Кости при гомоцистинурии - лучевая диагностика
Добавил пользователь Morpheus Обновлено: 31.01.2026
Рентгенограмма, МРТ при синдромах Марфана и Элерса-Данлоса
а) Определения:
• Синдром Марфана (МФ): семейное заболевание, обусловленное пороком развития соединительной ткани с поражением опорно-двигательного аппарата, глаз и сосудов, но с вариабельной фенотипической экспрессией
• Синдром Элерса-Данлоса (ЭД): наследственное заболевание соединительной ткани с различными фенотипическими проявлениями (множество синдромов)
б) Визуализация:
1. Непропорциональное удлинение конечностей (МФ, ЭД):
• Особенно кистей, стоп (арахнодактилия) в 89% случаев
2. Слабость связочного аппарата (МФ, ЭД):
• Патологическое искривление возможно в нескольких областях:
о Чаще возникает сгибательная деформация 5-го пальца (90°)
о Рекурвация коленного сустава, высокое стояние надколенника
• Реже нестабильность запястья
• Плоская стопа, вальгусная деформация большого пальца, молоткообразная деформация пальцев стопы
• Нестабильность суставов, вероятно обусловливающая хронические состояния:
о Повторяющаяся субклиническая травма
о Выпоты, гемартроз (ЭД>МФ)
о Ранний остеоартрит (ЭД>МФ)
• Вывихи суставов (наколенник, тазобедренный сустав, нижняя челюсть, ключица, пальцы) ЭД>МФ
• Протрузия вертлужной впадины
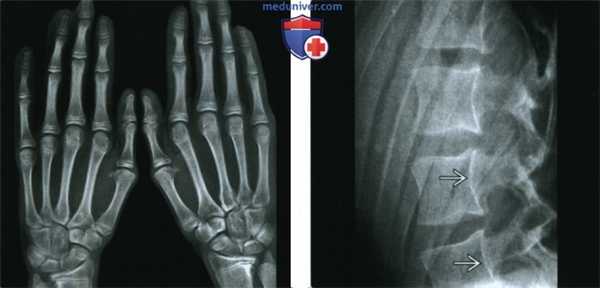
(Слева) Рентгенография кистей в ЗП проекции: определяется арахнодактилия при отсутствии других изменений. Наблюдается аномальный метакарпальный индекс.
(Справа) Рентгенография в боковой проекции, этот же пациент: определяется неровность задней поверхности тел позвонков. Плотность кости нормальная. На уровне позвонков L5-S1 был выявлен двусторонний спондилолизис с IV степенью спондилолистеза (изображение не представлено). Аномалии позвоночника в сочетании с арахнодактилией могут наблюдаться как при синдроме Марфана, так и при синдроме Элерса-Данлоса: в этом случае диагностирован синдром Марфана.
3. Мягкие ткани: различные проявления при МФ, ДМ:
• МФ: худощавость, атрофия мышц, скудная жировая клетчатка
• ЭД: подкожные кальцификации (некроз жировой ткани) в сочетании с повышенной частой гетеротопической оссификации
• При МРТ определяется слабость связочного аппарата, разрывы сухожилий, тендинопатия
4. Позвоночник (МФ, ЭД):
• Сколиоз (40-60%): картина схожа с идиопатическим сколиозом
• Неровность задней поверхности тел позвонков с эктазией твердой мозговой оболочки (63%):
о Также могут наблюдаться расширение отверстий, нарушение морфологии крестца
• Спондилолизис со спондилолистезом
• Подвывих атланто-аксиального сочленения (редко)
5. Грудная клетка (МФ, ЭД):
• Воронкообразная или килевидная деформация грудной клетки
6. Другие признаки:
• Плотность кости (МФ, ЭД): нормальная
• Эпифизиолиз головки бедренной кости (МФ, ЭД): ↑ частота встречаемости
в) Дифференциальная диагностика синдромов Марфана и Элерса-Данлоса:
1. Гомоцистинурия:
• Схожие признаки: непропорциональность длины конечностей, арахнодактилия и гиперподвижность суставов
• Клиническим признаком для дифференциального диагноза является задержка умственного развития
• Диффузная остеопения наблюдается при гомоцистеинурии, но не при других состояниях
• Большее число контрактур суставов
• Различный характер поражения глаз: двусторонний нижний вывих хрусталиков, возникающий в раннем детском возрасте
• Иной характер поражения сосудов: тромбоэмболия
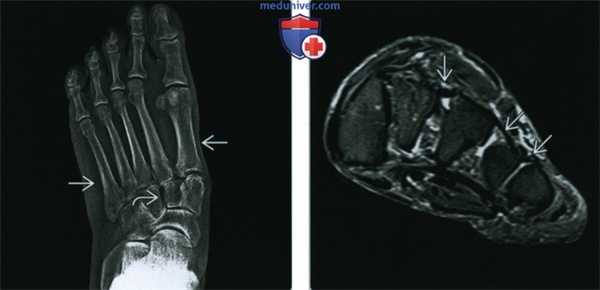
(Слева) Рентгенография в ПЗ проекции: плоская стопа, обусловленная болезнью Марфана. Наблюдается пронация переднего отдела стопы, определяемая по отклонению оснований плюсневых костей. Также имеется просвет между клиновидными костями, что говорит об ослаблении связок.
(Справа) МРТ Т2ВИ, режим подавления сигнала от жира, корональный срез: у этого же пациента определяется растяжение межклиновидных и межплюсневых связок В, что приводит к аномальному просвету между клиновидными и плюсневыми костями. На других изображениях (не представлены) определялись разрывы сухожилий, все это вносит вклад в формирование клинически плоской стопы, характерной для болезни Марфана.
г) Патология. Общая характеристика:
• Генетика:
о Синдром Марфана: обычно аутосомно-доминантный тип наследования; в 20-30% обусловлен спонтанными мутациями:
- Мутации в локусе (15q15-15q21) длинного плеча XV хромосомы (MFS1)
о ЭД: сложная классификационная система:
- Множество генетических дефектов, влияющих на синтез коллагена (не менее 19 фокусов минимум в 12 генах)
д) Клинические особенности:
1. Проявления:
• Типичные признаки/симптомы:
о Синдром Марфана:
- Высокий рост, худощавость (> 95-го перцентиля)
- Непропорциональная длина конечностей относительно туловища):
Кисти, стопы > нижние конечности > верхние конечности
- Поражение сосудов:
Кистозный медионекроз с расслоением и разрывом аорты
Расширение проксимального отдела восходящей аорты → недостаточность аортального клапана и расширение венечных пазух
Медионекроз основной легочной артерии (менее часто)
Недостаточность аортального и митрального клапанов
- Поражение глаз:
Патологическое изменение подвешивающих связок → двусторонний вывих хрусталиков (в верхнем направлении) в 57% случаев
Также наблюдаются страбизм и отслоение сетчатки
о Диагностика синдрома Элерса-Данлоса основана на наличии клинической триады:
- Хрупкость кожных покровов:
Кожу можно приподнять с формированием высокой складки; со временем складки становятся постоянными; кожа легко рубцуется
- Гипермобильность суставов
- Хрупкость сосудов:
Желудочно-кишечные, кожные и бронхолегочные кровотечения
Спонтанное расслоение аорты или крупных сосудов, смерть от потери крови
- Встречается поражение глаз, но двусторонний вывих хрусталиков встречается менее часто, чем при МФ
2. Демография:
• Возраст:
о Симптомы часто не проявляются до детского возраста
• Эпидемиология:
о Синдром Марфана: 4-6 на 100000 новорожденных
3. Течение и прогноз:
• Средний возраст смерти при синдроме Марфана 28 лет; смерть вызвана сердечно-сосудистыми заболеваниями
4. Лечение:
• Устранение проблем, связанных с сердечно-сосудистой системой и органами зрения
• Реконструкция нестабильных слабых связок
е) Список использованной литературы:
1. Hammarstedt JE et al: Arthroscopic ligamentum teres reconstruction of the hip in Ehlers-Danlos syndrome: a case study. Hip Int. 0, 2015
Кости при гомоцистинурии - лучевая диагностика
Кости, суставы при менингококковой инфекции (менингококцемии) - лучевая диагностика
а) Терминология:
• Инфекция, вызванная Neisseria meningitidis
• Признаки и симптомы поражения опорно-двигательной системы во время инфицирования:
о Теносиновит
о Артрит:
- Острый преходящий полиартрит: боль и тугоподвижность развиваются одновременно с петехиальной сыпью
- Гнойный артрит (часто коленного сустава), развивающийся через пять дней от начала заболевания (5-10%)
• Аномалии развития скелета, развивающиеся у детей, переживших тяжелую форму заболевания:
о Раннее сращение зон роста, часто в центральной части, обусловливающее картину по типу конических эпифизов/мета-физов
о Расщепление эпифиза, аномальная форма → изогнутость и угловые деформации
о Укорочение и неравная длина конечностей
о Кости полиостозные, но не симметричные: нижняя > верхняя конечность
(Слева) Рентгенография в ПЗ проекции: определяется аномально раннее сращение центральной и медиальной частей зон роста дистального конца бедренной кости и проксимального конца большеберцовой кости. Это обусловило аномальную форму эпифизов и варусную деформацию коленного сустава. Противоположный коленный сустав не изменен.
(Справа) Рентгенография в ПЗ проекции, этот же пациент: определяется схожая аномалия с поражением зоны роста дистального конца большеберцовой кости, что обусловило более выраженную коническую форму эпифиза. Таранная кость также изменена и имеет округлую головку. (Слева) Рентгенография в ПЗ проекции: нормальный правый тазобедренный сустав у этого же пациента. Это говорит о том, что эта аномалия не является диффузной метафизарной или эпифизарной дисплазией.
(Справа) Рентгенография левого тазобедренного сустава в ПЗ проекции: аномально расширенная зона роста и расщепление эпифиза, что привело к варусной деформации. В целом такая картина характерна как для эмболии, так и для осложнений менингококцемической коагулопатии и последующего сосудистого инсульта с поражением всей конечности. В этом случае анамнез указывал на последний их этих процессов.
в) Дифференциальная диагностика:
• Дифференциальная диагностика конических эпифизов:
о Эмболия
о Генерализованная инфекция
г) Патология:
• Этиология костных аномалий: время и картина позволяют считать роль инфекции маловероятной; считается, что они обусловлены поражением сосудов
д) Клинические особенности:
• Различная тяжесть течения: от доброкачественного и бессимптомного до молниеносного и смертельного:
о Лихорадка, потрясающий озноб, кожная сыпь, петехии, миалгии
• Молниеносное течение (синдром Уотерхауса-Фредериксена):
о Гипотензия, спутанность сознания, тахипноэ, периферический цианоз, диссеминированное внутрисосудистое свертывание (ДВС):
- ДВС → диффузная кровоточивость слизистых оболочек, окклюзия мелких сосудов с некрозом кожи, мозга, почек и надпочечников
Гомоцистинурия
Гомоцистинурия – наследственный дефект метаболизма, первичным звеном которого выступает нарушение обмена серосодержащих аминокислот, приводящее к поражению нервной, костно-мышечной и сердечно-сосудистой систем. Гомоцистинурия сопровождается умственной отсталостью, судорожным синдромом, подвывихом хрусталиков, катарактой, глаукомой, атрофией зрительных нервов, деформацией грудной клетки, сколиозом, арахнодактилией, артериальными и венозными тромбозами. Диагностика гомоцистинурии включает медико-генетическое консультирование, биохимическое исследование крови и мочи, офтальмологическое обследование, рентген-диагностику костной системы. Терапия гомоцистинурии проводится с учетом формы заболевания и включает диетическое питание, прием витаминов группы В.
Общие сведения
Гомоцистинурия – генетически обусловленная энзимопатия, характеризующаяся нарушением обмена незаменимой аминокислоты метионина, повышением уровня гомоцистина в биологических жидкостях и тканях, приводящим к повреждению органов и систем. Начало изучению заболевания положено в 1962 г. Частота гомоцистинурии в популяции составляет 1 случай на 200 000 новорожденных. Течение гомоцистинурии сопровождается нервно-психическими нарушениями, глазной патологией, изменениями со стороны опорно-двигательного аппарата, склонностью к тромбоэмболиям. В связи с многообразием последствий метаболических расстройств гомоцистинурия рассматривается с позиций различных медицинских дисциплин, главным образом, генетики, педиатрии, неврологии, офтальмологии, ортопедии.
Причины гомоцистинурии
Гомоцистинурия обусловлена аутосомно-рецессивным типом наследования. На сегодняшний день известны 4 типа метаболических нарушений, которые могут лежать в основе патологии, в связи с чем выделяют следующие биохимические варианты заболевания:
- Гомоцистинурия I – обусловлена отсутствием или снижением активности фермента цистатионин-бета-синтазы (классическая гомоцистинурия).
- Гомоцистинурия II – обусловлена отсутствием или снижением активности фермента N5, N10-метилентетрагидрофолат-редуктазы.
- Гомоцистинурия III – обусловлена низкой активностью фермента N5-метилентетрагидрофолата.
- Гомоцистинурия IV – обусловлена отсутствием или снижением активности фермента гомоцистеин трансметилазы, вызванным дефектом синтеза метилкобаламина.
Непосредственные патогенетические механизмы патологии связаны с нарушением метаболизма незаменимой аминокислоты метионина. Метаболические процессы контролируются рядом ферментов, при инактивации которых происходит энзиматический блок: в крови и тканях накапливается промежуточный продукт обмена метионина – гомоцистин, который экскретируется с мочой; при этом также уменьшается содержание цистатионина и цистина. Возможной причиной нарушения метаболического пути также может служить гиповитаминоз В6 и В12, а также фолиевой кислоты.
Высокие концентрации метионина и гомоцистина оказывают повреждающее действие на внутреннюю стенку артерий, что сопровождается усилением агрегации тромбоцитов и созданием условий для тромбообразования. Кроме этого, отмечается токсическое действие гомоцистина на нервную, соединительную и другие ткани. В зависимости от особенностей патогенеза различают две формы гомоцистинурии – пиридоксинзависимую (витамин B6-чувствительную) и пиридоксинрезистентную (витамин B6-нечувствительную), что определяет выбор метода лечебного воздействия на организм.
Симптомы гомоцистинурии
Проявления гомоцистинурии нарастают постепенно. Дети рождаются без каких-либо специфических отклонений. В течение первого года жизни развивается умеренно выраженная гипотрофия. Попытки устранить отставание в весе и росте за счет дополнительного введения в рацион белка в виде кефира или творога лишь усугубляют течение заболевания: нарастает дефицит массы тела, нарушается сон, ребенок становится раздражительным и плаксивым, отмечается позднее закрытие родничков, деформации конечностей, задержка психомоторного развития.
Обычно ярко выраженная клиника гомоцистинурии развивается в течение первых 10 лет жизни, однако часто диагноз становится очевидным уже в раннем детском возрасте. К этому времени у ребенка появляются высокоспецифичные глазные симптомы: подвывих хрусталиков, выраженная близорукость, дрожание радужки (иридодонез). Несколько позднее присоединяются астигматизм, глаукома, катаракта, отслойка сетчатки, атрофия зрительных нервов. Часто гомоцистинурии сопутствуют умственная отсталость, нарушения мышечного тонуса, гиперкинезы, судорожный синдром, поведенческие нарушения. Поражение опорно-двигательного аппарата включает килевидную деформацию грудной клетки, арахнодактилию, кифосколиоз, остеопороз, искривление голеней, полую стопу или плоскостопие, готическое нёбо. Практически половина пациентов с гомоцистинурией сталкивается с артериальными тромбозами (окклюзией церебральных, коронарных, почечных и периферических сосудов), а также венозными тромбозами (ТЭЛА).
Лица, страдающие гомоцистинурией, имеют определенные фенотипические черты: высокий рост, диспропорциональное телосложение (тонкие удлиненные конечности и укороченное туловище), голубые глаза, редкие светлые волосы. У них часто встречаются эритематозные пятна в области скуловых дуг, телеангиэктазии. Внешние проявления гомоцистинурии обладают определенным сходством с синдромом Марфана, однако для последнего не характерно снижение интеллекта и ряд других проявлений.
Диагностика гомоцистинурии
Пациенты с подозрением на гомоцистинурию должны направляться к медицинскому генетику для анализа генеалогических данных и проведения молекулярно-генетической диагностики. Диагноз устанавливается с помощью биохимического исследования крови и мочи: при гомоцистинурии в моче, плазме крови, ликворе обнаруживаются значительные количества гомоцистина, повышение содержания метионина при сниженном уровне цистина. В биоптатах кожи и печени выявляется специфический ферментативный дефект.
Рентгенологическое исследование трубчатых костей и позвоночника обнаруживает системный остеопороз. На ЭЭГ регистрируются нарушения биоэлектрической активности головного мозга, иногда пароксизмального характера. Консультация офтальмолога позволяет подтвердить характерные для гомоцистинурии нарушения со стороны зрительной системы. Также больные дети нуждаются в наблюдении и оценке развития со стороны педиатра, невролога, ортопеда, психиатра. Дифференциальная диагностика гомоцистинурии осуществляется с синдромом Марфана, последствиями родовой травмы и внутриутробных инфекций, другими энзимопатиями.
Лечение гомоцистинурии
Лечебная тактика зависит от формы заболевания (В6-зависимой или В6-резистентной) и во многом схожа с лечением фенилкетонурии. При В6-резистентной форме гомоцистинурии необходимо соблюдение низкобелковой диеты, основанной на ограничении поступления в организм метионина. Рацион пациентов должен состоять, главным образом, из растительной пищи при исключении или значительном снижении употребления продуктов животного происхождения. Для возмещения потребности в незаменимых аминокислотах назначаются специальные аминокислотные смеси, лишенные метионина (XMET Analog, XMET Maxamum, XMET Maxamaid, XMET Homidon). При В6-зависимой гомоцистинурии активность фермента удается активизировать назначением больших доз пиридоксина гидрохлорида.
При любых формах гомоцистинурии снижению уровня гомоцистина в биологических жидкостях способствует назначение фолиевой кислоты, бетаина. Для минимизации риска тромбозов показан постоянный прием ацетилсалициловой кислоты в низкой дозировке. По показаниям больным назначаются гепатопротекторы, ноотропы, препараты кальция, железа; проводятся курсы массажа, лазерной акупунктуры и рефлексотерапии, ЛФК.
Прогноз гомоцистинурии
Выявление заболевания на доклинической стадии, раннее начало лечения и соблюдение лечебной диеты позволяют отсрочить или предотвратить инвалидизирующие осложнения (интеллектуальные нарушения, параличи, атрофию зрительных нервов, легочное сердце, тяжелую артериальную гипертонию, инсульты, инфаркты внутренних органов и др.). В семьях, где есть носители гена гомоцистинурии, необходимо проведение инвазивной пренатальной диагностики с определением активности фермента в культуре клеток ворсин хориона или амниотической жидкости. В отношении детей с гомоцистинурией актуальны вопросы специализированного обучения, профессиональной ориентации, социальной адаптации, диспансерного наблюдения специалистов.
Гомоцистинурия у детей
Гомоцистинурия – наследственное заболевание из группы аминоацидопатий, обусловленное нарушением метаболизма серосодержащих аминокислот, в первую очередь метионина. Относится к классу редких (орфанных) заболеваний.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
13-15 октября, Алматы, "Атакент"
600 брендов, более 150 компаний-участников из 20 стран.
Новинки рынка стоматологии. Цены от производителей
Классификация
- гомоцистинурия, обусловленная нарушением активности 5-метилтетрагидрофолат-гомоцистеин-метилтрансферазы (метаболический блок 3)
- гомоцистинурия, обусловленная нарушением активности N(5,10)-метилентетрагидрофолатредуктазы (метаболический блок 4)
Этиология и патогенез
Заболевание вызвано дефицитом фермента цистатион-β-синтазы (CbS), участвующего в преобразовании метионина в цистеин, проявляется при наличии гомозиготных или компаунд-гетерозиготных мутаций в гене CBS, локализованном на длинном плече хромосомы 21 (21q22). Тип наследования аутосомно - рецессивный.
При классической гомоцистинурии вследствие недостаточности цистатион-β-синтазы нарушается цикл преобразования (метилирования и деметилирования) серосодержащей аминокислоты метионина.
Энзим цистатион-β-синтаза (CbS) является пиридоксин (витамин В6) зависимым ферментом, поэтому в классической гомоцистинурии выделяют В6-зависимую и В6-резистентную формы заболевания (метаболический блок 1 на рисунке 1: В6 – зависимая форма). Возможно также нарушение активности следующего фермента в этой метаболической цепи – цистатионазы, который обусловливает В6-резистентную форму заболевания (метаболический блок 2),
Две следующие формы гомоцистинурии связаны с генетически детерминированными дефектами реметилирования метионина, возникающими вследствие нарушения активности 5-метилтетрагидрофолат-гомоцистеин-метилтрансферазы (3-я форма, метаболический блок 3 на рисунке 1) и блоком фермента N(5,10)-метилентетрагидрофолатредуктазы(4-я форма, метаболический блок 4 на рис. 1). Указанные две формы сопровождаются не повышением, а снижением концентрации метионина в крови. В педиатрической практике чаще встречаются две первые формы болезни, именуемые классической гомоцистинурией [1,2,11, 14].
Эпидемиология
Средняя частота в общей популяции не определена из-за отсутствия повсеместного неонатального скрининга, есть данные, что она составляет от 1: 58 000 до 1: 335 000, в странах Ближнего Востока (Катар) от 1: 1 800 до 1:8 000. Ген локализован на длинном плече хромосомы 21, в локусе 21q22.1.
Диагностика
Задержка психомоторного развития, умственная отсталость, снижение зрения, скелетные деформации (вальгусная установка коленных суставов, кифосколиозы, воронкообразная или килевидная деформации грудной клетки), кардиоваскулярная патология, частые переломы у детей старшего возраста.
В анамнезе возможны указания на родственный брак, наличие сибсов с аналогичными клиническими признаками, наличие у близких родственников ранних инфарктов/инсультов.
Фенотипические черты больных: мягкие слегка вьющиеся светло-русые волосы, нежный румянец на щеках, голубой цвет радужной оболочки, высокий рост, астеническое телосложение, длинные тонкие конечности, арахнодактилия кистей и стоп. Часто обнаруживается вальгусная установка коленных суставов, кифосколиозы, воронкообразная или килевидная деформации грудной клетки. Наряду с этим, встречаются формы болезни, при которых изменения опорно-двигательного аппарата минимальны или полностью отсутствуют.
Характерна патология глазного аппарата: сублюксация (люксация) хрусталиков, часто осложняющийся вторичной глаукомой, нередко имеющей злокачественное течение, миопия, атрофия зрительных нервов, катаракта и отслойка сетчатки.
Сердечно-сосудистые нарушения обусловлены развитием тромбоэмболий в артериальных сосудах среднего и мелкого калибра.
Тромбозы возникают преимущественно у пациентов подросткового и молодого возраста, являются главной причиной инфаркта миокарда или инсульта с формированием очаговой неврологической симптоматики. Интеллект больных с гомоцистинурией чаще снижен: IQ (коэффициент интеллектуального развития) колеблется от 32 до 85 ед. (норма 85-115 ед.). У детей с В6 – зависимой формой гомоцистинурии умственное развитие может быть нормальным.
Тяжесть клинических проявлений болезни при В6 - резистентной форме более выражена, чем при В6-зависимой.
• Для диагностики заболевания в качестве скринингового теста рекомендовано использовать пробу на серосодержащие аминокислоты – качественная реакция с цианиднитропруссидом: при окрашивании мочи в интенсивный свекольный цвет проба считается положительной [2].
• Для диагностики классической гомоцистинурии рекомендовано количественное определение метионина, гомоцистина и цистина в сыворотке (плазме) крови и мочи методом тандемной масс-спектрометрии (ТМС) [3,13,15].
Комментарии: для классической гомоцистинурии характерны повышение уровня метионина и появление гомоцистина в сыворотке крови, снижение цистина в сыворотке крови и моче (таблица 3). Важно, что концентрация гомоцистеина и гомоцистина в плазме должна быть определена у пациента, не получающего пиридоксин (в том числе в составе поливитаминов) в течение двух недель.
Впервые гомоцистинурия была описана в 1962 году в Северной Ирландии: Карсон и Нилл (Carson и Neill) наблюдали двух светловолосых голубоглазых братьев с выраженной задержкой развития и офтальмологическими нарушениями. Спустя десятилетие, в 1972 году, Фриман и его коллеги описали другой вариант заболевания, обусловленный дефицитом фермента метилентетрагидрофолатредуктазы. Авторы обследовали 15-летнюю чернокожую девушку, у которой задержка нервно-психического развития была выраженам слабее, но в течение последних двух лет наблюдались прогрессирующие психические нарушения. Через 12 лет Шух (Schuh) и его соавторы описали случай гомоцистинурии у грудного ребенка с выраженной задержкой психомоторного развития и признаками мегалобластической анемии.
Гомоцистинурия — наследственное нарушение обмена метионина, наследуемое по аутосомно-рецессивному типу. Метионин — незаменимая серосодержащая аминокислота, которая используется для создания непротеиногенной аминокислоты гомоцистеина, синтеза белков (взаимодействие метионина с тРНК является необходимым для создания первой пептидной связи будущего белка) и ряда других важных соединений, благодаря тому, что является донором метильной группы.
В зависимости от того, на каком уровне происходит нарушение обмена метионина, выделяют несколько форм заболевания:
- Классическая гомоцистинурия — обусловлена дефицитом фермента цистатион-бета-синтазы (CbS), лимитирующего синтез цистатиона из гомоцистеина. Ген CBS (кодирующий вышеупомянутый энзим) локализован на длинном плече 21 хромосомы в локусе 1q21-q22.1. Его различные мутации способны привести к развитию гомоцистинурии двух разных фенотипов: пиридоксин (витамин B6) зависимого и пиридоксин (витамин B6) независимого. Средняя частота встречаемости данной формы заболевания до сих пор точно не установлена, т. к. неонатальный скрининг на гомоцистинурию проводится не во всех странах (есть данные, что она составляет 1: 58–1:335000, в странах Ближнего Востока 1:1800–1:8000).
- Гомоцистинурия, обусловленная дефицитом фермента метилентетрагидрофолатредуктазы (MTHFR). Причинный ген картирован на коротком плече 1 хромосомы в локусе 1p36.22. Заболевание встречается крайне редко, его частота не установлена.
- Гомоцистинурия, обусловленная нарушением метаболизма кобаламина (Сbl). Имена эта форма была описана в 1984. Заболевание встречается крайне редко, частота его не установлена. Существует несколько типов гомоцистинурии, связанных с кобаламинами, к настоящему времени наиболее известны:
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип G, CblG). Ген локализован на коротком плече 5 хромосомы.
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип Е, CblE). Ген локализован на длинном плече 1 хромосомы.
Патофизиология
При классическом варианте гомоцистинурии из-за дефекта CbS происходит накопление гомоцистеина и его метаболитов в крови и моче. Гомоцистеин синтезируется во всех органах и является токсичным веществом в первую очередь для эндотелия и нейронов. Его токсичность обусловлена, во-первых, наличием реактивной сульфгидрильной группы, которая способна к окислению при физиологическом pH в присутствии кислорода, что ведет к появлению активных форм кислорода и дальнейшему перекисному окислению липидов, повреждению ДНК и апоптозу. Во-вторых, гомоцистеин способен формировать дисульфидные связи со свободной сульфгидрильной группой цистеина в белках, нарушая функцию последних. В-третьих, один из метаболитов гомоцистеина, гомоцистеина тиолактон, соединяясь с лизином протеинов, провоцирует формирование токсических полимеров, агрегатов или амилоида, что является фактором риска нейродегенерации. Помимо этого, белки, присоединившие гомоцистеина тиолактон, становятся антигенном и способствуют развитию иммунной реакции, являющейся важным модулятором атерогенеза.![]()
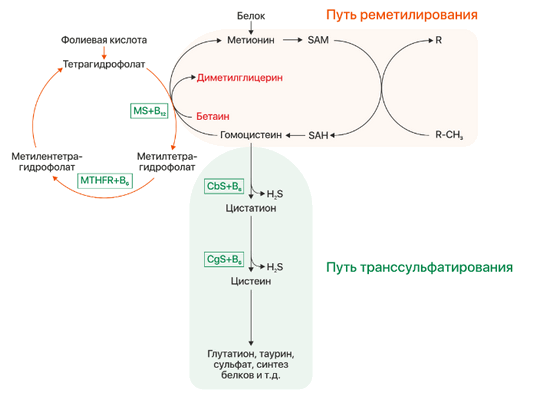
Рисунок 1 | Метаболизм гомоцистеина. CbS — цистатион-бета-синтаза, MS — метионинсинтаза, MTHFR — метилентетрагидрофолатредуктаза, SAM — S-аденозинметионин, SAH — S-аденозингомоцистеин.
Несмотря на то, что синтез гомоцистеина происходит практически повсеместно в организме, его утилизация реализуется преимущественно в почках и печени двумя путями: реметилированием и транссульфарацией (см. рис. 1). В сосудистой ткани и коже за неимением ферментов другого пути происходят реакции реметилирования, остальные ткани осуществляют детоксикацию гомоцистеина обоими путями в том или ином соотношении.
При повышение уровня клеточного метионина осуществляется путь транссульфарации: происходит синтез гомоцистеина, который в свою очередь конвертируется в цистатион под действием фермента CbS и кофермента витамина B6. Затем из цистатиона под действием цистатион-гамма-лиазы (CgL) и пиридоксина образуется цистеин. При низком же уровне метионина происходит реакция реметилирования: гомоцистеин принимает метильную группу от метилтетрагидрофолата, превращаясь в метионин. Данная реакция происходит в присутствии фермента метионинсинтазы и кофермента метилкобаламина (витамин B12, CblG), поэтому при нарушении обмена кобаламинов (Cbl) будет развиваться клиника гомоцистинурии. Синтез донора метильной группы регулируется ферментом метилентетрагидрофолатредуктазой (MTHFR), его дефект будет так же вести к развитию заболевания.Клиническая картина
Клиническая картина гомоцистинурии зависит от формы заболевания и может иметь различную степень выраженности.
При классической гомоцистинурии больные дети имеют мягкие, вьющиеся светлые волосы, румянец на щеках, голубой цвет глаз и «морфаноидный» внешний вид — высокий рост, астеничное телосложение, длинные конечности, арахнодактилия. Помимо этого, возможно развитие кифосколиоза с воронкообразной или килевидной деформацией грудной клетки, вальгусной установки коленных суставов, умеренного остеопороза и переломов у детей старшего возраста. Наиболее типичным поражением глаз являются подвывих (вывих) хрусталика, миопия и вторичная глаукома, также встречаются дети с атрофией зрительных нервов и отслойкой сетчатки. Интеллект детей снижен значительно, задержка развития обращает на себя внимание еще в младенчестве. Неврологическая симптоматика весьма полиморфна и меняется с возрастом (см. табл. 1). Изменения в сердечно-сосудистой системе проявляются развитием тромбоэмболии в сосудах мелкого и среднего калибра, что является главной причиной инсультов и ранней смерти. B6-зависимая гомоцистинурия обычно протекает мягче, чем B6-независимый вариант.Для больных гомоцистинурией, вызванной дефицитом MTHFR и нарушением обмена Cbl, характерны микроцефалия, умеренное отставание в психомоторном развитии. При отсутствии лечения возможно резкое нарастание нарушений в нервной системе, иногда приводящей к смерти. У больных, заболевание которых в детстве протекало мягко, с минимальной выраженностью клиники, в подростковом и взрослом возрасте происходит резкое ухудшение состояния. При этом изменение неврологической симптоматики происходит трехфазно: после периода нормального развития в раннем детстве (фаза 1), у детей старшего возраста обнаруживается микроцефалия и психомоторные нарушения (фаза 2), затем происходит стремительное нарастание психо-неврологических симптомов (фаза 3), иногда сопровождающееся дыхательной недостаточностью, что может привести к смерти.
Помимо этого, при нарушении обмена Cbl развивается мегалобластическая анемия, что является важным признаком для дифференциальной диагностики.Таблица 1 | Неврологическая симптоматика в различных возрастных периодах.
Нарушение CbS Нарушение обмена Cbl Нарушение MTHFR Неонатальный период, первый год жизни Острые неврологические нарушения - + + Неонатальные приступы - - + Задержка развития + + + Гидроцефалия - +/- + Нистагм - + - Раннее и позднее детство Инсульт + + + Нервно-психическая задержка развития, обычно трехфазная - + + Спастический тетрапарез - + + Психиатрическая симптоматика + + + Нистагм - + - Подростковый возраст, взрослые Нервно-психическая задержка развития - + + Комбинированная дегенерация спинного мозга - + + Инсульт + + + Миоклония - + + Необъяснимые острые психиатрические симптомы + + + Нистагм - + - Примечания: CbS — цистатион-бета-синтаза, Cbl — кобаламин, MTHFR — метилентетрагидрофолатредуктаза.
Диагности ка
В странах, где неонатальный скрининг включает исследование на гомоцистинурию при помощи теста Гатри или тандемной масс-спектрометрии (подобно фенилкетонурии) — диагностика значительно упрощена. Но к сожалению, таких стран немного и Россия не в их числе.
Заподозрить заболевание у новорожденных детей и детей раннего возраста помогают неврологические и офтальмологические нарушения. При этом важно дифференциировать гомоцистинурию от болезни Морфана в виду схожести фенотипических проявлений. Главные отличия: тип наследования (гомоцистинурия наследуется по аутосомно-рецессивному типу, а болезнь Морфана — по аутосомно-доминантному) и нарушение аминокислотного спектра сыворотки крови (при гомоцистинурии). Помимо этого, у детей с болезнью Морфана наблюдаются менее тяжелое поражение глаз, наличие аневризмы аорты, отсутствие интеллектуальных нарушений и остеопороза.
В качестве скринингового метода обнаружения метионина возможно использование качественной реакции с цианиднитропруссидом — при наличие серосодержащих аминокислот моча окрасится в свекольный цвет. Для дифференциальной диагностики между формами заболевания определяют уровень метионина, гомоцистеина и цистеина в плазме крови и моче (см. табл. 2), а также используют молекулярно-генетические методы.Таблица 2 | Изменения уровней метионина, гомоцистеина и цистеина при различных формах гомоцистинурии.
Общий гомоцистеин плазмы Метионин плазмы Цистеин плазмы и мочи Нарушение CbS ↑↑ ↑ ↓ Нарушение обмена Cbl ↑↑ ↓ ↑ Нарушение MTHFR ↑↑ ↓ / N ↑ Примечания: CbS — цистеин-бета-синтаза, Cbl — кобаламин, MTHFR — метилентетрагидрофолатредуктаза.
При подтверждении диагноза классическая гомоцистинурия (гомоцистинурия, обусловлена дефицитом фермента CbS)” проводят тест с витамином B6 для определения фенотипического варианта заболевания, что важно для последующего лечения.
Методы лечения
Выбор метода лечения зависит от формы заболевания.
При классической B6-зависимой гомоцистинурии рационально назначение пиридоксина, начиная с малых доз (200–250 мг/день для новорожденных и детей раннего возраста, 400–500 мг/день для детей позднего возраста и взрослых). Доза подбирается индивидуально и принимается в течение нескольких недель под контролем маркеров заболевания. Важно ограничить прием больших доз витамина B6 в течение длительного времени, т. к. он начинает оказывать токсический эффект и может вызвать респираторные нарушения, периферическую нейропатию и рабдомиолиз.Больным с B6-независимой гомоцистинурией показана строгая ограничительная диета: уровень метионина снижается за счет уменьшения потребления белковых продуктов. Для обеспечения физиологической концентрации метионина и цистеина, а также предотвращения белково-энергетической недостаточности, возможно применение аминокислотных смесей с низким содержанием метионина и высоким цистеина.
Бетаин наряду с метилтетрагидрофолатом (см. рис. 1) является донором метильной группы для гомоцистеина. Его прием при B6-резистентной форме гомоцистинурии обеспечивает альтернативный путь синтеза метионина и уменьшает уровень гомоцистеина в крови.
Прием витамина B12, кофактора метионинсинтазы, возможен при всех формах гомоцистинурии. Его натуральная форма (гидроксикобаламин) более эффективна, чем синтетическая (цианкобаламин). При классической гомоцистинурии витамин B12 назначается per os в малых дозах под контролем уровня витамина в плазме крови для предотвращения дефицита кобаламинов. При заболевании, вызванном дефицитом кобаламинов, витамин B12 применяется инъекционно ежедневно.
Назначение фолиевой кислоты (витамин B9) при классической гомоцистинурии (per os 1–5 мг/день ) предотвращает развитие фолатной недостаточности. При дефекте обмена Cbl длительный прием больших доз (5–30 мг/день) витамина B9 компенсирует фолатную ловушку и вызванные ею гематологические изменения (при недостаточности витамина B12 нарушается цикл фолатов и количество фолиевой кислоты в клетке постепенно уменьшается). При недостаточности фермента MTHFR также назначаются большие дозы фолиевой кислоты (5–45 мг/день) и рибофлавин (витамин B2), который является кофактором фермента и может стабилизировать мутантный энзим (см. рис. 1).
Читайте также:
- гомоцистинурия, вызванная дефицитом метилкобаламина (кобаламин тип G, CblG). Ген локализован на коротком плече 5 хромосомы.