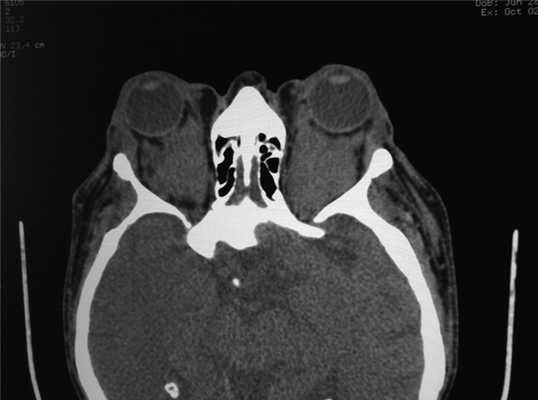
Рис. 3. Двусторонний экзофтальм. В ретробульбарной клетчатке орбит мягкотканые образования с неровными контурами полностью выполняющие полости глазниц.
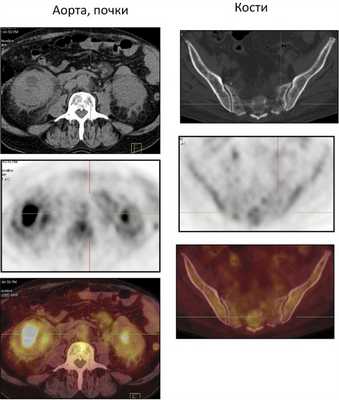
Рис. 4. ПЭТ/КТ брюшной полости (почки, аорта, кости таза).
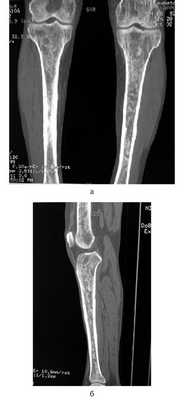
Рис. 5. Множественные остеосклеротические очаги в бедренных (а) и большеберцовых (б) костях.
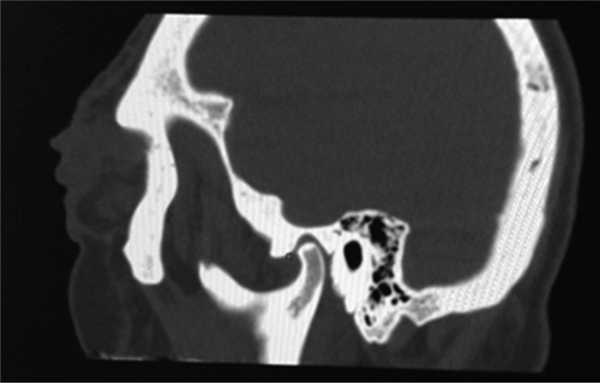
Рис. 6. Остеосклеротические очаги в плоских костях черепа.
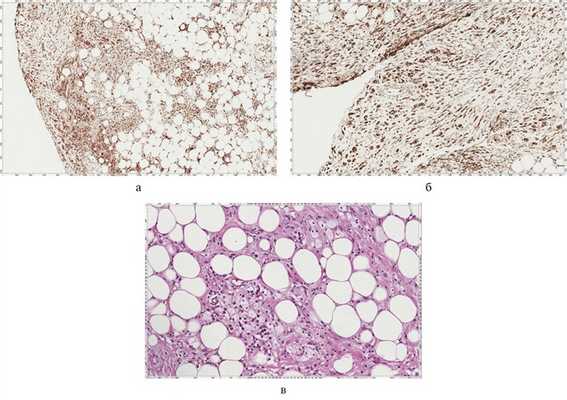
Рис. 7. CD68+. а — реакция с антителами к CD68, множество гистиоцитов, ув. 40; б — ув. 100; в — множественные плазматические клетки, гистиоциты, клетки типа ксантомных среди полиморфно-клеточного инфильтрата, окраска гематоксилином и эозином.
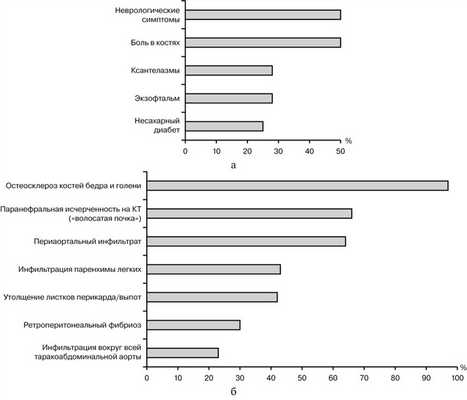
Рис. 8. Наиболее частые поражения при БЭЧ (по [15]). а — жалобы, данные обследования (в нашем случае имелись только ксантелазмы и экзофтальм); б — результаты визуализирующих исследований (в нашем случае выявлены поражения всех перечисленных локализаций за исключением инфильтрации паренхимы легких).
Несмотря на появление в последнее десятилетие работ, посвященных детальным описаниям клинических, рентгенорадиологических, лабораторных изменений при БЭЧ [5, 32] и различных проявлений при IgG4-CЗ [30, 33—37], в дифференциальной диагностике этих заболеваний сохраняются трудности. Они связаны с чрезвычайным разнообразием клинических проявлений и локализаций органных поражений, неспецифичностью современных рентгенорадиологических и лабораторных исследований [6, 17, 32—38], необходимостью использования инвазивных вмешательств для получения адекватного биопсийного материала (при поражении забрюшинного пространства, органов средостения, гепатобилиарного тракта и т. д.), зависимости выраженности гистологических изменений от локализации биопсий, размеров образца ткани и выраженности фиброзных изменений [39]. Оба заболевания чаще дебютируют в возрасте от 40 до 80 лет с медианой возраста 55 лет при постановке диагноза БЭЧ и медианой возраста 57 лет при IgG4-CСЗ, с превалированием заболевания у лиц мужского пола в соотношении 3,5:1 [20, 32], за исключением IgG4-CЗ поражающих слюнные и слезные железы, когда заболевание дебютирует в возрасте от 13 до 77 лет (медиана 42,4 года) в равных пропорциях у лиц мужского и женского пола [19, 34, 35] c описанием случаев развития поражений IgG4 глазниц у детей младше 5 лет [40, 41]. БЭЧ, дебютирующее с поражения костей, и IgG4-CЗ с поражения слюнных желез и области глазниц диагностируются раньше, так как разработанные малоинвазивные хирургические вмешательства позволяют получить достаточное количество материала для морфологического подтверждения характера заболевания и исключения сходных заболеваний, поражающих эти области [6, 20, 35, 40—41]. Дифференциальный диагноз можно провести только на основании всестороннего клинико-лабораторного обследования с обязательным гистологическим и иммуногистологическим исследованием, выполненным опытным морфологом. В нашем случае заболевание дебютировало с конституциональных нарушений и рентгенорадиологических признаков идиопатического ретроперитонеального фиброза. Отсутствие гистологической верификации диагноза привело к длительной терапии ГКС и дальнейшему прогрессированию заболевания с развитием псевдотумора глазниц. В связи с недостаточностью терапии ГКС, которые эффективны при IgG4-CCЗ [34, 35], и с целью гистологической верификации диагноза выполнена лапароскопия с последующей морфологической и иммуногистохимической оценкой биоптата. Первоначальная ошибочная трактовка морфологических изменений побудила использовать в терапии заболевания моноклональные антитела в комбинации с эндоксаном как наиболее эффективной в лечении псевдотумора глазниц, связанного с IgG4 [29—31]. Несмотря на положительное влияние терапии на конституциональные проявления заболевания и ретроперитонеальный фиброз, прогрессирование псевдотумора глазниц и наличие ксантелазм век явилось стимулом для дополнительного обследования больного с целью исключения генерализованного гистиоцитоза. Остеосклеротические поражения костей имеются практически у 100% больных с БЭЧ, однако, как и в нашем случае, у 50% больных они протекают без клинических проявлений [6]. Обнаружение при проведении ПЭТ, КТ нижних конечностей и сцинтиграфии костей остеосклеротических изменений, характерных для гистиоцитозов, позволило предположить наличие в нашем случае БЭЧ. Пересмотр препаратов с ИГХИ и выполненные молекулярные исследования позволили диагностировать заболевание с отрицательной мутацией BRAFV600E. Следует отметить, что, несмотря на развитие новых современных ультразвуковых, рентгенорадиологических (КТ, МСКТ, МРТ и т. д.) методов визуализации поражений головы, грудной клетки, брюшной области), необходима обязательная гистологическая и иммуногистологическая верификация диагноза согласно рекомендациям экспертов по диагностике этих заболеваний, опубликованным после проведенных Международных симпозиумов, посвященных изучению различных аспектов этих патологических состояний [6, 28]. Выполненное в полном объеме обследование больного согласно опубликованным рекомендациям привело к правильной постановки диагноза в нашем случае.
Читайте также: