Кровотечения при синдроме Элерса-Данло - диагностика, лечение
Добавил пользователь Morpheus Обновлено: 29.01.2026
Для цитирования: Шостак Н.А., Правдюк Н.Г., Магомедова Д.Н. Гипермобильный синдром: клиника, диагностика, подходы к терапии. РМЖ. 2009;4:288.
Гипермобильный синдром (ГС) – системное заболевание соединительной ткани, которое характеризуется гипермобильностью суставов (ГМС), сочетается с жалобами со стороны опорно–двигательного аппарата и/или внутренними и внешними фенотипическими признаками дисплазии соединительной ткани, при отсутствии какого–либо другого ревматического заболевания.
Эпидемиологические показатели ГС варьируют от 0,6 до 31,5% и зависят от возраста, этнических характеристик обследуемых и степени оценки ГМС. Особенностью ГС является склонность к семейной агрегации и наследование по женской линии, частота встречаемости данного синдрома уменьшается с возрастом [1,2].
Первое описание ГМС принадлежит Kirk, Ansell и Bywaters (1967 г.) [3]. Авторами был предложен термин «гипермобильный синдром», отражающий феномен гипермобильности суставов, сочетающийся с дисфункцией опорно–двигательного аппарата (подвывихи, артралгии). Позже стало известно, что ГМС ассоциируется с внешними фенотипическими признаками ДСТ, сходными с маркерами дисплазии при дифференцированных синдромах (синдром Марфана, Элерса–Данло и др.), а «гипермобильный синдром» стал рассматриваться в рамках нозологической формы. Однако генетическая основа ГС до настоящего времени остается неизвестной. В ряде исследований было показано, что у пациентов с гипермобильным типом синдрома Элерса–Данло и ГС имеются мутации в генах, кодирующих неколлагеновые молекулы Tenascin–X, при этом отмечается снижение уровня сывороточного Tenascin–X в обеих группах гетерозиготных лиц женского пола [4]. Идентификация мутаций в Тenascin–X является важной моделью изучения генетической основы ГС.
В основе патогенеза ГС лежит наследственный дефект коллагена, сопровождающийся гиперрастяжимостью и уменьшением механической прочности соединительнотканных структур (в т.ч. связок, энтезисов, сухожилий), приводящим к подвывихам и микротравматизации суставного аппарата (в т.ч. позвоночника).
Клинические проявления ГС многообразны и включают как суставные, так и внесуставные признаки. Среди поражений опорно–двигательного аппарата наиболее часто встречаются артралгии с локализацией в коленных и голеностопных суставах, ассоциированные с физической нагрузкой. Причиной болевого синдрома является изменение чувствительности проприорецепторов к нагрузке опорных суставов на фоне суставной гипермобильности. Дебют артралгий приходится на молодой возраст, преимущественно лиц женского пола. Подвывихи суставов (в основном голеностопных и коленных) типичны для пациентов с ГС. Появление в клинической картине рецидивирующего синовита опорных суставов создает трудности в диагностической оценке заболевания. Характерной особенностью синовита является непосредственная связь с травмой и/или избыточной нагрузкой, а также отсутствие системной воспалительной реакции. У лиц с ГС нередко от-мечаются дорсалгии с локализацией в поясничном и грудном отделах позвоночника, возникающие во второй половине дня, после длительных статических нагрузок, уменьшающиеся в горизонтальном положении, нередко сочетающиеся со спондилолистезом или спондилолизом. Недостаточность соединительнотканного фиксирующего аппарата позвоночника под воздействием неблагоприятных факторов (длительная нефизиологическая поза, разность в длине нижних конечностей, ношение сумки на одном плече) влечет за собой компенсаторное развитие деформаций позвоночника (сколиоз) с последующим перенапряжением мышечно–связочных структур позвоночника и появлению болевого синдрома. Периартикулярные поражения (тендиниты, эпикондилит, энтезопатии, бурситы, туннельные синдромы) у пациентов с ГС возникают в ответ на непривычную нагрузку или минимальную травму. Наиболее полная картина клинических проявлений и потенциальных осложнений ГС представлена в таблице 1.
В настоящее время изучению потенциальных осложнений ГС отводится немаловажная роль. Так, в работе зарубежных авторов показана ассоциация меж-ду ГМС и остеоартрозом на уровне шейного отдела позвоночника, 1 пястно–фалангового и коленного суставов у женщин старше 30 лет [6]. Kraus et al., высказали предположение о протективном эффекте гипермобильности на развитие ОА межфаланговых суставов кистей [7]. В ходе клинических наблюдений на кафедре факультетской терапии им. акад. А.И. Нестерова (РГМУ) прослежена взаимосвязь дебюта дорсалгий в подростковом возрасте, ассоциированных с дегенеративной болезнью диска, у пациентов с ГС. В клинической картине ГС артралгии коленных суставов диагностировались у 42% больных [8].
В основе диагностики ГС лежит оценка генерализованной ГМС по критериям Бейтона [9] (табл. 2).
Для установления гипермобильности общепринятой является балльная оценка: 1 балл означает патологическое переразгибание в одном суставе на одной стороне. Максимальная величина показателя, учитывая двухстороннюю локализацию – 9 баллов (8 – за 4 первых пункта и 1 – за 5–й пункт). Показатель от 4 до 9 баллов расценивается, как состояние гипермобильности.
Диагностические критерии ГС представлены в таблице 3 и именуются Брайтоновскими критериями (1998) [9].
Малые критерии ГС были дополнены в ходе работ А.Г. Беленького (2004 г.) и включают пролапс митрального клапана, полую стопу, браходактилию, деформацию грудной клетки, сандалевидную щель стопы, сколиоз, Hallux valgus [10]. Гипермобильный синдром диагностируется при наличии 2 больших критериев или 1 большого и 2 малых критериев, или 4 малых. Достаточно 2 малых критериев, если родственник 1 линии родства имеет признаки соединительнотканной дисплазии (табл. 3).
Лечение ГМС не требует назначения специальных мероприятий при отсутствии жалоб. При умеренных артралгиях показано ограничение физических нагрузок, исключение игровых видов спорта. При упорных болях в одном или нескольких суставах используют эластичные ортезы, обеспечивающие искусственное ограничение объема движений. Немаловажную роль играет укрепление окружающих болезненный сустав мышц с помощью изометрических упражнений, обеспечивающих оптимизацию локальной биомеханики и, как следствие, исчезновение болей. В качестве симптоматической медикаментозной терапии при артралгиях, дорсалгиях показан прием нестероидных противовоспалительных препаратов (НПВП) по требованию, при развитии реактивных синовитов показано курсовое лечение НПВП.
Одним из современных представителей этой группы лекарственных средств является мелоксикам (Амелотекс и др.), избирательно блокирующий ЦОГ–2 при минимальном воздействии на ЦОГ–1, что обеспечивает хороший противовоспалительный и анальгетический эффект с оптимальным профилем безопасности. Мелоксикам полностью всасывается при внутримышечном введении, максимальная концентрация его в плазме достигается через 60 минут. Период полувыведения препарата составляет в среднем 20 часов, что определяет однократность его суточного приема. Устойчивая концентрация в плазме достигается через 3–5 дней после начала использования. Кроме того, мелоксикам хорошо проникает в синовиальную жидкость, где его концентрация составляет 50% от концентрации в плазме крови. Наличие инъекционной формы мелоксикама позволяет использовать принцип ступенчатой терапии при дорсалгиях и реактивных синовитах: показано внутримышечное введение препарата в суточной дозе 15 мг (в течение 3 дней) с последующим переходом на пероральную терапию мелоксикамом в той же дозе (15 мг 1 раз в сутки после еды) в течение 7–10 дней.
Учитывая патогенетическую основу несостоятельности соединительной ткани и системный характер проявлений ГС, одним из важных направлений терапии является коррекция нарушенного метаболизма коллагена (препараты магния), что является существенным фактором профилактики возможных осложнений соединительнотканной дисплазии [11].
В заключение необходимо отметить, что проявления ГС носят, как правило, прогрессирующий характер и лежат в основе формирования соматической патологии, что требует своевременной клинической оценки и лечебно–профилактических мероприятий.
Литература
1. Beighton P., Solomon L., Soskolne C.L. Articular mobility in an African population. Ann Rheum Dis.1973; 32:413–418.
2. Al–Rawi Z.S., Al–Aszawi A.J., Al–Chalabi T. Joint mobility among university students in Iraq. Br J Rheumatol. 1985; 24:326–331.
3. Kirk J.A., Ansell B.M., Bywaters E.G. The hypermobility syndrome. Musculoskeletal complaints associated with generalized joint hypermobility. Ann Rheum Dis 1967;26:419–25.
4. Zweers M.C., Bristow J., Steijlen P.M. et al. Haploinsufficiency of TNXB is associated with hypermobility type of Ehlers–Danlos syndrome. Am J Hum Genet 2003; 73:214–17.
5. Simpson M.R. Benign Joint Hypermobility Syndrome: Evaluation, Diagnosis, and Management //JAOA. 2006; 389–97.
6. Moskowitz, Roland W. Osteoarthritis // Lippincott Williams & Wilkins 2006:239.
7. Kraus V.B., Li Y.J., Martin E.R. et al. Articular hypermobility is a protective factor for hand osteoarthritis. Arthritis Rheum 2004; 50:2178–2183.
8. Правдюк Н.Г. Клинико–инструментальная характеристика дорсопатий у лиц молодого возраста: Дис. … канд. мед. наук. – Москва, 2007. – 129 с.
9. Grahame R., Bird H.A., Child A. The revised (Brighton 1998) criteria for the diagnosis of benign joint hypermobility syndrome (BJHS). J Rheumatol 2000;27: 1777–9.
10. Беленький А.Г. Гипермобильность суставов и гипермобильный синдром: распространенность и клинико–инструментальная характеристика: Дис. … д–ра мед. наук. – Москва, 2004. – 249 с.
11. Кадурина Т.И. Наследственные коллагенопатии (клиника, диагностика, лечение и диспансеризация). / Спб.: Невский диалект, 2000. – 271 с.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Кровотечения при синдроме Элерса-Данло - диагностика, лечение
Тромботическая тромбоцитопеническая пурпура (болезнь Мошковица) - диагностика, лечение
Тромботическая тромбоцитопеническая пурпура (болезнь Мошковица) — геморрагическое заболевание, характеризующееся развитием тромбоцитопении потребления в связи с интенсивной агрегацией тромбоцитов, вторичным неиммунным гемолизом и распространенной окклюзией мелких артерий и артериол с ишемическими изменениями нервной системы, почек и других органов; впервые описано Е. Moschcowitz в 1924 г.
Тромботическая тромбоцитопеническая пурпура обычно возникает в возрасте 30-40 лет, в 2 раза чаще у женщин.
В патогенезе основную роль играет спонтанная агрегация тромбоцитов в сосудистом русле вследствие освобождения из сосудистой стенки тромбомодулина, тканевого активатора плазминогена и фактора Виллеб-ранда. Индукторами повреждения эндотелиальных клеток могут быть различные факторы (вирусы, токсины, химические агенты, в том числе медикаменты). В результате происходит тромбирование мелких артериальных сосудов агрегатами тромбоцитов, которые впоследствии трансформируются в гиалиновые тромбы.
Тромботическая тромбоцитопеническая пурпура в большинстве случаев характеризуется острым началом и быстрым развитием симптомокомплекса, включающего:
1) лихорадку;
2) геморрагический синдром петехиально-пятнистого типа (подкожные кровоизлияния, носовые, десневые и желудочно-кишечные кровотечения, реже кровохарканье);
3) различные изменения неврологического статуса (дезориентация, диплопия, атаксия, тремор, судороги, гемипарезы и гемиплегии, в тяжелых случаях — кома);
4) тромбоцитопению различной степени (от 10 до 100•10 9 /л);
5) неиммунную гемолитическую анемию средней и тяжелой степени (уровень гемоглобина в пределах 40-80 г/л);
6) поражение почек (протеинурия, эритроцитурия и цилиндрурия, иногда умеренные нарушения функции почек).
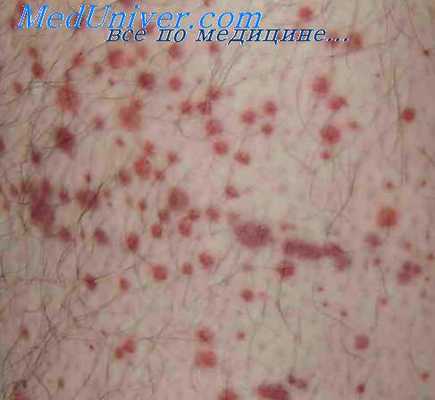
Сыпь при тромбоцитопенической пурпуре
Основным методом лечения тромботической тромбоцитопенической пурпуры является плазмаферез с массивными трансфузиями донорской плазмы (2-3 литра в сутки). Использование дезагрегантов, гепарина, глюкокортикостероидов не оказывает существенного эффекта.
Внедрение экстракорпоральных методов значительно улучшило прогноз заболевания: ранее летальность превышала 90%, в настоящее время при своевременно начатом лечении более 80% пациентов выздоравливают.
Очень схож с тромботической тромбоцитопенической пурпурой гемолитико-уремический синдром. Болезнь поражает новорожденных и детей младшего возраста и проявляется лихорадкой, тромбоцитопенией, микроангиопатической гемолитической анемией, артериальной гипертензией, нередко развивается острая почечная недостаточность. В отличие от тромботической тромбоцитопенической пурпуры при гемолитико-уремическом синдроме процесс носит локальный характер: гиалиновые тромбы обнаруживаются только в афферентных артериолах и клубочках почки, неврологические проявления редки.
Лечение не разработано; при своевременном гемодиализе смертность от острой почечной недостаточности составляет всего 5%, но у 10-50% пациентов остаются нарушения функции почек.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Элерса - Данлоса
Синдром Элерса-Данлоса – наследственная системная соединительнотканная дисплазия, обусловленная недостаточным развитием коллагеновых структур. В зависимости от клинического типа синдром Элерса-Данлоса может проявляться гипермобильностью суставов, необычайной ранимостью и растяжимостью кожи, склонностью к кровоизлияниям и кровотечениям, деформациями позвоночника и грудной клетки, миопией, косоглазием, птозом внутренних органов и пр. При диагностике синдрома Элерса-Данлоса учитываются клинические данные, результаты биопсии кожи и генотипирования; возможна пренатальная диагностика патологии. Лечение синдрома Элерса-Данлоса сводится к соблюдению щадящего режима, белковой диеты, симптоматической терапии.
Общие сведения
Синдром Элерса-Данлоса (несовершенный десмогенез, гиперэластическая кожа), наряду с несовершенным остеогенезом, синдромом Марфана и другими заболеваниями, относится к наследственным коллагенопатиям. Синдром Элерса-Данлоса неоднороден и включает в себя гетерогенную группу наследственных поражений соединительной ткани (соединительнотканных дисплазий), связанных с нарушением биосинтеза белка коллагена. Проявления синдрома Элерса-Данлоса носят системный характер и затрагивают опорно-двигательный аппарат, кожу, сердечно-сосудистую, зрительную, зубочелюстную и другие системы. Поэтому синдром Элерса-Данлоса представляет практический интерес не только для генетики, но и травматологии и ортопедии, дерматологии, кардиологии, офтальмологии, стоматологии.
Сложность верификации и наличие легких форм затрудняет получение точных сведений об истинной распространенности синдрома Элерса-Данлоса; частота диагностированных среднетяжелых случаев составляет 1:5 000 новорожденным, тяжелых форм - 1:100 000.
Причины синдрома Элерса-Данлоса
Различные варианты синдрома Элерса-Данлоса различаются по типу наследования, первичным молекулярным и биохимическим дефектам. Однако в основе всех клинических форм лежат мутации генов, обусловливающие количественную или структурную патологию коллагена. На сегодняшний день молекулярные механизмы синдрома Элерса-Данлоса установлены не для всех форм заболевания.
Так, известно, что I тип синдрома характеризуется снижением активности фибробластов, усилением синтеза протеогликанов, отсутствием ферментов, отвечающих за нормальный биосинтез коллагена. Синдром Элерса-Данлоса IV типа связан с недостаточностью продукции коллагена III типа; при VI типе заболевания имеет место недостаточность фермента лизилгидроксилазы, участвующего в гидроксилировании лизина в молекулах проколлагена. VII тип обусловлен нарушением превращения проколлагена I типа в коллаген; X тип - патологией плазменного фибронектина, участвующего в организации межклеточного матрикса и т. п.
Патоморфологическая картина при различных типах синдрома Элерса-Данлоса характеризуется истончением дермы, нарушением ориентации и потерей компактности коллагеновых волокон, разрастанием эластических волокон, увеличением числа сосудов и расширением их просвета.
Классификация синдрома Элерса-Данлоса
Всего выделяют 10 типов синдрома Элерса-Данлоса, различающихся по генетическому дефекту, характеру наследования и клиническим проявлениям. Рассмотрим основные из них:
I тип синдрома Элерса-Данлоса (классический тяжелого течения) – наиболее частый вариант заболевания (43% случаев) с аутосомно-доминантным типом наследования. Ведущим симптомом является гиперэластичность кожи, растяжимость которой по сравнению с нормой увеличена в 2-2,5 раза. Характерна гипермобильность суставов, носящая генерализованный характер, деформации скелета, повышенная ранимость кожи, склонность к наружным кровотечениям, образованию рубцов, плохому заживлению ран. У части больных выявляется наличие моллюскоподобных псевдоопухолей и варикозного расширения вен нижних конечностей. Беременность у женщин с I типом синдрома Элерса-Данлоса часто осложняется преждевременными родами.
II тип синдрома Элерса-Данлоса (классический мягкого течения) – характеризуется вышеописанными признаками, но выраженными в меньшей степени. Растяжимость кожи превосходит нормальную лишь на 30%; гипермобильность отмечается преимущественно в суставах стоп и кистей; кровоточивость и наклонность к рубцеванию незначительны.
III тип синдрома Элерса-Данлоса – имеет аутосомно-доминантное наследование, доброкачественное течение. Клинические проявления включают генерализованную повышенную подвижность суставов, скелетно-мышечные деформации. Остальные проявления (гиперэластичность и рубцевание кожи, геморрагии) минимальны.
IV тип синдрома Элерса-Данлоса – встречается редко, протекает тяжело; может наследоваться различными путями (доминантно или рецессивно). Гиперэластичность кожи незначительна, отмечается повышенная подвижность только суставов пальцев рук. Ведущим проявлением данного типа заболевания является геморрагический синдром: склонность к образованию экхимозов, спонтанных гематом (в т. ч. во внутренних органах), разрывам полых органов и сосудов (в т. ч. аорты). Сопровождается высокой летальностью.
V тип синдрома Элерса-Данлоса – имеет Х-сцепленное рецессивное наследование. Характеризуется повышенной растяжимостью кожи, умеренно выраженными гипермобильностью суставов, кровоточивостью и ранимостью кожи.
VI тип синдрома Элерса-Данлоса - наследуется по аутосомно-рецессивному типу. Кроме гиперэластичности кожи, наклонности к кровотечениям, повышенной подвижности суставов, имеются мышечная гипотония, тяжелый кифосколиоз, косолапость. Характерной чертой синдрома Элерса-Данлоса VI типа является глазной синдром, проявляющийся близорукостью, кератоконусом, косоглазием, глаукомой, отслойкой сетчатки и т. д.
VII тип синдрома Элерса-Данлоса (артроклазия) - наследуется как аутосомно-доминантно, так и аутосомно-рецессивно. Клиническую картину определяет низкий рост пациентов и гиперподвижность суставов, приводящая к частым привычным вывихам.
VIII тип синдрома Элерса-Данлоса – преимущественно наследуется аутосомно-доминантно. Ведущую роль в клинике играет хрупкость кожи, выраженный периодонтит, приводящий к ранней потере зубов.
X тип синдрома Элерса-Данлоса – характеризуется аутосомно-рецессивным наследованием; умеренной гиперэластичностью кожи и гипермобильностью суставов, стриями (полосовидной атрофией кожи), нарушением агрегации тромбоцитов.
XI тип синдрома Элерса-Данлоса – имеет аутосомно-доминантный тип наследования. У больных отмечаются рецидивирующие вывихи плечевых суставов, вывихи надколенника, встречается врожденный вывих бедра.
IX тип (Х-спепленный вариант вялой кожи) в настоящее время исключен из классификации синдрома Элерса-Данлоса. В современном варианте классификации синдрома Элерса-Данлоса рассматривается 7 основных типов заболевания:
- классический (типы I и II)
- гипермобильный (тип III)
- сосудистый (тип IV)
- кифосколиоз (тип VI)
- артроклазия (тип VIIB)
- дермоспараксис (тип VIIC)
- недостаток тенасцина-X
Симптомы синдрома Элерса-Данлоса
Ввиду того, что подробная характеристика различных типов синдрома Элерса-Данлоса дана выше, в настоящем разделе обобщим основные проявления заболевания. Поскольку соединительная ткань присутствует практически во всех органах, проявления синдрома Элерса-Данлоса носят системный, генерализованный характер.
Ведущим в клинической картине является кожный синдром: гиперэластичность кожи, которая легко собирается в складку и оттягивается. На ощупь кожа бархатистая, нежная, слабо фиксированная с подлежащими тканями, морщинистая на ладонных и подошвенных поверхностях. Гиперэластичность кожи при синдроме Элерса-Данлоса отмечается с рождения или дошкольного возраста, с годами имеет тенденцию к снижению.
Кроме, гиперрастяжимости, характерна повышенная ранимость, хрупкость кожи, обнаруживающаяся в возрасте старше 2-3-х лет. Минимальная травматизация приводит к образованию длительно не заживающих ран, на месте которых спустя время формируются атрофичные или келоидные рубцы, псевдоопухоли.
Суставные проявления синдрома Элерса-Данлоса представлены гипермобильностью (разболтанностью) суставов, которая может носить локальный (например, переразгибание межфаланговых суставов) или генерализованный характер. Суставной синдром проявляется с началом ходьбы ребенка, что приводит к повторным подвывихам и вывихам. С возрастом гипермобильность суставов обычно уменьшается.
Со стороны сердечно-сосудистой системы у детей с синдромом Элерса-Данлоса нередко выявляются врожденные пороки сердца, пролапс митрального клапана, аневризмы сосудов головного мозга, варикоз. Отмечается склонность к кровотечениям - экхимозам, гематомам различной локализации, носовым, десневым, маточным, желудочно-кишечным кровотечениям.
Глазные проявления синдрома Элерса-Данлоса могут включать гиперэластичность кожи век, миопию, птоз, косоглазие, разрывы роговицы и глазного яблока при минимальных механических повреждениях, спонтанную отслойку сетчатки.
Изменения скелета при синдроме Элерса-Данлоса характеризуются воронкообразной или килевидной деформацией грудной клетки, сколиозом, кифозом, косолапостью, неправильным прикусом, частичной адентией. Висцеральные нарушения представлены птозом внутренних органов, пупочными, паховыми, диафрагмальными грыжами, рецидивирующим спонтанным пневмотораксом, дивертикулезом кишечника и др. Умственное развитие детей с синдромом Элерса-Данлоса обычно соответствует возрасту.
Диагностика синдрома Элерса-Данлоса
Диагностика синдром Элерса-Данлоса проводится медицинским генетиком на основании генеалогических данных, анамнеза, клинического анализа, молекулярно-генетических исследований. Предварительно синдром Элерса-Данлоса может быть заподозрен при наличии больших диагностических критериев (гипермобильности суставов, гиперэластичности кожи, склонности к кровотечениям) и дополнительных малых (хрупкости кожи, патологии сердца, сосудов, глаз и т. д.).
Некоторые формы заболевания требуют проведения биопсии кожи для гистологического, гистохимического, электронно-микроскопического исследования.
Наличие в семье больных синдромом Элерса-Данлоса является показанием к медико-генетическому консультировании и проведению инвазивной пренатальной диагностики.
Больные с различными типами синдром Элерса-Данлоса могут нуждаться в наблюдении и обследовании детским травматологом-ортопедом, детским кардиологом, детским офтальмологом, детским стоматологом, сосудистым хирургом.
Лечение синдрома Элерса-Данлоса
Эффективная специфическая терапия синдрома Элерса-Данлоса не разработана. Детям требуется создание щадящего режима, исключающего излишнюю травматизацию суставов и кожи; ограничение физических нагрузок; соблюдение белковой диеты с включением в рацион костных бульонов, заливных блюд, студня. Обязательны регулярные курсы массажа, лечебной физкультуры, физиотерапии (магнитотерапии, электрофореза, лазеропунктуры).
Медикаментозная терапия синдрома Элерса-Данлоса включает применение аминокислот (карнитина), витаминов (С, Е, D, группы В), хондроитина сульфата, глюкозамина, минеральных комплексов (препаратов кальция и магния), метаболических препаратов (рибоксин, АТФ, коэнзим Q10) повторными курсами до1-1,5 мес. 2-3 раза в год.
При синдроме Элерса-Данлоса может быть показано хирургическое лечение: реконструкция грудной стенки, удаление псевдоопухолей, коррекция ВПС и пр.
Прогноз синдрома Элерса-Данлоса
На качество и продолжительность жизни больных синдромом Элерса-Данлоса влияет тип заболевания. Наиболее серьезный прогноз имеет IV тип синдрома Элерса-Данлоса – летальный исход может наступить вследствие разрывов сосудов, внутренних органов и кровотечений. Наличие синдрома I типа существенно ограничивает качество жизни. Относительно благоприятно протекание II—III типов болезни.
В целом, наличие синдрома Элерса-Данлоса сопряжено со множеством социальных трудностей, ограничивает полноценную физическую активность и выбор профессии.
Синдром Меллори-Вейса ( Желудочно-пищеводный разрывно-геморрагический синдром )
Синдром Меллори-Вейса — линейные разрывы слизистой кардиоэзофагеальной зоны, возникшие на фоне рвоты, позывов на рвоту, икоты. Проявляется наличием крови в рвотных массах, эпигастральными или загрудинными болями, артериальной гипотензией, тахикардией. Диагностируется с помощью эзофагогастроскопии, обзорной рентгенографии брюшной полости. Для лечения применяется гемостатическая, кровезаместительная терапия, противорвотные препараты, сердечные аналептики, ингибиторы протонной помпы, Н2-гистаминоблокаторы, антациды. При необходимости выполняется эндоскопический гемостаз, терапевтическая эмболизация, гастротомия для ушивания повреждений.
МКБ-10
Впервые клиника разрывно-геморрагического синдрома была описана в 1929 году американскими патологами Дж.К. Меллори и С. Вейсом. В настоящее время заболевание является одной из ведущих причин неязвенных кровотечений из верхних отделов пищеварительного тракта. Распространенность патологии достигает 5-10%. Болезнь Меллори-Вейса выявляется преимущественно у 45-60-летних пациентов, злоупотребляющих спиртными напитками. У мужчин разрывы желудочной и пищеводной слизистой возникают в 7 раз чаще, чем у женщин. У 79-80% больных поражается эзофагогастральный переход, у 16-17% — стенка пищевода, у 3-5% — кардиальная оболочка. Длина разрывов обычно составляет 0,4-4,5 см. В 77-78% случаев повреждения являются единичными, в 22-23% — множественными.
Причины
Продольные разрывы слизистой в области пищеводно-желудочного перехода возникают при локальном повышении давления у пациентов со сниженной резистентностью эпителиального слоя. Предпосылками к развитию разрывно-геморрагического гастроэзофагеального синдрома служат патологические процессы, при которых повреждаются эпителиоциты или наблюдается повышенное кровенаполнение сосудов верхних отдела ЖКТ: асептическое воспаление слизистой при частом употреблении спиртных напитков, воспалительные заболевания ЖКТ (эзофагиты, гастриты), длительный прием НПВС, кортикостероидов, скользящая грыжа пищеводного отверстия диафрагмы, расширение пищеводных вен при портальной гипертензии у больных с гепатитами, жировым гепатозом, фиброзом, циррозом печени. Непосредственными причинами болезни Меллори-Вейса являются:
- Рвота, неукротимая икота. У 80-85% пациентов развитие рвоты связано с алкогольным опьянением. Провоцирующими факторами также становятся рвота беременных, диспепсические расстройства при патологии пищеварительного тракта (язвенной болезни, панкреатите, холецистите), отравления, уремия.
- Длительный интенсивный кашель. В редких случаях разрывно-геморрагический синдром провоцируется острыми и хроническими респираторными заболеваниями. Линейное повреждение слизистой пищевода, верхних отделов желудка может осложнить коклюш, ОРВИ, хронический бронхит, бронхиальную астму.
- Ятрогенные воздействия. Повреждение стенки пищевода, желудка возможно при грубом выполнении эндоскопических манипуляций (гастроскопии, эзофагогастродуоденоскопии), введении желудочного зонда. Иногда разрывы слизистой возникают при проведении сердечно-легочной реанимации.
В спорадических случаях повышение давления, приводящее к разрыву эпителиального слоя, вызывается другими факторами — подъемом тяжестей, интенсивными физическими нагрузками с резким напряжением мышц брюшного пресса, тупой травмой живота. Крайне редко заболевание осложняет течение судорожного синдрома при эпилепсии, опухолях головного мозга, энцефалопатиях, менингите, энцефалите, эклампсии.
Патогенез
Пусковым моментом разрыва пищеводно-желудочной слизистой обычно становится многократная рвота, резкое повышение абдоминального давления при переполненном желудке или кардиоэзофагеальном спазме, реже — прямые механические воздействия. Возникновение избыточного давления в кардиальном отделе желудка способствует перерастяжению стенки органа. При морфологической несостоятельности эпителия, вызванной воспалительными процессами, растянутая слизистая желудка, пищевода разрывается в наиболее истонченном или патологически измененном участке. Обычно разрыв распространяется не глубже эпителиального и подслизистого слоя. В тяжелых случаях повреждается мышечная, серозная желудочная либо адвентициальная пищеводная оболочки с выходом агрессивного содержимого в средостение или брюшную полость.
Классификация
- I стадия. Повреждение слизистой желудочно-пищеводного перехода и дистальной трети пищевода. Встречается у 36-37% пациентов. В большинстве случаев кровотечение прекращается спонтанно.
- II стадия. Дефекты расположены в той же зоне, однако их глубина достигает подслизистого слоя. Выявляется у 52-53% больных. Обычно проводится консервативная гемостатическая терапия.
- III стадия. Глубокие зияющие разрывы с вовлечением мышечной оболочки и интенсивным кровотечением. Наблюдаются в 9-11% случаев. Необходим эндоскопический или хирургический гемостаз.
- IV стадия. Редко диагностируемое тяжелое повреждение с разрушением всех оболочек гастроэзофагеального участка ЖКТ. Осложняется медиастинитом, перитонитом, пневмотораксом.
Симптомы синдрома Меллори-Вейса
Клинические проявления заболевания обычно развиваются на фоне многократной рвоты. Основным признаком синдрома является выделение ярко-красной крови с рвотными массами (гематемезис), которое может иметь различную интенсивность – от нескольких капель до профузного кровотечения. Возникает резкая боль в эпигастральной области или за грудиной. Вследствие кровопотери у больного формируется острый анемический синдром, для которого характерны головокружение, бледность кожных покровов, мелькание «мушек» перед глазами, падение артериального давления, значительное учащение сердцебиения. При массивном кровотечении возможна потеря сознания.
Осложнения
Острая кровопотеря при симптомокомплексе Меллори-Вейса может привести к развитию геморрагического шока с тяжелыми нарушениями микроциркуляции, изменениями реологических свойств крови, прогрессирующей гипоксией. При отсутствии лечения шок переходит в декомпенсированную стадию, сопровождающуюся полиорганной недостаточностью. Наиболее тяжелым осложнением синдрома является тотальный разрыв стенки брюшного отдела пищевода, распространяющийся выше уровня диафрагмы. При этом у пациента возникает приступ одышки, цианоз кожи, сильнейшие боли в грудной клетке. Такое осложнение, известное как синдром Бурхаве, в 20-40% случаев заканчивается летальным исходом. Попадание содержимого желудка в средостение, полость брюшины провоцирует развитие медиастинита, перитонита.
Диагностика
Постановка диагноза при синдроме Мэллори-Вейса может быть затруднена, что обусловлено стремительным нарастанием клинической картины и необходимостью оказания пациенту экстренной медицинской помощи. Диагностика заболевания предполагает комплексное инструментальное обследование пищеварительного тракта для выявления первопричины кровавой рвоты. Наиболее информативными являются:
- Эзофагогастроскопия. Введение гибкого эндоскопа через ротовую полость позволяет оценить состояние эпителиальной оболочки верхних отделов ЖКТ и обнаружить линейные разрывы, которые обычно локализованы в области перехода пищевода в желудок. С помощью визуального осмотра удается установить глубину поражения стенки пищевода или желудка.
- Обзорная рентгенография брюшной полости. Проведение рентгенологического исследования информативно при подозрении на разрыв полого органа. Основной признак перфорации – наличие свободного газа в полости брюшины (симптом «серпа»). На рентгенограмме также можно обнаружить другие болезни ЖКТ, которые являются первопричиной патологии Меллори-Вейса.
В клиническом анализе крови определяются изменения, характерные для анемического синдрома — уменьшения содержания эритроцитов и гемоглобина, снижение показателя гематокрита. Для исключения хронического кишечного кровотечения проводится реакция Грегерсена, позволяющая обнаружить скрытую кровь в кале. При выраженном диспепсическом синдроме может выполняться бактериологический посев кала для выявления патогенных микроорганизмов.
Дифференциальная диагностика синдрома осуществляется с легочным кровотечением, отеком легких, сердечной астмой, кровотечением из язвы желудка, варикозным расширением пищеводных вен, острым гастроэнтеритом, кишечными инфекциями, распадом опухоли желудка или пищевода, синдромом Рандю-Ослера. Кроме осмотра хирурга и гастроэнтеролога пациенту могут потребоваться консультации гематолога, инфекциониста, пульмонолога, кардиолога, гематолога, гепатолога.
Лечение синдрома Меллори-Вейса
Пациент подлежит неотложной госпитализации в хирургический стационар. На начальном этапе больному обеспечивается покой, холод на область желудка, при позывах на рвоту применяются блокаторы дофаминовых и серотониновых рецепторов с противорвотным эффектом. Назначается консервативное лечение и малоинвазивные манипуляции, направленные на остановку кровотечения, восполнение объема циркулирующей крови. При резком падении АД терапию дополняют введением средств для поддержания гемодинамики. Пациентам с болезнью Меллори-Вейса показаны:
- Инфузионная терапия. При умеренной кровопотере проводятся внутривенные вливания коллоидных и кристаллоидных растворов. При массивном кровотечении переливается эритроцитарная масса или взвесь, нативная и свежезамороженная плазма, реже — донорская кровь.
- Гемостатические препараты. Для медикаментозного гемостаза используют стимуляторы свертывающей системы крови. Эффективность кровоостанавливающей терапии повышается при парентеральном введении препаратов кальция, синтетических аналогов витамина К.
- Эндоскопический гемостаз. При продолжающемся кровотечении с помощью эндоскопа обкалывают место повреждения средствами с сосудосуживающим эффектом, вводят склерозанты, лигируют или клипируют сосуды. Возможно выполнение аргоноплазменной или электрокоагуляции.
- Терапевтическая эмболизация. Для прекращения кровотечения из поврежденных сосудов в них под контролем ангиографии вводят смесь эмболов с физиологическим раствором. Альтернативным методом является редко применяемое внутриартериальное вливание жировых суспензий.
Баллонная зондовая тампонада используется ограниченно из-за возможного усугубления разрывов. Важным условием быстрого восстановления поврежденной стенки является угнетение желудочной секреции при помощи ингибиторов протонной помпы, блокаторов Н2-гистаминорецепторов. Прием секретолитиков дополняют назначением невсасывающихся антацидов, препаратов коллоидного висмута. Хирургические методы лечения геморрагического разрывного синдрома показаны при неостанавливающихся или рецидивирующих кровотечениях, глубоких дефектах, полном разрыве пищеводной или желудочной стенки. Рекомендованным вмешательством является гастротомия с прошиванием надрывов, кровоточащих сосудов, ушиванием дефектов, иногда — перевязкой левой желудочной артерии.
Прогноз и профилактика
Исход патологического состояния зависит от величины кровопотери и тяжести основного заболевания пациента. В 90% случаев кровотечение останавливается самопроизвольно или консервативными способами. Прогноз синдрома относительно неблагоприятный при потере больше 10% ОЦК и наличии сопутствующей патологии. Меры профилактики при заболевании Меллори-Вейса заключаются в отказе от злоупотребления алкоголем, своевременном устранении провоцирующих факторов, выявлении и лечении болезней желудочно-кишечного тракта, соблюдении техники проведения инвазивных медицинских манипуляций на пищеводе, желудке.
2. Лечение разрывно-геморрагического синдрома (синдрома Маллори-Вейсса) в специализированном Центре: автореферат диссертации/ Чередников Е.Е. – 2011.
Синдром Конна (первичный гиперальдостеронизм) - симптомы и лечение
Что такое синдром Конна (первичный гиперальдостеронизм)? Причины возникновения, диагностику и методы лечения разберем в статье доктора Лукьянова Сергея Анатольевича, хирурга-эндокринолога со стажем в 18 лет.
Над статьей доктора Лукьянова Сергея Анатольевича работали литературный редактор Маргарита Тихонова , научный редактор Сергей Федосов и шеф-редактор Лада Родчанина
Определение болезни. Причины заболевания
Синдром Конна (первичный гиперальдостеронизм) — это гормональное расстройство, при котором надпочечники в избыточном количестве вырабатывают гормон альдостерон. Проявляется повышением артериального давления.
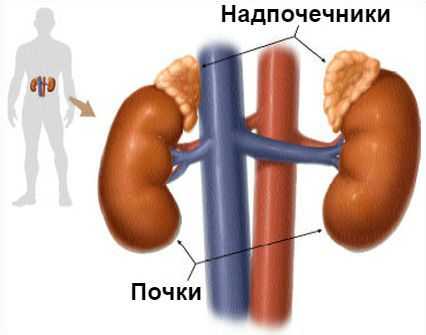
Основная задача альдостерона состоит в регулировании уровня натрия и калия в крови. Если этого гормона становится слишком много, организм начинает терять калий и удерживать слишком много воды, из-за чего объём крови и артериальное давление увеличиваются.
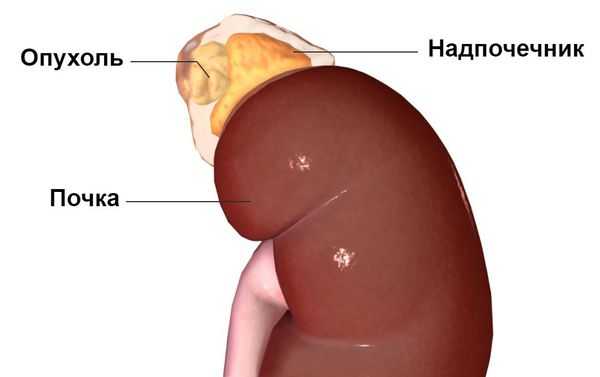
Первичный гиперальдостеронизм может быть вызван гиперактивностью либо одного, либо обоих надпочечников, т. е. быть односторонним или двусторонним. Одностороннее заболевание обычно вызывается альдостерон-продуцирующей аденомой (доброкачественной опухолью ), гиперплазией (усиленным ростом клеток) или, реже, раком одного надпочечника. На развитие этих причин влияют генетические факторы. Двустороннее заболевание обычно вызывает двусторонняя гиперплазия, т. е. усиленный рост клеток обеих желёз . Причиной такой гиперактивности надпочечников могут быть редкие генетические синдромы: семейный гиперальдостеронизм 1-го и 2-го типа.
Наиболее часто синдром Конна встречается у людей 30-50 лет, преимущественно среди женщин — в 60-70 % случаев. Изначально он считался крайне редким заболеванием. В настоящее время установлено, что это одна из наиболее распространённых причин симптоматической артериальной гипертензии. Последние исследования показывают, что данное заболевание встречается у 5-15 % пациентов с гипертонией. Но так как пациентов, устойчивых к лечению артериальной гипертензии, редко направляют для обследования к эндокринологу, синдром Конна, по-видимому, часто остаётся недиагностированным [1] .
Хотя первичный гиперальдостеронизм всё ещё является значительной диагностической проблемой, его распознавание имеет решающее значение, поскольку артериальную гипертонию при этом заболевании можно полностью вылечить с помощью хирургического вмешательства (в отличие от гипертонической болезни, когда пациенты вынуждены пожизненно принимать гипотензивные препараты).
При обнаружении схожих симптомов проконсультируйтесь у врача. Не занимайтесь самолечением - это опасно для вашего здоровья!
Симптомы синдрома Конна
Высокое давление является постоянным и зачастую единственным симптомом первичного гиперальдостеронизма. Оно наблюдается в 75-98 % случаев, вызывает головные боли, помутнение зрения и головокружение [2] [4] .
Повышение артериального давления может быть как незначительным, так и очень высоким. Причём высокие цифры при синдроме Конна обычно трудно контролировать, поэтому пациенты вынуждены принимать по четыре препарата для снижения давления или даже больше.
Часто у больных могут наблюдаться гипертонические кризы, при которых верхнее (систолическое) давление поднимается выше 250 мм рт. ст [3] . Клиническая картина таких кризов при синдроме Конна не отличается от обычного гипертонического криза: пациентов беспокоит головная боль, тошнота, рвота, головокружение, мелькание мушек перед глазами, избыточная потливость, боли в области сердца, учащённый пульс, чувство страха, звон в ушах и др.

Гипокалиемия (низкий уровень калия в крови) — второй по частоте симптом первичного гиперальдостеронизма. Его выявляют у 9-37 % больных [5] . Гипокалиемия может вызвать такие симптомы, как усталость, онемение, учащённое мочеиспускание, жажда, судороги и мышечная слабость. Увеличение объёма мочи (полиурия) и ночные пробуждения из-за позыва к мочеиспусканию (ноктурия) являются результатом нарушения работы почек. Иногда гипокалиемия может быть вызвана приёмом диуретиков. Уровень натрия в этом случае может быть нормальным или умеренно повышенным. Отёчный синдром, в отличие от других заболеваний почек, при этом встречается редко.
Все перечисленные жалобы часто встречаются и при других заболеваниях. Заподозрить именно синдром Конна можно, если у пациента:
- очень высокое артериальное давление, плохо поддающееся лекарственной терапии;
- семейный анамнез первичного альдостеронизма;
- высокое артериальное давление в возрасте до 40 лет;
- высокое артериальное давление и опухоль в одном из надпочечников;
- высокое артериальное давление и низкий уровень калия;
- высокое артериальное давление и апноэ во время сна.
Патогенез синдрома Конна
В связи с высоким уровнем альдостерона почки начинают сильнее всасывать натрий, задерживать воду и выводить калий из организма. Повышенное всасывание натрия почками, в свою очередь, увеличивает объём плазмы, что является первичным инициирующим механизмом гипертонии. Все эти изменения могут вызвать воспаление и отёк тканей с последующим развитием фиброза в жизненно важных органах, таких как сердце, почки и сосудистая сеть. В результате этого в дальнейшем развивается хроническая почечная недостаточность, мерцательная аритмия, инсульт, ишемическая болезнь сердца и застойная сердечная недостаточность.
Первичный гиперальдостеронизм может быть семейным (наследственным) и спорадическим (ненаследственным). Семейный вариант заболевания вызывает один из 4-х генетических вариантов гиперальдостеронизма, которые передаются по наследству. Также он может возникать спорадически, т. е. от случая к случаю. Генетические формы болезни связаны с мутацией генов, спорадические — с развитием альдостерон-продуцирующей аденомы или двусторонней гиперплазии надпочечников. До 2011 года причина возникновения этих аденом была не известна. Позже врачи установили, что в возникновении этих заболеваний особую роль также играют различные мутации [14] .
Не менее 6 % всех случаев первичного гиперальдостеронизма передаётся по наследству. Первый тип вызывается химерным геном, который содержит определённую последовательность структурных элементов ДНК гена 11β-гидроксилазы (CYP11B1). Этот ген регулируется АКТГ — адренокортикотропным гормоном, поэтому при первом типе заболевания также нарушается синтез глюкокортикоидов — других гормонов надпочечников. Тяжесть гиперальдостернизма может быть различной, отмечается раннее начало заболевания и высокая частота инсульта.
Клиническое течение второго типа неотличимо от спорадической формы болезни. Он диагностируется тогда, когда по крайней мере у двух членов семьи установлен этот диагноз. Генетическая основа данного типа остаётся неизвестной.
Третий тип ассоциируется с мутациями в гене KCNJ. Данные мутации приводят к нарушению транспорта калия и увеличению проводимости для натрия. В результате этого клеточная мембрана деполяризуется и вызывает открытие зависимых от каналов ионов кальция, который затем активирует выработку альдостерона. Для этого типа характерен дебют заболевания в раннем возрасте.
Недавно был описан новый, четвёртый тип семейного гиперальдостеронизма. Его вызывает мутация в гене CACNA1H, который кодирует работу кальциевых каналов. Этот тип также часто развивается ещё в детском возрасте и протекает с высоким артериальным давлением.
Спорадические формы гиперальдостеронизма (ненаследственные), тоже обусловлены различными мутациями, но уже соматическими (возникающими в течение жизни). Это в основном мутации усиления функции в генах, кодирующих ионные каналы или транспортёры (KCNJ5, CACNA1D, ATP1A1 и ATP2B3), которые приводят к избыточной продукции альдостерона [6] .
Достижения в области секвенирования генома человека (направленного на определение последовательности структурных элементов в молекуле ДНК) значительно продвинули наше понимание патогенеза первичного гиперальдостеронизма и показали, что чрезмерная выработка альдостерона при этой наиболее распространённой форме эндокринной гипертензии не так проста, как считалось ещё каких-то 10 лет назад.
Классификация и стадии развития синдрома Конна
Первичный гиперальдостеронизм классифицируется на основании анатомических характеристик и физиологических реакций.
Анатомически синдром Конна можно разделить на неопластический (аденомы, карциномы и эктопический гиперальдостеронизм) и неопухолевый (односторонняя или двусторонняя гиперплазия надпочечников).
Физиологически выделяют две формы заболевания:
- Ангиотензин чувствительная форма. К ней относится двусторонняя гиперплазия надпочечников и редкие формы односторонней аденомы. Они частично зависят от гормональной системы человека, которая регулирует артериальное давление.
- Ангиотензин нечувствительная форма. К ней относятся большинство альдостерон-продуцирующих аденом, рак или гиперплазия одного надпочечника. Они полностью не зависят от гормональной системы, регулирующей артериальное давление.
Частота встречаемости различных типов заболевания:
- Альдостерон-продуцирующие аденомы — 60 %.
- Первичная двухсторонняя гиперплазия надпочечников — 40 %.
- Односторонняя гиперплазия надпочечников — менее 1 %:
- альдостерон-продуцирующие ненадпочечниковые опухоли;
- альдостерон-продуцирующий рак коры надпочечника;
- глюкокортикоид-зависимый гиперальдостеронизм [7] .
Осложнения синдрома Конна
Повышенное артериальное давления и низкий уровень калия при синдроме Конна становятся причинами развития других проблем [8] . Так, гипокалиемия может привести к слабости, нарушению сердечного ритма, мышечным судорогам, чрезмерной жажде или мочеиспусканию, а постоянно высокое давление — к проблемам с сердцем и почками, в том числе инфаркту, инсульту и почечной недостаточности.
Риск возникновения сердечно-сосудистых проблем у пациентов с первичным гиперальдостеронизмом гораздо выше, чем у людей, которые имеют только высокое артериальное давление. Чтобы избежать возможных последствий синдрома Конна, больным с гипертонией, не поддающейся лечению, следует обратиться к эндокринологу для проведения обследования.
Диагностика синдрома Конна
Первичный гиперальдостеронизм диагностируется путём измерения в крови уровня альдостерона и ренина (гормона, вырабатываемого почками). Для более точной диагностики анализы необходимо сдавать утром. При синдроме Конна уровень альдостерона будет высоким, а уровень ренина — низким. При этом уровень калия может быть низким или нормальным.
Если результаты этих анализов будут положительными, то пациенту могут назначить дополнительные тесты для подтверждения диагноза. Они направлены на снижение количества альдостерона. Это можно сделать несколькими способами: ввести определённое лекарство, выполнить внутривенное вливание солевых растворов или ввести дополнительную соль с помощью диеты. Если после этих тестов уровень альдостерона останется высоким, а уровень ренина снизится, то диагноз подтверждается.
Альдостерон-рениновое соотношение (АРС). Этот анализ крови является скрининговым тестом, то есть он проводится при подозрении на гиперальдостеронизм. В настоящее время АРС является самым надёжным тестом по выявлению синдрома Конна. Однако различные методики определения этого соотношения часто приводят к ложноположительным или ложноотрицательным результатам. Об этом обязательно нужно помнить. Поэтому даже при высоком АРС, которое указывает на первичный гиперальдостеронизм, для подтверждения диагноза может потребоваться дополнительное тестирование.
Существует ряд правил по выполнению этого анализа:
- кровь необходимо брать в утренние часы (с 8:00 до 10:00);
- перед забором крови пациенту необходимо спокойно посидеть 5-10 минут;
- если у пациента есть гипокалиемия, то перед проведением анализа уровень калия необходимо нормализовать. Для этого в течение трёх дней перед забором крови рекомендуют соблюдать диету без ограничения соли, при которой пациенту нужно употреблять минимум 5-6 г поваренной соли в день;
- приём препаратов, которые могут повлиять на уровень альдостерона, необходимо отменить (например диуретики, противовоспалительные средства и др.) [9] .
Тест на подавление каптоприлом. Этот анализ крови измеряет реакцию альдостерона на каптоприл — лекарство, используемое для лечения высокого артериального давления. При отсутствии первичного гиперальдостеронизма уровень альдостерона в крови снизится более чем на 30 % от исходного.
24-часовая экскреция альдостерона с мочой. Для проведения этого теста пациент в течение пяти дней придерживается диеты с высоким содержанием соли, а затем сдаёт анализ мочи, собранной за сутки. Если уровень альдостерона в моче будет высоким, то наличие первичного гиперальдостеронизма будет подтверждено.
Тест на подавление физиологическим раствором. В этом тесте пациент получает солевой раствор через капельницу, затем ему измеряют уровень альдостерона и ренина. Первичный гиперальдостеронизм может быть подтверждён, если уровень альдостерона в крови после такой солевой нагрузки останется высоким, а уровень ренина понизится.
Для пациентов с доказанным первичным гиперальдостеронизмом следующим шагом является выяснение, каким заболеванием он вызван, — односторонним или двусторонним. Это очень важно, так как лечение каждого из них отличается. Компьютерная или магнитно-резонансная томография позволяют увидеть, есть ли опухоль в надпочечнике. Если пациент младше 40 лет и у него есть опухоль только в одном надпочечнике, то в этом случае можно приступать к лечению. Если пациент старше 40 лет и/или у него либо нет опухоли, либо есть опухоль, но в обоих надпочечниках, то возникает необходимость в проведении специального теста — селективного забора венозной крови из центральной вены надпочечников. В ходе этого теста хирург или рентгенолог берёт кровь непосредственно из вен обоих надпочечников, чтобы определить, какой именно орган вырабатывает патологическое количество гормона.
Лечение синдрома Конна
Лечение синдрома Конна зависит от его основной причины.
У пациентов с односторонней аденомой надпочечника единственным способом лечения является адреналэктомия — удаление опухоли вместе с надпочечником. Эта операция обычно выполняется ретроперитонеоскопически, т. е. через несколько очень маленьких разрезов со стороны спины. После односторонней адреналэктомии почти у 100 % пациентов снижается артериальное давление и нормализуется уровень калия в крови. При этом более чем у 50 % пациентов артериальная гипертензия полностью излечивается (без приёма лекарственных препаратов). Причинами стойкого повышения артериального давления после адреналэктомии может быть сопутствующая гипертоническая болезнь неизвестной причины и пожилой возраст пациентов.
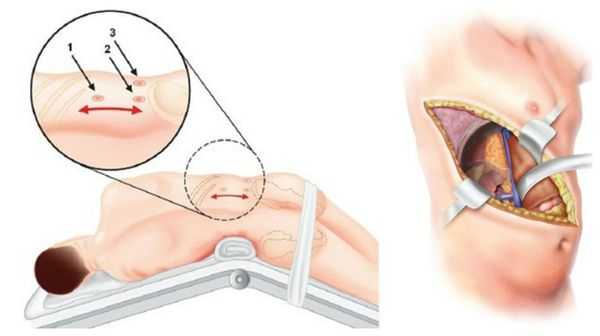
В крайних случаях проводится открытая адреналэктомия. Во время такой операции выполняется широкий разрез через грудную, брюшною полость и диафрагму. По сравнению с таким вмешательством, эндоскопическая адреналэктомия снижает сроки пребывания в стационаре и сочетается с меньшим количеством осложнений. Уже через неделю при эндоскопической операции пациент может вернуться к работе и обычным физическим нагрузкам.
Медикаментозное лечение рекомендуется пациентам с крайне высоким уровнем риска оперативного лечения (например при тяжёлой сердечной недостаточности). Поскольку односторонняя эндоскопическая адреналэктомия может полностью избавить пациента от медикаментозного лечения, её необходимо выполнять во всех остальных случаях. При этом важно, чтобы такое лечение выполнял хирург-эндокринолог, имеющий большой опыт проведения эндоскопических адреналэктомий.
Перед операцией артериальное давление и уровень калия в крови необходимо привести в норму. Если достичь этого в короткие сроки не удаётся, то операцию следует отложить.
После операции следует измерять уровень активности альдостерона и ренина в плазме крови, чтобы сделать предварительные выводы об эффективности оперативного лечения. Также в послеоперационном периоде нужно отменить приём добавок калия, прекратить приём спиронолактона и, при необходимости, уменьшить антигипертензивную терапию. Обычно артериальное давление улучшается или нормализуется через 1-6 месяцев после односторонней адреналэктомии, но у некоторых пациентов оно может оставаться повышенным до 1 года.
Для пациента с двусторонней гиперплазией лучшим способом лечением является консервативное — приём препарата под названием спиронолактон, который блокирует действие альдостерона. Кроме того, пациент должен придерживаться диеты с низким содержанием соли [10] .
Прогноз. Профилактика
Заболеваемость и смертность при синдроме Конна в первую очередь связаны с гипокалиемией и артериальной гипертензией. В случае своевременной диагностики болезнь излечивается полностью.
Причиной смерти могут стать сердечно-сосудистые осложнения. Установлено, что факторами риска развития этих осложнений при первичном гиперальдостеронизме являются гипокалиемия, односторонний первичный гиперальдостеронизм и высокий уровень альдостерона в плазме (не менее 125 пг/мл) [13] . Так, гипокалиемия, особенно тяжёлая, вызывает нарушения сердечного ритма, которые могут привести к летальному исходу.
Другими осложнениями гиперальдостеронизма являются инфаркт миокарда, цереброваскулярные болезни и сердечная недостаточность. Также у пациентов с синдромом Конна с большей вероятностью, чем у пациентов с гипертонией, развиваются острые коронарные синдромы, гипертрофия левого желудочка и инсульт [12] .
Существуют доказательства того, что хронический гиперальдостеронизм при отсутствии повышенного кровяного давления (как это происходит при вторичном гиперальдостеронизме) также связан с повышенным риском сердечных осложнений, включая ишемические, гипертрофические и фиброзные повреждения [11] .
Профилактика синдрома Конна предполагает своевременное лечение болезней, которые могут его вызвать. Пациентам с длительно существующей или трудно контролируемой артериальной гипертензией рекомендуется проходить скрининг на гиперальдостеронизм. В зоне риска находятся молодые люди до 40 лет и те, чьи родители страдают гипертонией.
Читайте также: