Краниосиностоз - классификация, причины
Добавил пользователь Дмитрий К. Обновлено: 30.01.2026
Синдромы с краниосиностозом - методы диагностики, лечения по Европейским рекомендациям
Синдромные краниосиностозы представляют собой гетерогенную группу патологических состояний, характеризующихся связью раннего слияния нескольких черепных швов и различных врожденных пороков развития, особенно лица и конечностей. Отличительной чертой синдромных черепно-лицевых дизостозов является вовлечение как мозгового черепа (свода и основания), так и лицевого черепа (орбитальная и средняя зоны лицевого скелета). Свод черепа характеризуется множественными синостозами швов и гипоплазией верхней челюсти. До сих пор неизвестно, следует ли часто ассоциированные аномалии развития головного мозга и динамики ликвора, выявляемые при многих из этих состояний, рассматривать как первичные или вторичные проявления.
Вероятно, количество и время слияния швов могут играть прямую роль в фенотипе, по крайней мере, при некоторых из этих патологических состояний, о чем свидетельствует более часто встречающееся грыжевое выпячивание миндалин мозжечка при синдроме Крузона чем при синдроме Аперта, последний из которых характеризуется поздним слиянием ламбдовидного шва по сравнению с первым.
При этих пороках часто нарушен венозный отток из полости черепа, с развитием вторичной гипоплазии задней черепной ямки и различной степени повышения давления ликвора с расширением субарахноидального или желудочкового пространства. Существует несколько гипотез для объяснения этих изменений: венозная обструкция может быть вторичной по отношению к аномалии роста костей, расстройство может быть первично вызвано ростом диспластического основания черепа или в связи с нарушением венозного оттока у плода и нарушением нормального созревания дренажа задней ямки.
Кроме того, при некоторых этих состояниях, в связи с недостаточным развитием средней трети лица появляется экзофтальм с риском повреждения роговицы даже при небольшой травме. В дальнейшем недоразвитие дыхательных путей, в ряде случав, может вызывать изменения функции внешнего дыхания, из которых самыми тяжелыми являются ночные апноэ. Многообразное участие различных структур и черепно-лицевых функций оправдывает общий термин «фациокраниостенозы», указывающий на конкретные ассоциации костных аномалий черепа и лицевого скелета и подчеркивает трудности в их лечении.
а) Генетика краниосиностозов. Были выявлены некоторые мутации, ответственные за синдромные краниосиностозы. Обычно они включают три из четырех факторов роста, преимущественно с аутосомно-доминантным типом наследования. При синдромных синостозах один генный дефект порождает различные клинические фенотипы. Предполагается разница во взаимодействии с компонентами внеклеточной матрицы, что могло бы объяснить разнообразия фенотипов при общих дефектах гена.

б) Основные синдромы. Краткий обзор основных синдромных краниосиностозов приведен в таблице ниже.
1. Краниосиностоз при синдроме Крузона (черепно-лицевой дизостоз). Синдром Крузона — аутосомно-доминантный синдром, впервые описанный Крузоном в 1912 г. На него приходится около 4,8% случаев краниосиностозов при рождении. Распространенность была оценена как 1:25000 родов. Синдром проявляется краниосиностозом и гипоплазией лица. Фенотип краниосиностоза индивидуально отличен, но в большинстве случаев участвуют коронарные швы. Фенотип лица характерен гипертелоризмом, короткой верхней губой и относительным прогнатизмом нижней челюсти с обратным прикусом. Экзофтальм связан с ретрузией лобной области и верхних челюстей.
Эти особенности можно увидеть при рождении, но обычно они появляются в возрасте двух лет с постепенным ухудшением. Тем не менее, существуют некоторые врожденные формы, при которых гипоплазия верхней челюсти является более заметной. Больные страдают от затруднения дыхания и сильного экзофтальма, что может привести к ухудшению смыкания век. Вентрикуломегалия встречается практически всегда, иногда прогрессирующая. Мальформация Киари 1 при этом синдроме, встречается примерно в 70% случаев, может ассоциироваться с сирингомиелией и осложнять хирургическое лечение. Этот порок может быть связан с небольшим размером задней черепной ямки и преждевременным слиянием ламбдовидного шва во время первых двух лет жизни.
Ген, ответственный за синдром Крузона расположен на длинном плече хромосомы 10. Более 30 мутаций FGFR2 гена (экзон IIIа и IIIс) были идентифицированы, и они выявляются приблизительно у 60% пациентов.
Мутация в FGFR2 также может привести к синдрому Джексона-Вейсса, во многом сходному с синдромом Крузона. Тем не менее, пострадавшие субъекты также предъявляют увеличенные большие пальцы ног и тарзометаnарзальное сращение.
Мутации в FGFR3 могут привести к определенной форме синдрома Крузона, связанной с кожными аномалиями (акантокератодермия).
2. Краниосиностоз при синдроме Пфайффера (акроцефалосиндактилия тип V). Синдром был описан в 1964 г. Пфайффером. Частота синдрома Пфайффера была оценена в 1:200000. Он наследуется аутосомно-доминант-ным путем с полной пенетрантностью и варьирующей экспрессией. Синдром Пфайффера — брахицефалия, мембранная синдактилия рук и ног с увеличенными и наклоненными большими пальцами рук и ног. Могут выявляться брахидактилия, анкилоз локтевых суставов и различные висцеральные пороки развития.
В настоящее время синдром делится на три подтипа. Подтип 1 является классической и самой легкой формой, с бикоронарным синхондрозом, ведущим к брахицефалии и плоскому лицу, гипертелоризмом и слабой синдактилией с широкими большими пальцами. Подтипы 2 и 3 являются спорадическими и более тяжелыми, с выраженным экзофтальмом и аномалиями мозга, часто, но не всегда связаны с высокой летальностью. Череп в форме листа клевера характеризуется, хотя и не всегда, подтипом 2 с гидроцефалией и маленькой задней черепной ямкой и аномалией Киари 1 типа. Череп в форме листа клевера — признак очень плохого прогноза.
Синдром Пфайффера является генетически гетерогенным. Он может быть связан с мутацией в гене FGFR1 или возникать в связи с несколькими типами мутаций в FGFR2. Мутации одного и того же гена, FGFR2, таким образом, могут привести к трем типам фациокраниосиностозов, т. е. Крузона, Джексона-Вейсса и Пфайфера давая основания для предположения об участии в патогенезе и других факторов.
3. Краниосиностоз при синдроме Апера (акроцефалосиндактилии тип I). Синдром Апера является врожденным синдромом, описанным в 1906 г., связывающим фациокраниосиностоз с синдактилией на конечностях. Заболеваемость составляет примерно 1:50000 живорожденных. В большинстве случаев является спорадическим из-за отцовской de novo мутации в экзоне IIIа FGFR2 гена. Описано и аутосомно-доминантное наследование.
Краниостеноз является бикоронарным и затрагивает продольной шов. Лицо большое, с гипертелоризмом и экзорбитизмом, нос по типу клюва попугая, прикус перевернутый. Синдактилия может быть полной или с сохранением большого пальца и/или мизинца. Также часто наблюдается подвывих шейных позвонков. Церебральные пороки, в основном с участием мозолистого тела и лимбической структуры, могут быть выявлены с помощью МРТ. На аутопсии было найдено ненормальное развитие височной доли с дисгенезией парагиппокампальной области. Размер желудочков часто увеличен, вентрикуломегалия практически никогда не бывает прогрессирующей. Различная степень умственной отсталости связана с синдромом Апера, около одной пятой пациентов с коэффициентом интеллекта ниже 50, однако также сообщается и о людях с нормальным интеллектом.
Высокий уровень психических нарушений связан с аномалиями мозга, в том числе обонятельных-лимбико-септо-каллозальных структур, гипоплазией белого вещества, аномалиями пирамидных путей и непрогрессирующей «ассиметричной» вентрикуломегалией.
Тем не менее, может быть вовлечен любой шов с асимметрией лица, необычной формой уха, с частичной синдактилией пальцев рук и ног (второй и третий пальцы, третий и четвертый пальцы с небольшими дистальными фалангами). Большой палец, как правило, увеличен, но не искривлен. Птоз, симметричный или нет, наблюдается почти во всех случаях. Этот синдром может быть чрезвычайно разнообразным в своих клинических проявлениях, таким образом, обследование членов семьи имеет первостепенное значение для выявления потенциальных носителей. Геном, ответственным за синдром Сетре-Хотцена, является TWIST, расположенный на хромосоме 7. Умственная отсталость является редкостью.
5. Краниосиностоз при синдроме Карпентера (акроцефалосиндактилии тип II). Этот синдром, аутосомно-рецессивный, крайне редкий и характеризуется акроцефалией, синдактилией мягких тканей рук, синдактилией и полидактилией ног. Некоторые авторы описывают его с ожирением и гипогонадизмом.
6. Краниосиностоз при синдроме Лежена-Мюнке. Синдром Лежена-Мюнке (Lajeunie-Muenke) характеризуется одно- или двусторонним коронарным синостозом, недоразвитием верхней челюсти, гипертелоризмом и птозом. Передача аутосомно-доминантная. У некоторых пациентов, синдром связан со скелетными аномалиями такими, как наперсткообразные фаланги среднего пальца, конические эпифизы и/или неврологические нарушения, а именно нейросенсорная потеря слуха или умственная отсталость. Несмотря на переменный фенотип, этот синдром был связан с уникальной мутацией в гене FGFR3, Pro 250-Arg.
в) Функциональные аспекты. Слияние нескольких швов, которое встречается при синдромном синостозе, приводит к худшему прогнозу интеллектуального развития, усугублению состояния и повышению внутричерепного давления, чем при моношовных синостозах. Более того, аномалии морфологии лица и лицевого роста также могут привести к недостаточной защите глаз, препятствиям дыханию, неправильному прикусу и скученности зубов, которые часто требуют конкретных процедур.
Повышение внутричерепного давления (ВЧД) является, по сути, частым для синдромного краниосиностоза. Повышенное ВЧД не только вторично по отношению к синостозу, но и встречается также в сопровождении гидроцефалии и венозных аномалий.
Нарушения зрения могут следовать за повышением ВЧД, но они также могут быть вторичными по отношению к экзофтальму, типичному при этих синдромах. Стоит отметить, что атрофия зрительного нерва и потеря зрения наблюдается в основном при синдроме Крузона.
Частота отставания умственного развития определяется типом синдрома и сопутствующими пороками развития мозга (в основном нарушения развития прозрачной перегородки). Умственная отсталость часто встречается при синдроме Апера, который представляется наиболее серьезным состоянием, и у некоторых пациентов с синдромом Пфайффера, особенно у пациентов с деформацией черепа в виде листа клевера. С другой стороны, умственная отсталость при синдроме Крузона встречается редко. В общих чертах, когнитивные функции лучше после ранней хирургической коррекции и в случае хорошей психосоциальной среды ребенка.
г) Принципы лечения синдромных краниосиностозов. Кранио-фациальная хирургия выполняет задачи как функционального, так и эстетического характера. Первый год жизни имеет первостепенное значение для лечения с целью снижения аномально повышенного внутричерепного давления и, в конечном итоге, нормализации потока ликвора. В самом деле, в первый год, мозг растет очень быстро, и результаты интеллектуального развития гораздо лучше у больных, оперированных на ранних сроках. Дополнительные преимущества раннего хирургического вмешательства включают более легкую реконструкцию костей и высокий потенциал их роста, обеспечивающий «физиологическое» заполнение возможных костных дефектов, которые следуют за хирургической коррекцией.
Классическое лечение фациокраниосиностоза включает в себя переднюю реконструкцию черепа, как первый шаг, и коррекцию лица в качестве второго шага.
Передний доступ позволяет заниматься как проблемой аномального ВЧД, так и надглазничной рецессией, следовательно, защитить глаза и зрительные функции. Лобно-орбитальное вытяжение может быть выполнено с использованием флотирующей техники или горизонтального вытяжения у детей старше 6-месячного возраста. Улучшение состояния лицевого черепа может быть достигнуто путем остеотомии по типу Le Fort III. В отдельных случаях могут выполняться фронтофациальные моноблочные перемещения с одновременной мобилизацией орбиты и лицевых костей. В настоящее время для достижения адекватного вытяжения лица предпочтительно использовать внутренние или внешние дистракторы.
Как правило, вытяжение лица должно быть отложено до тех пор, пока окончательно не сформируются зубные ряды и не возникнет стабильная окклюзия. Тем не менее, в отдельных случаях, особенно в случае нарушения дыхания, может быть необходимо раннее лицевое вытяжение. В других случаях перед хирургическим вмешательством должна быть наложена трахеостома.
Раннее расширение заднего черепного свода может также быть выполнено как первый шаг для снижения внутричерепного давления, предполагая фронтофациальную коррекцию до старшего возраста ребенка. Это может быть достигнуто, в частности, при затылочном уплощении, когда задняя ямка чрезмерно мала. Процедура обычно приводит к постепенному улучшению венозного оттока из черепа. Раннее заднее расширение может быть достигнуто как увеличением теменно-затылочного костного лоскута, так и применением пружинной краниопластики у детей с открытым лямбдовидным швом. Расширение БЗО также может быть необходимо в некоторых случаях с симптомной мальформацией Киари 1 типа.
Расширение желудочков, встречающееся при синдромном краниосиностозе также может потребовать лечения. Часто это связано с грыжей миндаликов различной степени, и расширение БЗО может стать необходимым. В других случаях, с механическим препятствием току ликвора, эндоскопическая вентрикулоцистерностомия может обеспечить необходимый контроль ВЧД. Тем не менее, в ряде случаев синдромного краниосиностоза венозный отток нарушается. Прогрессирующее открытие коллатерального венозного канала может привести к отсутствию гидроцефалии. Тем не менее, некоторым пациентам может потребоваться установка вентрикулоперитонеального шунта.
Во всех случаях требуется несколько видов хирургического вмешательства. Тесное сотрудничество между пластическим хирургом и нейрохирургом, а также детским нейроанестезиологом является обязательным условием получения хорошего функционального и косметического исхода.
Умственная отсталость у детей с синдромными краниосиностозами в зависимости от возраста на момент операции. Распространенность непрогрессирующей вентрикуломегалии и гидроцефалии при синдромных краниосиностозах. Виды операций при синдромных фациокраниосиностозах. Три поколения пациентов с синдромом Крузона. Слева: МРТ (сагиттальная проекция) мозга у ребенка с синдромом Крузона.
Обратите внимание на акроцефальную форму черепа и сопутствующие деформации церебральных структур,
расширение желудочков и каудальное смещение миндалин мозжечка (мальформация Киари I типа).
Справа: ангиография в венозной фазе. Отметьте призанки нарушения венозного оттока, сочетающегося с коллатеральными дренажами (стрелка). Слева: интраоперационная КТ головы у ребенка с синдромом Крузона.
Видно смещение лица кзади и обратный прикус.
Справа: тот же ребенок во время дистракции после фронто-фациального расширения.
Обратите внимание на коррекцию смещения лица. Ребенок с синдромом Пфайффера. Ухудшение дыхания вынудило к выполнению трахеостомии. Широкий большой палец руки и огромный большой палец ноги характерны для синдрома Пфайффера. Деформация черепа в виде листа клевера. Ребенок с синдромом Апера: типичная деформация лица в сочетании с синдактилией. Сагиттальная (слева) и коронарная (справа) МРТ мозга у ребенка с синдромом Апера.
Отметим расширение желудочков, тонкое мозолистое тело, аномалии височной доли и отсутствие тонзиллярных выпячиваний.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Краниосиностоз - классификация, причины
Краниосиностоз является преждевременным слиянием черепных швов, что приводит к формированию ненормальной формы черепа. Аномальные формы черепа были описаны Гиппократом и Галеном. Вирхов, однако, был первым, кто соотнес аномальную форму черепа с преждевременным слиянием черепных швов. Степень морфологических и функциональных проблем варьирует от количества пострадавших швов и наиболее выражена в синдромных случаях. Одношовные краниосиностозы, как правило, считаются только косметической проблемой. Тем не менее, особое внимание уделялось появлению и развитию функциональных проблем именно при одношовном кранисиностозе. Повышение внутричерепного давления (ВЧД), встречается у 33% пациентов.
Когнитивная дисфункция, особенно, развитие речи и способности к обучению, встречаются в различной степени при всех типах одношовного синостоза. Проблемы с самооценкой возникают в 10%.
О хирургии краниосиностоза было впервые сообщено Lannelongue в 1890 г. С тем пор появилось множество описаний хирургической техники. В прошлом эти вмешательства сопровождались высоким уровнем инвалидизации и смертности. Началось обсуждение оправданности вмешательства, особенно при одношовных краниосиностозах. Междисциплинарный подход значительно улучшил лечение этих пациентов.
По мере развития хирургической техники и особенно анестезии инвалидизация и смертность свелись к минимуму. В настоящее время операция по одношовному синостозу вполне приемлема не только за счет снижения инвалидизации, но и за счет возможности выявления функциональных проблем. Текущие дискуссии связаны с оптимальными сроками и хирургическими методами для улучшения морфологических, а также функциональных результатов у пациентов с краниосиностозом.
а) Классификация краниосиностоза. Краниосиностоз относится к ненормальному закрытию одного или более швов. Развивающаяся в итоге форма головы получается за счет ограничения роста перпендикулярно пораженному шву и компенсаторного роста параллельно неповрежденным швам. Тип краниосиностоза обычно называют по конкретно полученной форме головы. Например, скафоцефалия или ладьевидный череп развивается при сагиттальном синостозе, и тригоноцефалия, или треугольный череп, результат метопического синостоза. Дальнейшая классификация дифференцирует между первичными и вторичными краниосиностозами, синдромными или несиндромными случаями и простыми или сложными краниосиностозами.
Большинство случаев первичные или идиопатические. Вторичный краниосиностоз связан с известными основными заболеваниями. Например, такие нарушения обмена веществ, как синдром Пфаундлера-Гурлер, недостаточность питания, как рахит и действие тератогенных факторов, например, вальпроата натрия или гормонов щитовидной железы. К хорошо известным причинам вторичного краниосиностоза относятся механические факторы, такие как внутриутробные ограничения головы плода и отсутствие роста мозга при микроцефалии.
Краниосиностоз может быть синдромным, происходящим в контексте других дисморфизмов, или несиндромным. Описано более ста синдромов, при в которых встречается краниосиностоз. Чаще всего встречаются синдромы Крузона и Апера. Несиндромный краниосиностоз обычно включает в себя один шов, в то время, как синдромный краниосиностоз чаще затрагивает несколько швов. В данной главе будут рассмотрены несиндромные краниосиностозы.
Причины вторичных краниосиностозов.
б) Распространенность. Распространенность краниосиностоза в целом составляет около 1:2500. Среди пациентов специализированного центра на синдромные случаи приходится около 15-20%, в то время как на несиндромные приходится 80—85%. Среди несиндромных краниосиностозов сагиттальный синостоз встречается чаще всего, далее метопический синостоз, одно-и двусторонний коронарный синостоз, ламбдовидный синостоз и тяжелые формы множественного поражения. Скафоцефалия и тригоноцефалия чаще встречаются у мальчиков, в то время как брахицефалия и плагиоцефалия у девочек. Семейные случаи описаны при любом типе синостозов и составляют около 7-8% сагиттальных и метопических синостозов и даже 30% при бикоронарных синостозах.
в) Патогенез краниосиностоза. Во время развития свода черепа плода, кости развиваются из мембранных «зачатков» черепа в определенное время. Из этих центров костей, окостенение распространяется в радиальном направлении. Эта закономерность видна на черепе детей. Начало венечного шва видно еще в 16 недель беременности на постоянном месте, в середине будущего шва, где впервые встречаются две теменных и лобные кости. Впоследствии развивается шов в краниальном и каудальном направлении. Синостоз венечного шва также может быть прослежен до 16 недели развития, со слиянием двух костей свода черепа, начинающихся в этом же месте при нормальном развитии швов. Слияние костей распространяется также как и формирование швов. Это слияние на самом деле означает недоразвитие шва.
Затылочная кость формируется не перепончатым, а эндохондральным окостенением, как крылья клиновидной кости, височные кости и основание черепа.
Рост черепа очень важен в первые три года жизни. В основном рост происходит в швах между костными пластинами. В центре шва поддерживается популяция остеопрогениторных клеток. Часть этих клеток начинают путь остеогенной дифференцировки. Во время движения от пролиферирующей популяции при текущей дифференцировке, эти секретирующие кость остеобласты достигают костных границ и способствуют расширению кости. Нарушение баланса между распространением, дифференцировкой и апоптозом остеобластов вызывает преждевременное окостенение шва, а тем самым и его синостоз. Были обнаружены некоторые факторы, влияющие на этот баланс, такие как подлежащая твердая мозговая оболочка (ТМО) и давление, которое оказывает на ТМО растущий головной мозг и ликворное давление.
Без непосредственного контакта с твердой мозговой оболочкой костные швы срастутся. В экспериментах, в которых швы поворачивали и перемещали в другие места на ТМО, шов начинал закрываться. В экспериментах на крысах, твердая мозговая оболочка помогает выявить местные различия в экспрессии остеогенных цитокинов и активации остеобластов, связанные с вышележащими швами.
Сигнальный путь, по которому ТМО влияет на рост швов все более проясняется. Например, факторы роста фибробластов (ФРФ) играют важную роль. Привязка к рецепторам фактора роста фибробластов 2 (ФРФР-2), который является одним из четырех рецепторов ФРФ и лигандом ФРФ4, вызывает слияние эмбриологических швов. Мутация гена, кодирующего ФРФР-2 вызывает непрерывную активацию рецепторов, независимо от наличия ФРФ лиганда. Эта мутация тем самым модифицирует сигнальный путь. Мутации в ФРФР генах встречаются при болезнях Крузона и Аперта, но также и у некоторых пациентов с очевидно изолированным синостозом одного шва. Другие типы мутаций поражают TWIST и MSX2 гомеобоксные гены и могут быть найдены при синдроме Saethre-Chotzen, бостонском типе краниосиностоза и в небольшом проценте случаев несиндромного синостоза.
Все эти гены участвуют в пролиферации и созревании мезенхимальных и костных клеток.
Факторы роста фибробластов (ФРФ) играют важную роль.
В экспериментах Киршнера показано, как экологические факторы могут играть определенную роль. Он смог применить давление на основные кости черепа плода крысы, индуцировав изменение выработки фетального бета-TGF в кости и нижележащую ТМО на уровне швов. У сдавленных плодов повышалась бета l-TGF-иммунореактивность, в то время как TGF-бета-З-иммунореактивность по сравнению с несдавленными плодами снижалась.
Дифференциация кости имеет место и в других областях кроме шва. Этот рост, очевидно, зависит от ТМО. Остеогенные свойства незрелой костной ткани более выражены, чем в зрелых костях. Этим объясняется способность заращения черепа у маленьких детей до двухлетнего возраста, в отличие от старших детей. Интересно, что этот потенциал исцеления свода наиболее выражен в период развития мозга и активного расширения внутричерепных пространств. Это привело группу Longaker к гипотезе, что механическое напряжение растущего мозга может вызвать усиление экспрессии остеогенных факторов. Они обнаружили, что при воздействии механического растяжения повышается регуляция TGF-бета-1 мРНК и FGF-2 белка в незрелых клетках твердой мозговой оболочки крысы. Эта оболочка играет важную роль в росте костей свода черепа, при этом дефекты черепа связаны с дефектами в ТМО.
В возрасте до шести лет рост черепа в основном достигается за счет резорбции кости на внутренней поверхности черепа и нарастания костной ткани на внешней поверхности. Роль швов, очевидно, с этого момента минимальна, хотя часто швы остаются открытыми значительно дольше. Метопический шов является единственным, который закрывается в течение первого года жизни.
г) Функциональные проблемы при несиндромных краниосиностозах. Функциональные проблемы, связанные с краниосиностозом, включают повышенное внутричерепное давление, задержку развития и проблемы со зрением.
Внутричерепная гипертензия имеет место при несиндромном, а также при одношовном краниосиностозе. Систематическое инвазивное измерение показало подъем ВЧД (среднее ВЧД выше 15 мм рт.ст.) у 15-20% пациентов и пограничное ВЧД (среднее ВЧД между 10 и 15 мм рт.ст.) у 38% пациентов.
Повышенное ВЧД связано с числом пострадавших швов. Частота подъема ВЧД достигает 30-60% у пациентов с брахицефалией и комплексным синостозом. Развитие повышенного ВЧД является прогрессирующим состоянием, так как заболеваемость увеличивается с возрастом. Без хирургического вмешательства частота удваивается после первого года жизни. Среди пациентов с неисправленной скафоцефалией частота после первого года возрастает четырехкратно. Renier обнаружил статистически значимую связь между предоперационным повышенным ВЧД и снижением уровня IQ. Эта связь была более очевидна у пожилых пациентов, из чего следует, что влияние на интеллектуальные функции зависит от продолжительности краниосиностоза и сопутствующего ВЧД.
Гидроцефалия, которая является более частой этиологией повышенного ВЧД при синдромных краниосиностозах, редко наблюдается при несиндромных краниосиностозах. В этих случаях гидроцефалию обычно объясняют случайными расстройствами.
Задержка когнитивного развития хорошо известна при синдромных краниосиностозах. Все больше подтверждений того, что несиндромные краниосиностозы также связаны с проблемами развития. Ряд исследований показал небольшую, но значимую задержку развития для всех типов одношовного синостоза. У детей старшего возраста особенно выражены проблемы собучениеми речью. Большинство исследований не показывает отношения между пострадавшим швом, хирургическими аспектами и задержкой развития. В сериях, выявивших такую связь, отмечена низкая частота задержки развития и расстройствов обучения у больных со скафоцефалией (39%), у пациентов с тригоноцефалией (57%) или плагиоцефалией (51-61%).
При одношовном синостозе возможны задержки не только когнитивного развития, но и моторики. Доля детей с отклонениями в развитии и поведенческими проблемами на первой консультации увеличивается с возрастом. Это может быть связано с вредным влиянием сохраняющегося краниосиностоза. Однако некоторые авторы сообщают об увеличении с возрастом доли детей в той же когорте с проблемами в развитии, независимо от операции. Эти данные позволяют предположить, что задержка развития скорее первична, чем вторична относительно краниосиностоза. Raybaud и Di Rocco предложили гипотезу, согласно которой при синдромном краниосиностозе кроме механической деформации мозга, вызванной деформацией черепа, нарушением белого вещества, может произойти мутация гена, вызывающего синостозирование шва.
L1САМ представляет собой адгезивную молекулу, участвующую в развитии белого вещества, ее мутации связаны с заболеваниями белого вещества головного мозга. Было показано, что L1CAM не может выполнять свою роль без тесного взаимодействия с FGFR.
Большинство авторов не находят положительного влияния операции на эти задержки в развитии, хотя Bellew и Cohen обнаружили, что моторное развитие улучшилось. Shimoji описывает послеоперационное улучшение поведенческих реакций после операции по поводу тригоноцефалии у детей старшего возраста. Тем не менее, пациенты, оперированные в возрасте до одного года, имеют лучшие показатели развития, чем дети оперированные в старшем возрасте, таким образом, операция не уменьшает задержку развития, но предотвращает дальнейшее ухудшение.
Среди первичных нарушений зрения встречаются астигматизм и расходящееся косоглазие, которые встречаются при плагиоцефалии. Папиллярный отек редко встречается при несиндромном краниосиностозе (1-3% при одно-шовном синостозе), с частотой до 4,3-20% при комплексных синостозах. В нашем исследовании мы обнаружили папиллярный отек до операции в 6,7% при сагиттальном синостозе и в 2,4% при метопическом синостозе.
Недавно Ricchi описал корковые нарушения зрения при несиндромном краниосиностозе. У пациентов с плагиоцефалией аномальные движения глаз и дефекты поля зрения были обнаружены у 6 и 5 из 11 пациентов, соответственно. У 11 из 15 пациентов имелись аномалии фиксированного смещения, которые вызывали дисфункцию теменной коры. Авторы приходят к выводу, что присутствие этих нарушений зрения связано с типом краниосиностоза и предполагают, что причиной этих нарушений является прямое давление на кору.
Доля детей с нормальным психическим уровнем развития в зависимости от возраста на момент первого обращения. Доля детей с нормальным психическим результатом после лечения в зависимости от возраста на момент операции. Когнитивное развитие при краниосиностозе
Учебное видео урок швы и роднички головки плода - плод как объект родов
Краниосиностоз
Краниосиностоз – это заболевание, основным симптомом которого является деформация мозгового отдела черепа, возникающая вследствие преждевременного зарастания костных швов. Клиника включает в себя деформации черепа, симптомы внутричерепной гипертензии, патологию зрительного нерва, отставание в психическом развитии. Редко заболевание сопровождается аномалиями костей лицевого черепа. Диагностика заключается в оценке степени зарастания черепных швов и определении костных дефектов путем физикального обследования, рентгенографии, КТ и МРТ. Основное лечение – ранняя хирургическая коррекция формы костей черепа.

Общие сведения
Краниосиностоз – это патологическое состояние в педиатрии, возникающее на фоне раннего зарастания черепных швов, характеризующееся деформацией черепной коробки и нарушением развития тканей головного мозга. В среднем распространенность разных форм заболевания в станах СНГ составляет 0,03-3,5% от всех новорожденных. Мужской пол более склонен к развитию данной патологии. Наиболее распространенный вариант – моносиностоз. Чаще всего наблюдается преждевременное зарастание сагиттального шва (скафоцефалия) – 50-65% от всех краниосиностозов. Самой редкой и прогностически неблагоприятной является синдромальная форма, при которой имеется высокий риск летального исхода на первом году жизни ребенка. При своевременной диагностике и адекватном лечении в первые 6-9 месяцев жизни дальнейшее развитие пациента проходит без отклонений.
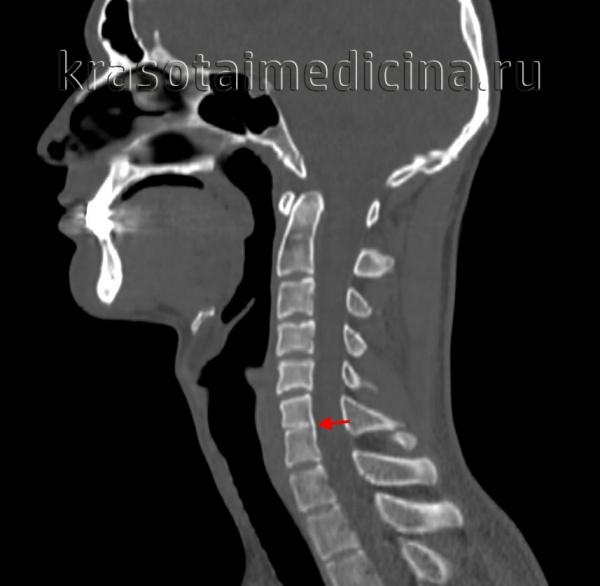
Причины краниосиностоза
Точная этиология краниосиностоза не установлена. Согласно выдвинутым теориям, данное заболевание может развиваться в результате внутриутробного нарушения гормонального фона ребенка, перинатальных травм и сдавливания костей черепа в полости матки. Также данная патология возникает при наследственных патологиях – синдроме Апера, синдроме Крузона и синдроме Пфайффера. Доподлинно известна одна из ведущих причин развития краниосиностоза – аномалия гена, отвечающего за образование рецепторов фактора роста фибробластов (FGFR типы I, II и III).
Патогенетически краниосиностоз обусловлен преждевременным синостозированием одного или сразу нескольких черепных швов: коронарного, сагиттального, лямбдовидного или метопического. На фоне этого, согласно закону Вирхова, возникает компенсаторный рост костной ткани в перпендикулярном направлении, из-за чего формируется деформация черепа. Полисиностоз (а зачастую – и моносиностоз) часто сопровождается внутричерепной гипертензией, которая может проявляться неврологическими нарушениями вследствие сдавливания коры головного мозга, венозным застоем глазного дна, отеком диска зрительного нерва, а при длительном течении – полной атрофией зрительного нерва и потерей зрения.
Классификация краниосиностоза
Краниосиностоз, согласно этиологическим факторам, разделяют на две группы:
- Синдромальный. В данном случае патология сочетается с другими врожденными пороками. Сюда относятся сцепленные с Х-хромосомой, моногенные, хромосомные и другие краниосиностозы. Например – комбинация синостоза с дисплазией костей лицевого черепа, синдром Смита-Лемли-Опица или рото-пальце-лицевой синдром.
- Несиндромальный. Это изолированная форма, которая возникает самостоятельно и не имеет сопутствующих заболеваний.
В зависимости от количества заросших черепных швов выделяют:
- Моносиностоз. Характеризуется поражением только 1 шва. В случае с коронарным и лямбдовидным швом зарастание может быть одно- или двухсторонним. Наиболее распространенная форма.
- Полисиностоз. В патологический процесс втягиваются 2-3 шва.
- Пансиностоз. При этой форме наблюдается сращивание всех костных швов черепа ребенка. Встречается крайне редко.
Симптомы краниосиностоза
Клинически краниосиностоз проявляется с момента рождения ребенка. Для всех форм характерны плагиоцефалия и раннее закрытие большого родничка (в норме это происходит в 12-18 месяцев). Только при полисиностозе или сопутствующей гидроцефалии он может оставаться открытым до 3-х летнего возраста. Также при краниосиностозах зачастую наблюдается повышение внутричерепного давления, которое может проявляться неврологическими нарушениями: беспокойством, интенсивным плачем, тошнотой и рвотой, нарушением сна, снижением аппетита, позитивным симптомом Грефе, судорогами.
Каждая из форм заболевания имеет характерные клинические особенности. Краниосиностоз стреловидного шва (скафоцефалия или ладьевидный череп) характеризуется увеличением переднезаднего размера головы ребенка при недостаточности ее ширины. Визуально определяется вытягивание черепа, «вдавливание» височных областей, «нависание» лба и затылочной части, сужение лица и приобретение им овальной формы. Пальпаторно над местом прохождения стреловидного шва выявляется костный гребень. В раннем возрасте возможна задержка психического развития.
Зарастание лямбдовидного шва чаще всего носит односторонний характер и проявляется уплощением затылочной области. Является трудно диагностируемой формой, поскольку плагиоцефалия практически незаметна под волосами, а неврологические нарушения минимальны. При взрослении пациента динамика заболевания практически отсутствует.
Коронарный или венечный краниосиностоз может быть как одно-, так и двухсторонним. Зарастание только одной половины шва сопровождается типичной деформацией черепа ребенка – уплощением лобной кости и верхней части глазницы с пораженной стороны. При этом противоположная половина компенсаторно «нависает». Со временем развиваются искривление носа в противоположную сторону, уплощение скулы, нарушение прикуса и косоглазие. Двухсторонний коронарный краниосиностоз проявляется широким, плоским и высоким лбом с уплощенными глазничными краями лобной кости, редко – башенной деформацией черепа (акроцефалией). Неврологические нарушения неспецифичны и аналогичны другим формам.
Нетопический краниосиностоз или тригоноцефалия характеризуется развитием треугольного лба с костным килем, проходящим от глабеллы до большого родничка. Также наблюдается гипотелоризм – смещение глазниц кзади с уменьшением межглазничного промежутка. Со временем происходит некоторое сглаживание костного гребня и нормализация формы лба. В половине случаев возникают нарушения зрения и отставание в психическом развитии.
Синдромальный краниосиностоз является самой редкой и тяжелой формой. Помимо плагиоцефалии отмечается дисплазия костей лицевой части черепа, из-за чего возникают дыхательная недостаточность, нарушение приема пищи и патология зрения. Характеризуется синостозом венечного шва и, как результатом – брахицефалической формой головы ребенка. Также возникают гипоплазии костей верхней челюсти, выпячивание глазных яблок из орбит, гипертелоризм. Часто наблюдается значительное расширение родничка и расхождение стреловидного шва. Без лечения у детей развивается выраженное отставание в психическом развитии, зачастую они погибают на протяжении первых 12 месяцев жизни от ОРВИ, осложнившихся пневмонией.
Диагностика краниосиностоза
Диагностика краниосиностоза базируется на физикальном осмотре и инструментальных методах исследования. Анамнез нередко малоинформативен, но его данные позволяют педиатру проследить динамику клинической симптоматики, если таковая имеет место. Важным моментом становится визуальный осмотр ребенка, который дает возможность обнаружить характерные деформации черепа, аномалии костей и т. д. Лабораторные анализы специфических изменений не выявляют и могут использоваться с целью определения генетической патологии или диагностики осложнений.
Обязательными являются инструментальные методы, позволяющие визуализировать костные деформации и оценить степень поражения тканей головного мозга. Сюда относятся нейросонография, рентгенография, компьютерная и магнитно-резонансная томография. Нейросонография используется с целью оценить состояние тканей головного мозга и размеры желудочков, выявить внутричерепную гипертензию. На рентгенограмме удается определить нарушения структуры костей, окостенение черепных швов, а при повышенном внутричерепном давлении – усиление пальцевых вдавлений. КТ и МРТ применяются для получения более информативных результатов. При подозрении на поражение зрительной системы проводится офтальмоскопия, позволяющая обнаружить поражение диска зрительного нерва. Рекомендованы консультации нейрохирурга и офтальмолога.
Дифференциальная диагностика краниосиностоза осуществляется с позиционной плагиоцефалией, родовой травмой новорожденных (кефалогематомой, подапоневротическим кровоизлиянием, переломом костей черепа), кистами головного мозга, рахитом и микроцефалией.
Лечение краниосиностоза
Основное лечение краниосиностоза – хирургическая коррекция костной деформации черепа. Оптимальное время для проведения оперативного вмешательства – первые 6-9 месяцев жизни ребенка. Данные сроки обусловлены тем, что в этом периоде наблюдается наиболее интенсивное развитие тканей головного мозга, которому может препятствовать деформация черепной коробки. Кроме того, кости черепа в этом возрасте быстро восстанавливают свою структуру без развития осложнений. Объем и техника операции зависят от формы краниосиностоза и сопутствующих патологий. В 2-3-х летнем возрасте коррекция проводится исключительно с целью ликвидировать косметический дефект. Помимо хирургического лечения осуществляется изменение рациона ребенка в соответствии с возрастными требованиями. При развитии интеркуррентных заболеваний показана медикаментозная терапия.
Прогноз и профилактика краниосиностоза
Прогноз для детей с краниосиностозом напрямую зависит от формы заболевания, своевременности диагностики и эффективности оперативного вмешательства. При качественном проведении лечебных мероприятий исход заболевания, как правило, благоприятный. Прогностически неблагоприятной принято считать синдромальную форму краниосиностоза.
Специфической профилактики для данной патологии не существует. Неспецифические меры подразумевают медико-генетическую консультацию семьи и планирование беременности, охрану здоровья женщины при вынашивании ребенка, рациональное питание, отказ от вредных привычек и исключение всех потенциальных этиологических факторов развития краниосиностоза.
Плагиоцефалия
Плагиоцефалия – это деформация головы, которая возникает внутриутробно или в первые месяцы жизни, характеризуется асимметрией либо искривленной косой формой черепа. С одной стороны череп выдается вперед, с другой – назад. Патология развивается вследствие сдавления головы или преждевременного окостенения швов. Часто сопровождается вторичными лицевыми деформациями. Диагностируется на основании данных внешнего осмотра, результатов рентгенографии, компьютерной томографии. В зависимости от причины развития плагиоцефалии требуются консервативные мероприятия или хирургическое вмешательство.
МКБ-10
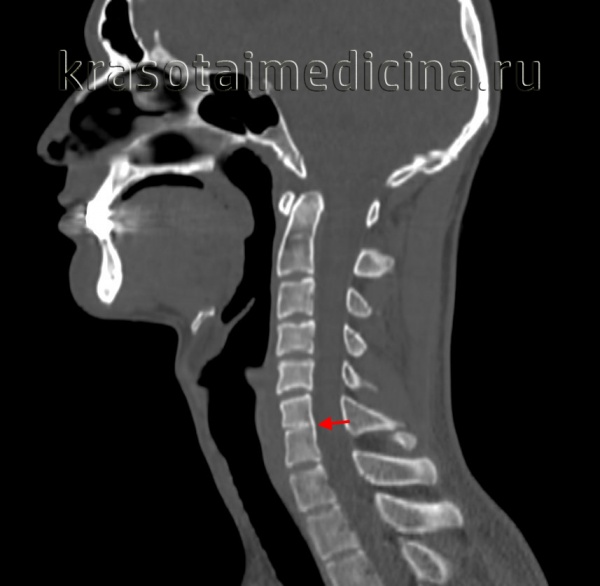

Плагиоцефалия – собирательный термин, включающий ассиметричные деформации головы различной этиологии. Достаточно широко распространена. По разным данным, обнаруживается у 1-5% детей младше 1 года. Патология, которая формируется на фоне сдавления, как правило, не влечет за собой негативных последствий, за исключением косметического дефекта, лечится консервативно. При образовании краниосиностозов возможно повышение внутричерепного давления, другие негативные последствия, требуются операции.
Причины плагиоцефалии
Голова ребенка может деформироваться в результате сдавления, которое происходит внутриутробно либо в раннем возрасте. Причинами сдавления после рождения являются кривошея, длительное пребывание в одном положении. У младенцев с кривошеей частота патологии достигает 30%. В остальных случаях распространенность в значительной степени определяется врачебными рекомендациями.
Так, в 90 годы прошлого века родителям стали советовать укладывать детей на спину, чтобы избежать развития синдрома внезапной детской смерти, малыши стали чаще лежать в этом положении не только во сне, но и во время бодрствования. В результате позиционная плагиоцефалия в тот период начала выявляться у каждого четвертого или пятого ребенка. Вероятность формирования данной разновидности деформации в каждом конкретном случае определяется активностью малыша, регулярностью изменения положения его тела родителями.
Раннее заращение швов между костями черепа (преждевременное образование синостозов) может быть спорадическим или передаваться по наследству. Семейные формы болезни составляют 8,4-9,5% от общего числа случаев синостоза. У пациентов обнаруживаются мутации генов TWIST, FGFR3 и FGFR2. Количество сращений варьируется. Девочки страдают чаще мальчиков. Более чем в 60% случаев синостоз выявляется справа.
Классификация
С учетом этиофакторов различают три варианта плагиоцефалии:
- Синостозная. Может протекать с заращением венечного, лямбдовидного, лобно-клиновидного, лобно-решетчатого шва.
- Компенсаторная. Лямбдовидный шов зарастает на одной стороне, лобная кость компенсаторно растет и выбухает с другой.
- Деформационная. Синостозы отсутствуют, деформация обусловлена позиционным сдавлением головы.
При вовлечении венечного шва формируется лобная плагиоцефалия, при поражении лямбдовидного – затылочная. Наиболее распространенным вариантом является сращение венечного шва, патология выявляется у 0,04-0,1% младенцев. Реже всего встречается синостоз лямбдовидного шва, частота составляет 1 случай на 150 тысяч детей.
Симптомы
Тяжесть плагиоцефалии существенно варьируется, у одних детей изменения едва заметны, у других бросаются в глаза из-за резкой асимметрии мозговой части черепа, выраженного смещения лицевых структур. Задняя часть головы справа или слева уплощена, передняя с другой стороны компенсаторно выдается кпереди. У ряда пациентов оси черепа смещаются на 20 и более градусов. Внешний вид больного в некоторой степени определяется характером патологии, что позволяет заподозрить определенный вид плагиоцефалии по результатам физикального обследования.
Синостозная лобная плагиоцефалия
Лоб со стороны синостоза сглажен, противоположная лобно-теменная зона выбухает. Из-за сопутствующего преждевременного закрытия других швов деформируется основание черепа. В результате форма клиновидной кости изменяется, сама кость смещается, что обуславливает характерные лицевые дефекты. Глазница с больной стороны укорачивается, западает и сдвигается кверху. Развивается экзофтальм.
На фоне вторичных нарушений прикрепления глазодвигательных мышц возникают проблемы со зрением. Ухо, нос, скуловые кости и челюсти также перемещаются. Основание носа и скула на стороне синостоза выстоят, ушная раковина поднимается кверху и располагается несколько кпереди. Подбородок и кончик носа смещаются в противоположную сторону.
Компенсаторная плагиоцефалия
Во время осмотра черепа во фронтальной плоскости обнаруживается выбухание темени со здоровой стороны, затылочной части и сосцевидного отростка – с больной. В результате голова при взгляде сверху выглядит трапециевидной. В отличие от других разновидностей плагиоцефалии, лицо не изменено.
Деформационная плагиоцефалия
Лоб выстоит справа, затылок – слева или наоборот. Из-за отсутствия сращений череп остается достаточно эластичным, поэтому его укорочение отсутствует. Наблюдается смещение всех структур по оси. Корень носа отклонен к выбуханию лба, глазная щель на этой стороне уменьшена, ухо сдвинуто кзади и книзу. Подбородок «уравновешивает» вышележащие структуры, смещаясь к сглаженной части лба.
У больных без кривошеи превалирует уплощение одной половины черепа. При наличии кривошеи асимметрия приобретает более выраженный двухсторонний характер. Вдавленность правой затылочной и левой лобной части говорят о левосторонней плагиоцефалии, сдавление левой затылочной и правой лобной зоны – о правосторонней. Отмечается смещение длинной оси черепа в сторону от центральной линии. Сверху голова выглядит, как скошенный параллелограмм.
Осложнения
По данным большинства исследований, патология чаще протекает благоприятно и не вызывает никаких последствий, кроме косметических дефектов. В отдельных источниках сообщается, что у некоторых пациентов с деформационной плагиоцефалией возникали незначительные затруднения при обучении в младших классах. Вместе с тем, существуют работы, в которых указывается на негативное влияние синостозного типа болезни на развитие детей в старшем возрасте.
У четверти больных были выявлены сложности при формировании речи и трудности в обучении. Почти у половины наблюдались функциональные проблемы различной степени выраженности. Многие авторы сообщают о риске возникновения внутричерепной гипертензии и неврологических нарушений.
Диагностика
Первичная диагностика врожденной плагиоцефалии осуществляется неонатологом, приобретенной – врачом-педиатром. Для дальнейшего обследования пациентов направляют к нейрохирургу. Особенности патологии определяют с учетом данных осмотра и результатов аппаратных исследований. Детям назначают рентгенографию и компьютерную томографию. На рентгенограммах выявляют нарушения костной структуры, окостенение швов. По данным КТ черепа у ребенка составляют детальное представление о форме и тяжести изменений. По результатам обследования определяют тактику лечения и необходимость оперативного вмешательства.
Лечение
Терапия позиционной плагиоцефалии
Хирургические вмешательства не требуются, коррекция осуществляется с использованием консервативных методов. Основную роль играют профилактические мероприятия, позволяющие предупредить формирование и усугубление перекоса. Родителям советуют регулярно переворачивать ребенка на живот за исключением времени сна. Многие младенцы с плагиоцефалией не любят данное положение тела, поэтому для предупреждения плача и попыток переворота внимание малыша советуют отвлекать общением и яркими игрушками.
Еще одной мерой, позволяющей предотвратить развитие и прогрессирование деформационной разновидности болезни, является поочередное укладывание то на один, то на другой бок. Поскольку дети не любят лежать лицом к стене, при сопротивлении и переворотах стоит поворачивать кроватку либо занимать внимание ребенка яркими предметами. При ранней диагностике перечисленных мероприятий достаточно, чтобы обеспечить восстановление нормальной формы головы в период активного роста черепа.
При неэффективности метода применяют корригирующий пластиковый шлем, который изготавливают индивидуально с учетом параметров головы пациента. Данное приспособление представляет собой полузакрытую конструкцию, которая уравнивает давление на различные части черепа и стимулирует его рост в нужном направлении. Ранее для этой цели использовали гипсовые повязки, но в настоящее время их накладывают редко из-за более высокого уровня дискомфорта для ребенка и сложностей в уходе для родителей.
Наибольшая результативность отмечается при проведении лечения в период от полугода до полутора лет – в это время мозг достаточно, но не слишком быстро растет, диаметр головы каждый месяц увеличивается примерно на полсантиметра, что позволяет скорректировать возникшее нарушение. После 1,5 лет методика малоэффективна, поскольку рост головы существенно замедляется. До 6 месяцев данный способ применять не рекомендуется, так как мозг растет очень быстро, ношение шлема может стать причиной задержки развития церебральных структур.
Лечение других форм плагиоцефалии
При незначительной выраженности компенсаторного типа заболевания коррекция не требуется. При взрослении волосы ребенка полностью скрывают косметический дефект. В тяжелых случаях показано объемное вмешательство – тотальная реконструкция черепа.
Из-за значительных эстетических дефектов (в том числе – лицевой части черепа), функциональных нарушений и угрозы развития осложнений синостозная плагиоцефалия рассматривается, как показание к оперативному лечению. Оптимальные сроки коррекции окончательно не определены. Одни специалисты считают, что наилучшим периодом для применения хирургических методик являются первые три месяца жизни. Другие указывают на отсутствие значимых различий между отдаленными результатами у пациентов шести месяцев и полутора лет.
Достижение удовлетворительного результата возможно в возрасте до 3 лет, но эффективность метода снижается по мере взросления. В ходе операции осуществляют реконструкцию лобных и верхнеорбитальных отделов черепа. Формируют костные лоскуты, перемещают верхний край глазницы. При необходимости под западающие части подкладывают рассасывающиеся пластины или костные вставки. Кости фиксируют минивинтами и минипластинами.
В послеоперационном периоде назначают анальгетики, проводят антибиотикотерапию. При необходимости для окончательного устранения деформации дополнительно используют пластиковые шлемы. Нерассасывающиеся конструкции в последующем удаляют. Осуществляют наблюдение для оценки особенностей развития и неврологического статуса ребенка, своевременного выявления рецидивов.
Прогноз
При адекватном консервативном лечении прогноз благоприятный. Форма черепа успешно корректируется, косметический дефект исчезает или становится минимально выраженным. Отдаленные результаты хирургических вмешательств в большинстве случаев хорошие. Лицевые деформации самопроизвольно устраняются после исправления перекоса головы. Смещение глазницы полностью исчезает или значительно уменьшается у 81% пациентов. Вероятность неудовлетворительного исхода операции с необходимостью повторной коррекции возрастает у больных с генетическими нарушениями.
Профилактика
Профилактика наследственной плагиоцефалии и искривлений в результате внутриутробного сдавления не разработана. Для предупреждения позиционных нарушений необходимо регулярно менять положение ребенка, следить, чтобы он достаточно лежал на животе и на обоих боках. При выявлении первых признаков болезни необходимо обратиться к врачу для своевременного устранения патологии.
1. Хирургическое лечение несиндромальных краниосиностозов у детей. Клинические рекомендации/ Ассоциация нейрохирургов России – 2015.
2. Семиотика детских болезней. Симптомы и синдромы поражения органов и систем. Учебно-методическое пособие/ Твардовский В.И. и др. – 2017.
Читайте также:
- Лечение атипической гиперплазии эндометрия (АГЭ)
- Тендовагинит и разрыв ахиллова сухожилия. Диагностика и лечение
- Дифференциальная диагностика глубокой депрессии. Трициклические антидепрессанты
- Механизмы развития метгемоглобинемии - патофизиология
- Пищевое отравление Yersinia enterocolitica и его лечение