Лабораторная диагностика миелодиспластического синдрома - анализы
Добавил пользователь Алексей Ф. Обновлено: 28.01.2026
Лабораторная диагностика миелодиспластического синдрома - анализы
Диагноз миелодиспластического синдрома (МДС) основывается на обнаружении качественных нарушений кроветворения в одном или нескольких ростках гемопоэза (дизэритропоэз, дизгрануломоноцитопоэз, дизмегакариоцитопоэз). Вариант заболевания определяется на основании анализов крови и костного мозга.
Анализ периферической крови при миелодиспластическом синдроме. У больных миелодиспластическим синдромом (МДС) выявляются различные варианты цитопении: в 60-70% случаев — панцитопения (анемия, лейкои нейтропения, тромбоцитопения), у 30-40% пациентов — двуростковая цитопения, у 5% — одноростковая цитопения.
Анемия наблюдается более чем у 90% больных, имеет нормо- или макроцитарный характер, сопровождается умеренным анизо- и пойкилоцитозом. Уровень ретикулоцитов обычно снижен. Качественные изменения эритроцитов (дизэритропоэз) характеризуются наличием мегалоцитов, эритроцитов с базофильной пунктацией, тельцами Жолли; могут встречаться единичные нормоциты.
Лейко- и нейтропения отмечаются у 60% больных миелодиспластическим синдромом (МДС). В 10-20% случаев количество лейкоцитов в норме или повышено (при хроническом миеломоноцитарном лейкозе). Дизгранулоцитопоэз проявляется прежде всего псевдопельгеровской аномалией (пельгеризацией), которая наблюдается более чем у 80% пациентов. Наряду с этим могут встречаться гиперсегментированные и двуядерные нейтрофилы, а также клетки, содержащие фрагменты ядра.
В цитоплазме нейтрофилов выявляется либо резкое снижение зернистости (вплоть до полной дегрануляции), либо, напротив, аномально большие гранулы. При определенных формах миелодиспластического синдрома (МДС) в периферической крови выявляются бластные клетки. Для хронического миеломоноцитарного лейкоза характерен абсолютный моноцитоз (более 1 • 10 9 /л).
У большинства пациентов в периферической крови выявляется тромбоцитопения. Дизмегакариоцитопоэз характеризуется появлением в периферической крови качественно измененных тромбоцитов (гигантские клетки с бедным грануломером), а также фрагментов мегакариоцитов.

Миелограмма при миелодиспластическом синдроме
При исследовании аспирата костного мозга в большинстве случаев выявляется его нормальная или повышенная клеточность. Уменьшение количества миелокариоцитов отмечается менее чем у 15% больных. Основное при анализе миелограммы у больных миелодиспластическим синдромом — выявление качественных изменений гемопоэза и подсчет количества бластных клеток.
В первую очередь страдает эритропоэз, что проявляется гиперплазией эритроидного ростка, признаками мегалобластоидного кроветворения, наличием гигантских эритроидных клеток (до 20 мкм и более), многоядерных нормоцитов с фрагментами ядра, тельцами Жолли, кариорексисом и пикнозом ядер; цитоплазма клеток содержит базофильную пунктацию и вакуолизацию.
У больных с рефрактерной анемией с кольцевыми сидеробластами (РАКС) увеличено количество сидеробластов, имеются их кольцевидные формы. Дизэритропоэз является самой ранней находкой при миелодиспластическом синдроме и доминирует при рефрактерной анемии (РА) и РАКС.
Количество бластных клеток в пунктате костного мозга зависит от формы миелодиспластического синдрома и колеблется от нормального количества до 20%.
Дизгранулопоэз характеризуется псевдопельгеровской аномалией, гипо- и агрануляцией цитоплазмы или, напротив, появлением аномально крупных гранул. Количество мегакариоцитов в норме или уменьшено. Признаки дизмегакариоцитопоэза включают появление микромегакариоцитов, клеток с однои двулопастными ядрами или с множеством отдельно расположенных ядер небольших размеров с гигантскими аномальными гранулами. Может отмечаться плазмоцитарная реакция костного мозга (4-8%).
Трепанобиопсия дает более полное представление о клеточности костного мозга и признаках дизмегакариоцитопоэза (микромегакариоциты обнаруживаются чаще, чем в миелограмме). Выявляются также признаки дизэритро- и дизгранулоцитопоэза.
Биохимигеские исследования не имеют патогномоничного значения для диагностики миелодиспластического синдрома. В ряде случаев отмечается снижение уровня пируваткиназы, увеличение фетального гемоглобина в крови и лизоцима — в крови и моче.
Цитогенетические нарушения отмечаются у 50% больных первичным миелодиспластическим синдромом и более чем у 80% пациентов со вторичным миелодиспластическим синдромом. Для первичного миелодиспластического синдрома характерны del 5q, +8 и -7. При вторичном миелодиспластическом синдроме чаще выявляются del 7q и del 5q.
Изолированные хромосомные аберрации могут иметь благоприятное (del 5q и del 20q), неблагоприятное значение (del 7q) либо существенно не влиять на прогноз. Выявление любых множественных (не менее 3) цитогенетических аномалий сопровождается неблагоприятным прогнозом.
Дифференциальный диагноз миелодиспластического синдрома (МДС)
Некоторые формы миелодиспластического синдрома (МДС) (РА и РАКС) необходимо дифференцировать от апластической и В12-дефицитной анемии. Цитологическое и гистологическое исследование костного мозга, выявляющее выраженную дисплазию клеток миелоидного ростка при отсутствии аплазии кроветворения, позволяют поставить правильный диагноз.
Трудности могут возникнуть при дифференциальной диагностике МДС и острого эритромиелоза (М6 по FAB-классификации острого лейкоза), поскольку для обоих заболеваний характерны выраженный анемический синдром, бицитопения или панцитопения, гиперплазия и дисплазия эритроидного ростка костного мозга. В то же время при остром эритромиелозе в крови определяются ретикулоцитоз и нормоцитоз, в миелограмме выявляется 20% и более бластных клеток, а также уродливые нормоциты и многоядерные эритробласты при отсутствии качественных изменений гранулоцитарного ряда.
Дифференциальный диагноз РАИБ с другими вариантами острых лейкозов основывается на количестве бластов в миелограмме (при миелодиспластическом синдроме — всегда менее 20%).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Острый миелоидный лейкоз
Острый миелоидный лейкоз – злокачественное заболевание системы крови, сопровождающееся неконтролируемым размножением измененных лейкоцитов, снижением количества эритроцитов, тромбоцитов и нормальных лейкоцитов. Проявляется повышенной склонностью к развитию инфекций, лихорадкой, быстрой утомляемостью, потерей веса, анемией, кровоточивостью, образованием петехий и гематом, болями в костях и суставах. Иногда выявляются изменения кожи и припухлость десен. Диагноз устанавливается на основании клинических симптомов и данных лабораторных исследований. Лечение – химиотерапия, трансплантация костного мозга.
МКБ-10
Общие сведения
Острый миелоидный лейкоз (ОМЛ) – злокачественное поражение миелоидного ростка крови. Неконтролируемая пролиферация лейкозных клеток в костном мозге влечет за собой подавление остальных ростков крови. В результате количество нормальных клеток в периферической крови уменьшается, возникают анемия и тромбоцитопения. Острый миелоидный лейкоз является самым распространенным острым лейкозом у взрослых. Вероятность развития болезни резко увеличивается после 50 лет. Средний возраст пациентов составляет 63 года. Мужчины и женщины молодого и среднего возраста страдают одинаково часто. В старшей возрастной группе наблюдается преобладание лиц мужского пола. Прогноз зависит от вида острого миелоидного лейкоза, пятилетняя выживаемость колеблется от 15 до 70%. Лечение осуществляют специалисты в области онкологии и гематологии.
Причины острого миелоидного лейкоза
Непосредственной причиной развития ОМЛ являются различные хромосомные нарушения. В числе факторов риска, способствующих развитию таких нарушений, указывают неблагоприятную наследственность, ионизирующее излучение, контакт с некоторыми токсическими веществами, прием ряда лекарственных препаратов, курение и болезни крови. Вероятность возникновения острого миелоидного лейкоза увеличивается при синдроме Блума (низкий рост, высокий голос, характерные черты лица и разнообразные кожные проявления, в том числе гипо- или гиперпигментация, кожная сыпь, ихтиоз, гипертрихоз) и анемии Фанкони (низкий рост, дефекты пигментации, неврологические расстройства, аномалии скелета, сердца, почек и половых органов).
Острый миелоидный лейкоз достаточно часто развивается у больных с синдромом Дауна. Прослеживается также наследственная предрасположенность при отсутствии генетических заболеваний. При ОМЛ у близких родственников вероятность возникновения болезни повышается в 5 раз по сравнению со средними показателями по популяции. Самый высокий уровень корреляции выявляется у однояйцевых близнецов. Если острый миелоидный лейкоз диагностируется у одного близнеца, риск у второго составляет 25%. Одним из важнейших факторов, провоцирующих ОМЛ, являются заболевания крови. Хронический миелоидный лейкоз в 80% случаев трансформируется в острую форму болезни. Кроме того, ОМЛ нередко становится исходом миелодиспластического синдрома.
Ионизирующее излучение вызывает острые миелоидные лейкозы при превышении дозы 1 Гр. Заболеваемость увеличивается пропорционально дозе облучения. На практике имеет значение пребывание в зонах атомных взрывов и аварий на атомных электростанциях, работа с источниками излучения без соответствующих защитных средств и радиотерапия, применяемая при лечении некоторых онкологических заболеваний. Причиной развития острого миелоидного лейкоза при контакте с токсическими веществами является аплазия костного мозга в результате мутаций и поражения стволовых клеток. Доказано негативное влияние толуола и бензола. Обычно ОМЛ и другие острые лейкозы диагностируются спустя 1-5 лет после контакта с мутагеном.
В числе лекарственных средств, способных провоцировать острые миелоидные лейкозы, специалисты называют некоторые препараты для химиотерапии, в том числе ингибиторы ДНК-топоизомеразы II (тенипозид, этопозид, доксорубицин и другие антрациклины) и алкилирующие средства (тиофосфамид, эмбихин, циклофосфамид, хлорамбуцил, кармустин, бусульфан). ОМЛ также может возникать после приема хлорамфеникола, фенилбутазона и препаратов мышьяка. Доля лекарственных острых миелоидных лейкозов составляет 10-20% от общего количества случаев заболевания. Курение не только повышает вероятность развития ОМЛ, но и ухудшает прогноз. Средняя пятилетняя выживаемость и продолжительность полных ремиссий у курильщиков ниже, чем у некурящих.
Классификация острого миелоидного лейкоза
Классификация острого миелоидного лейкоза по версии ВОЗ очень сложна и включает в себя несколько десятков разновидностей заболевания, разделенных на следующие группы:
- ОМЛ с типичными генетическими изменениями.
- ОМЛ с изменениями, обусловленными дисплазией.
- Вторичные острые миелоидные лейкозы, возникшие в результате лечения других заболеваний.
- Болезни с пролиферацией миелоидного ростка при синдроме Дауна.
- Миелоидная саркома.
- Бластная плазмацитоидная дендритноклеточная опухоль.
- Другие виды острого миелоидного лейкоза.
Тактика лечения, прогноз и продолжительность ремиссий при разных видах ОМЛ могут существенно различаться.
Симптомы острого миелоидного лейкоза
Клиническая картина включает в себя токсический, геморрагический, анемический синдромы и синдром инфекционных осложнений. На ранних стадиях проявления острого миелоидного лейкоза неспецифичны. Отмечается повышение температуры без признаков катарального воспаления, слабость, утомляемость, потеря веса и аппетита. При анемии присоединяются головокружения, обморочные состояния и бледность кожных покровов. При тромбоцитопении наблюдаются повышенная кровоточивость и петехиальные кровоизлияния. Возможно образование гематом при незначительных ушибах. При лейкопении возникают инфекционные осложнения: частые нагноения ран и царапин, упорные повторные воспаления носоглотки и т. д.
В отличие от острого лимфобластного лейкоза при остром миелоидном лейкозе отсутствуют выраженные изменения со стороны периферических лимфатических узлов. Лимфоузлы небольшие, подвижные, безболезненные. Иногда выявляется увеличение лимфатических узлов в шейно-надключичной области. Печень и селезенка в пределах нормы или незначительно увеличены. Характерны признаки поражения костно-суставного аппарата. Многие больные острым миелоидным лейкозом предъявляют жалобы на боли различной степени интенсивности в области позвоночника и нижних конечностей. Возможны ограничения движений и изменения походки.
В числе экстрамедуллярных проявлений острого миелоидного лейкоза – гингивит и экзофтальм. В отдельных случаях наблюдаются припухлость десен и увеличение небных миндалин в результате инфильтрации лейкозными клетками. При миелоидной саркоме (составляет около 10% от общего количества случаев острого миелоидного лейкоза) на коже пациентов появляются зеленоватые, реже – розовые, серые, белые или коричневые опухолевидные образования (хлоромы, кожные лейкемиды). Иногда при поражениях кожи обнаруживается паранеопластический синдром (синдром Свита), который проявляется воспалением кожных покровов вокруг лейкемидов.
В развитии острого миелоидного лейкоза выделяют пять периодов: начальный или доклинический, разгара, ремиссии, рецидива и терминальный. В начальном периоде острый миелоидный лейкоз протекает бессимптомно или проявляется неспецифической симптоматикой. В периоде разгара токсический синдром становится более выраженным, выявляются анемический, геморрагический и инфекционный синдромы. В период ремиссии проявления острого миелоидного лейкоза исчезают. Рецидивы протекают аналогично периоду разгара. Терминальный период сопровождается прогрессирующим ухудшением состояния больного и завершается летальным исходом.
Диагностика и лечение острого миелоидного лейкоза
Решающую роль в процессе диагностики играют лабораторные анализы. Используют анализ периферической крови, миелограмму, микроскопические и цитогенетические исследования. Для получения образца тканей выполняют аспирационную биопсию костного мозга (стернальную пункцию). В анализе периферической крови больного острым миелоидным лейкозом обнаруживается снижение количества эритроцитов и тромбоцитов. Количество лейкоцитов может быть как повышенным, так и (реже) пониженным. В мазках могут выявляться бласты. Основанием для постановки диагноза «острый миелоидный лейкоз» становится обнаружение более 20% бластных клеток в крови либо в костном мозге.
Основой лечения острого миелоидного лейкоза является химиотерапия. Выделяют два этапа лечения: индукцию и консолидацию (постремиссионную терапию). На этапе индукции выполняют лечебные мероприятия, направленные на уменьшение количества лейкозных клеток и достижение состояния ремиссии. На этапе консолидации устраняют остаточные явления болезни и предотвращают рецидивы. Лечебную тактику определяют в зависимости от вида острого миелоидного лейкоза, общего состояния больного и некоторых других факторов.
Наиболее популярная схема индукционного лечения – «7+3», предусматривающая непрерывное внутривенное введение цитарабина в течение 7 дней в сочетании с одновременным быстрым периодическим введением антрациклинового антибиотика в течение первых 3 дней. Наряду с этой схемой в процессе лечения острого миелоидного лейкоза могут применяться другие лечебные программы. При наличии тяжелых соматических заболеваний и высоком риске развития инфекционных осложнений в результате подавления миелоидного ростка (обычно – у больных старческого возраста) используют менее интенсивную паллиативную терапию.
Программы индукции позволяет добиться ремиссии у 50-70% пациентов с острым миелоидным лейкозом. Однако без дальнейшей консолидации у большинства больных наступает рецидив, поэтому второй этап лечения рассматривается, как обязательная часть терапии. План консолидационного лечения острого миелоидного лейкоза составляется индивидуально и включает в себя 3-5 курсов химиотерапии. При высоком риске рецидивирования и уже развившихся рецидивах показана трансплантация костного мозга. Другие методы лечения рецидивных ОМЛ пока находятся в стадии клинических испытаний.
Прогноз острого миелоидного лейкоза
Прогноз определятся разновидностью острого миелоидного лейкоза, возрастом больного, наличием или отсутствием миелодиспластического синдрома в анамнезе. Средняя пятилетняя выживаемость при разных формах ОМЛ колеблется от 15 до 70%, вероятность развития рецидивов – от 33 до 78%. У пожилых людей прогноз хуже, чем у молодых, что объясняется наличием сопутствующих соматических заболеваний, являющихся противопоказанием для проведения интенсивной химиотерапии. При миелодиспластическом синдроме прогноз хуже, чем при первичном остром миелоидном лейкозе и ОМЛ, возникшем на фоне фармакотерапии по поводу других онкологических заболеваний.
Миелодиспластический синдром
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Миелодиспластические синдромы (МДС) - эпидемиология, причины, клиника
Миелодиспластические синдромы (МДС) — биологически и клинически гетерогенная группа клональных заболеваний, характеризующихся дисплазией кроветворения с неэффективным гемопоэзом и цитопеническими синдромами периферической крови и различной вероятностью эволюции в острые миелоидные лейкозы.
Признаки дисплазии кроветворения нередко сопровождаются бластозом крови и костного мозга, однако количество бластных клеток всегда меньше 20% (при уровне бластов равном или более 20% ставится диагноз острого лейкоза). В медицинской литературе прежних лет миелодиспластический синдром (МДС) имел различные названия (малопроцентный острый лейкоз, предлейкоз, тлеющая лейкемия и др.).
Частота миелодиспластических синдромов (МДС) в популяции составляет 3-5 случаев на 100 000 населения в год и существенно увеличивается с возрастом (частота у лиц старше 70 лет достигает 20 случаев на 100 000 населения в год). Средний возраст больных на момент начала заболевания составляет 70 лет.
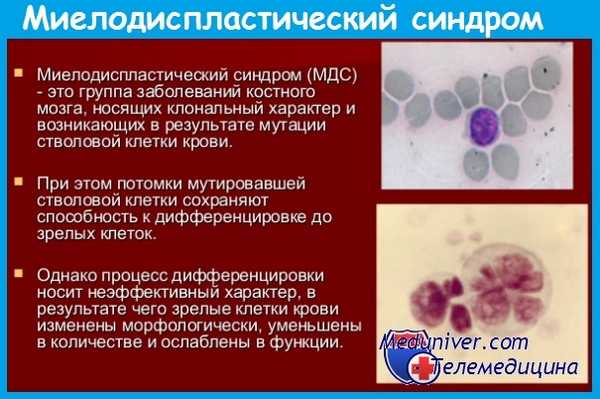
Этиология и патогенез миелодиспластических синдромов
К этиологическим факторам миелодиспластического синдрома (МДС) относятся ионизирующее излучение, цитостатические препараты, производные бензола и другие химические агенты (в том числе продукты табакокурения), генетические факторы.
Предшествующее лечение онкологических и онкогематологических заболеваний с использованием радиологических методов, алкилирующих средств (хлорамбуцил, циклофосфамид, мельфалан) и эпиподофиллотоксинов (этопозид, тенипозид) существенно повышает риск развития МДС, причем пик заболеваемости после лечения алкилирующими агентами отмечается через 5-10 лет, после эпиподофиллотоксинов — через 5 лет.
Длительное использование алкилирующих препаратов при лечении ревматических или других неопухолевых заболеваний также сопровождается высоким риском развития миелодиспластического синдрома (МДС). У детей с синдромом Швахмана-Дайемонда, анемией Фанкони и нейрофиброматозом 1-го типа частота развития миелодиспластического синдрома (МДС) выше, чем в общей популяции.
Как и другие гемобластозы, миелодиспластический синдром (МДС) имеет клональный патогенез. Родоначальницей клона является дефектная стволовая кроветворная клетка. В развитии заболевания имеют значение дефекты кроветворного микроокружения, приводящие к нарушению продукции цитокинов клетками стромы костного мозга и сопровождающиеся кумуляцией хромосомных повреждений и нарушением регуляции апоптоза.

Клиническая картина миелодиспластических синдромов
Симптоматика миелодиспластического синдрома (МДС) обусловлена наличием и выраженностью цитопении (чаще встречается панцитопения, реже — одно- и двуростковая цитопения). Основные клинические синдромы: анемический, геморрагический и инфекционных осложнений.
Наиболее часто первым признаком заболевания является анемия, проявляющаяся общей слабостью, одышкой при обычной физической нагрузке, головокружениями, сердцебиениями. Геморрагический синдром обусловлен тромбоцитопенией и качественными нарушениями клеток мегакариоцитарного ростка и обычно манифестирует подкожными кровоизлияниями, носовыми и десневыми кровотечениями, меноррагиями у женщин.
При глубокой тромбоцитопении могут возникать менометроррагии, желудочно-кишечные, почечные кровотечения, острые нарушения мозгового кровообращения. У 20-30% больных в клинической картине преобладают инфекционные осложнения, частота и выраженность которых связаны со степенью и длительностью нейтропении или агранулоцитоза: в более легких случаях возникают стрептодермии, стоматиты, синуситы, в тяжелых случаях — пневмонии, сепсис, причем возбудителями инфекционных осложнений часто являются условно-патогенная бактериальная, вирусная и грибковая микрофлора.
Увеличение лимфатических узлов, печени и селезенки наблюдается у 10-20% больных. Исключением является хронический миеломоноцитарный лейкоз, при котором спленомегалия отмечается почти у половины пациентов.
У 10% больных в начале заболевания клинические признаки отсутствуют и миелодиспластический синдром (МДС) обнаруживается случайно (при исследовании крови).
В 10-50% случаев (в зависимости от варианта миелодиспластического синдрома (МДС)) в исходе заболевания развиваются вторичные острые миелоидные лейкозы. В связи с резистентностью к цитостатической терапии и пожилым возрастом большинства пациентов ремиссии достигаются редко и обычно непродолжительны.
Миелодиспластический синдром у взрослых
1. Клинический протокол диагностики и лечения пациентов с заболеванием «миелодиспластический синдром» (далее-МДС) предназначен для оказания медицинской помощи в амбулаторных и стационарных условиях районных, областных и республиканских организаций здравоохранения, имеющих в своем составе гематологические отделения.
2. Возрастная категория: взрослое население.
3. Наименование нозологической формы заболевания (шифр по МКБ-10): миелодиспластический синдром - С92.1;
4. Определение: МДС - группа биологически и клинически гетерогенных клональных заболеваний, характеризующихся неэффективным гемопоэзом и цитопенией в периферической крови вследствие повышения апоптотической активности гемопоэтических предшественников с тенденцией к развитию костно-мозговой недостаточности или острого мие- лобластного лейкоза.

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 800 RUB / 4500 KZT / 27 BYN - 1 рабочее место в месяц

Автоматизация клиники: быстро и недорого!
- Подключено 300 клиник из 4 стран
- 1 место - 800 RUB / 4500 KZT / 27 BYN в месяц
Мне интересно! Свяжитесь со мной
13-15 октября, Алматы, "Атакент"
600 брендов, более 150 компаний-участников из 20 стран.
Новинки рынка стоматологии. Цены от производителей
Классификация
КЛАССИФИКАЦИЯ ЗАБОЛЕВАНИЯ
5. Классификация МДС, принятая Всемирной организацией здравоохранения (далее-ВОЗ) в 2008 году базируется на цитоморфологических, кариотипических и клинических признаках заболевания.
Классификация миелодиспластических синдромов
- 5 % бластов Палочки Ауэра отсутствуют
- 1 -10 9 /л моноцитов
*- хромосомные аномалии, которые рассматривают как предполагаемое свидетельство наличия МДС при стойкой цитопении неопределенного происхождения и при отсутствии абсолютных морфологических критериев МДС:
несбалансированные аномалии: - 7 или del(7q); - 5 или del(5q); i( 1 7q) или t(17p); - 13 или del(13q); del(l lq); del(12p) или t(12p); del(9q); idic(X)(ql3);
сбалансированные аномалии: t(l 1; 16)(q23;p 13.3); t(3;21) (q26.2;q22.1); t(l;3) (рЗб.З; q21.1); t(2;l 1) (p21;q23); inv(3) (q21q26.2); t(6;9)(p23;q34);
сложный кариотип (3 или более хрмосомных аномалий) с вовлечением вышеупомянутых нарушений.
Диагностика
КРИТЕРИИ ДИАГНОЗА МДС
6. Предварительные критерии (А).
6.1. Стабильная цитопения по > 1 из следующих клеточных линий:
эритроидной (уровень гемоглобина < 110 г/л);
нейтрофильной (количество нейтрофилов < 1,5х10 9 /л);
мегакариоцитарной (количество тромбоцитов < 100 х10 9 /л).
6.2. Исключение других гематологических и негематологических заболеваний - причин цитопении/дисплазии.
7. МДС-ассоциированные критерии (В).
7.1. Дисплазия в > 10% клеток эритроидной, нейтрофильной или мегакариоцитарной клеточных линиях костного мозга, либо наличие > 15% кольцевых сидеробластов.
7.2. От 5 до 19% бластных клеток в костном мозге.
7.3. Типичные хромосомные абберации по результатам стандартного цитологического исследования или флюоресцентной гибридизации in situ (+8, -7, 5q-, 20q- и др. согласно классификации)
8. Вспомогательные критерии (С) (для пациентов, имеющих критерии А, но не имеющих критерии В).
8.1. Аномальный иммунный фенотип эритроидных или миелоидных клеток костного мозга, указывающий на их клональное происхождение (по результатам проточной цитометрии).
8.2. Молекулярно-генетические признаки наличия клональной клеточной популяции в костном мозге (по результатам HUMARA исследования или биологического микрочипирования).
8.3. Значительное и стабильное снижение колониеобразующей активности костного мозга и/или периферической крови.
Диагноз устанавливается при наличии 2 предварительных критериев (А) и не менее чем одного МДС-ассоциированных критериев (В). Вспомогательные
критерии (С) используются при отсутствии критериев В и наличии у пациента признаков клональной миелоидной пролиферации. Критерии группы С не входят в обязательный стандарт диагностики МДС.
Диагноз «идиопатическая цитопения неопределенного значения» применяется для обозначения случаев цитопении по одной и более клеточным линиям в течение > 6 месяцев при отсутствии критериев МДС и других причин цитопении. Такие пациенты должны наблюдаться и обследоваться гематологом с интервалом 1-6 месяцев.
Диагностические критерии разработаны ICWG (International Consensus Working Group), 2007 г.
Алгоритм диагностики МДС включает в себя клинические и лабораторные исследования, мультидисциплинарный подход с привлечением смежных специалистов и последовательно проводится на базе учреждений здравоохранения различного уровня с соблюдением преемственности на всех этапах. Это обусловлено полиэтиологичностью и гетерогенностью проявлений при данной патологии, стремлением к рационализации использования специального диагностического оборудования, минимизации диагностических ошибок.
Этапы диагностики МДС:
| Этап | Задачи | Уровень медицинского учреждения и специалисты | Содержание |
| Первичного скрининга | Выявление пациентов с цитопе- ническими синдромами и клиническими проявлениями МДС; обоснование необходимости и направле-ние пациентов, нуждающихся в дополнительном обследовании, на следующий этап | Районный, городской (врачи любой специальности амбулаторно - поликлинических и стационарных учреждений здравоохранения) | Анамнез (химио- или лучевая терапия в прошлом, случаи МДС/ОМЛ в семье, рецидивирующие инфекционные заболевания или геморрагический синдром) Физикальное обследование (бледность, инфекционновоспалительные процессы, геморрагический синдром, спле- номегалия) Общий анализ периферической крови, с подсчетом количества тромбоцитов, лейкоцитарной формулы. Биохимический анализ крови (общий белок, альбумины, глобулины, мочевина, креатинин, билирубин, щелочная фосфатаза, ACT, АЛТ, ЛДГ, сывороточное железо) |
| Углубленного кли- нико - лабораторного и инструментального обследования | Углубленное клиниколабораторное и инструментальное обследование и выявление МДС - ассоциированных критериев; обосно-вание необходимости и направление па-циентов на следующий этап; диспансеризация пациентов с ранее верифицированным МДС | Областной, городской (гематоло гические отделения, кабинеты) | Общий анализ периферической крови, с подсчетом количества тромбоцитов, ретикулоцитов, лейкоцитарной формулы, морфологической оценкой. Биохимический анализ крови (сывороточный ферритин) Исключение реактивной дисплазии (мегалобластная анемия в результате дефицита витамина В12 и фолиевой кислоты, инфицирование ВИЧ, алкоголизм, недавнее цитотоксическое лечение, солидные злокачественные новообразования): маркеры вирусных гепатитов В и С, сифилиса, вируса иммунодефицита человека, ФГДС, колоноскопия, ультразвуковое исследование органов брюшной полости и малого таза, лимфатических узлов, щитовидной железы, рентгенография органов грудной полости. Аспирационная биопсия костного мозга: морфологическое исследование, иммунный фенотип, цитохимическое исследование эритрокариоцитов |
| Дифференциальной диагностики и подбора терапии | Клинико - лабораторное и инструментальное обследование пациентов с целью дифференциальной диагностики, верификации диагноза; подбор и коррекция индиви-дуальной про-граммы терапии; диспансеризация пациентов с ранее верифицированным МДС; создание базы и анализ данных пациентов с МДС для изучения проблемы МДС и усовершенствования оказания медицинской помощи | Областной, республи канский (гематоло гические отделения) | Исключение реактивной дисплазии: маркеры вируса Эпштейна - Барр, цитомегаловируса, определение в сыворотке крови уровня витамина В]2 и фолиевой кислоты. Определение в сыворотке крови уровня эритропоэтина Аспирационная биопсия костного мозга: морфологическое, цитогене- тическое исследования, иммунный фенотип, цитохимическое исследование эритрокариоцитов костного мозга Билатеральная трепанобиопсия передних или задних остей подвздошных костей Молекулярно-биологический анализ Клоногенный тест |
Лечение
КЛИНИЧЕСКИЕ ВАРИАНТЫ МДС
10. Определение клинического варианта МДС имеет значение для выбора тактики лечения.
10.1. 5ц-синдром: болеют преимущественно женщины, характерны вялотекущий характер заболевания, низкая вероятность трансформации в ОМЛ (10%), тяжелая макроцитарная анемия, нормальный или умерено сниженный уровень лейкоцитов и тромбоцитов, дисплазия мегакариоци- тарного ростка, отсутствие значительно повышения уровня бластных клеток в костном мозге; хороший ответ на леналидомид*.
10.2. Вторичный МДС: частота вторичного МДС нарастает в связи с успехами химиотерапии опухолей и воздействием загрязнения окружающей среды; для большинства пациентов характерны множественные хромосомные аберрации; прогноз хуже, чем при первичном МДС.
10.3. Гипопластический МДС:
до 15% случаев МДС характеризуются низкой клеточностью костного мозга при гистологическом исследовании (доля кроветворной ткани в препарате менее 30% у пациентов моложе 60 лет или менее 20% у пациентов 60 лет и старше);
дисплазия мегакариоцитов и клеток миелоидного ряда может отсутствовать;
возможны трудности в дифференциации от апластической анемии, для которой характерна более выраженная панцитопения, отсутствие типичных для МДС хромосомных аббераций и снижение содержания CD34+ клеток в костном мозге.
10.4. МДС с миелофиброзом: до 50% случаев всех вариантов МДС характеризуется фиброзом костного мозга (до 15% имеют выраженный фиброз); фиброз более характерен для вторичного МДС; характерны ги- перклеточность костного мозга, диффузный ретикулиновый фиброз его стромы и дисплазия не менее чем в 2 клеточных линиях; в периферической крови панцитопения, признаки клеточной дисплазии и лейкоэрит- робластоза; органомегалия нехарактерна; заболевание быстро прогрессирует; необходимо дифференцировать от острого мегакариобластного лейкоза, острого миелофиброза (острого панмиелоза с фиброзом), хронических миелопролиферативных заболеваний, метастатического рака, лимфом и волосатоклеточного лейкоза.
ЛЕЧЕНИЕ
11. Выбор терапии основан на диагнозе и группе риска по международной прогностической бальной системе (IPSS). В соответствии с международными рекомендациями для выбора терапевтической тактики пациентов с МДС подразделяют на 2 большие группы риска:
группу относительно низкого риска, включая в нее пациентов с низким и промежуточным 1 риском по системе IPSS;
группу высокого риска, включая в нее пациентов с промежуточным 2 и высоким риском по системе IPSS.
У пациентов из группы относительно низкого риска возможно применение только поддерживающей терапии либо терапии малой интенсивности. Интенсивная терапия показана пациентам группы высокого риска с учетом возраста, анамнеза заболевания, клинических проявлений, общего состояния и наличия признаков прогрессирования заболевания.
11.1. Поддерживающее лечение.
Поддерживающее лечение назначают с целью уменьшения проявлений заболевания и поддержания качества жизни. У пациентов из группы относительно низкого риска это может быть основным видом терапии.
11.1.1. Трансфузии донорских эритроцитов. Основным клиническим показанием для трансфузии донорских эритроцитов является не столько уровень гемоглобина, сколько степень адаптированности пациента к анемии.
11.1.2. Применение хелаторов железа.
Показаниями к применению хелаторов железа является переливание более 20-25 доз эритроцитной массы, уровень сывороточного ферритина более 2500 мкг/л, наличие дисфункции сердца (аритмия, сердечная недостаточность) и поражения печени.
Дефероксамин применяют в дозе 30-40 мг/кг в виде 12 часовых подкожных инфузий 5-7 раз в неделю (ночью). Дозу лекарственного средства снижают до 25 мг/кг при уровне ферритина < 2000 мкг/л. Необходимы контроль функции почек, аудиометрия и офтальмологический контроль до начала терапии и ежегодно на фоне ее проведения.
11.1.3. Трансфузии донорских тромбоцитов в стандартных дозировках показаны пациентам с глубокой тромбоцитопенией и петехиально - пятнистой кровоточивостью.
11.1.4. Эмпирическая антибактериальная и противогрибковая терапия лекарственными средствами широкого спектра действия показана пациентам с фебрильной нейтропенией. Профилактический прием антибактериальных и противогрибковых лекарственных средств показан лишь пациентам с рецидивирующими инфекционными осложнениями на фоне нейтропении.
11.2. Терапия малой интенсивности.
11.2.1. Эритропоэтин применяют в качестве терапии первой линии у пациентов группы относительно низкого риска с РА и РАИБ, частота трансфузий донорских эритроцитов у которых менее 2 доз в месяц и базальный уровень эритропоэтина в сыворотке крови менее 200 МЕ/л, в дозе 10 000 Ед подкожно в сутки ежедневно (40-60 000 ЕД 1-3 раза в неделю) в течение 6 недель.
11.2.2. Филграстим (далее - Е-КСФ) назначают пациентам резистентным к монотерапии эритропоэтином в дозе 1-2 мкг/кг подкожно в сутки ежедневно или 1-3 раза в неделю (в сочетании с эритропоэтином). При отсутствии ответа на терапию в течение 2-3 месяцев её прекращают. При наличии ответа постепенно снижают дозу эритропоэтина и Е-КСФ до минимально эффективной.
Возможно монотерапия Е-КСФ у пациентов с нейтропенией и рецидивирующими или резистентными к антибиотикотерапии инфекциями. Профилактическое применение препарата не целесообразно.
11.2.3. Эпигенетическую терапию применяют у пациентов группы высокого риска, у которых невозможно применение интенсивной терапии:
децитабин 20 мг/м в сутки внутривенно 5 дней ежемесячно 4-6 курсов.
11.2.4. Иммуносупрессивную терапию применяют преимущественно у пациентов с гипопластическим вариантом МДС.
Антитимоцитарный глобулин (далее-АТГ) 40 мг/кг в сутки внутривенно 4 дня.
Циклоспорин А в дозе 1-5 мг/кг/день в 2 приема не менее 6 месяцев. Дозу корригируют в соответствии с концентрацией препарата в сыворотке крови (не выше 400 мкг/мл), уровнем артериального давления, функциональным состоянием печени и почек.
11.2.5. Пациентам с РАИБ-1 и РАИБ-2, гипопластическим вариантом МДС показан мелфалан в дозе 2 мг/сутки перорально до получения клинико-гематологического эффекта.
11.3. Интенсивная терапия.
11.3.1. Пациентам группы высокого риска в возрасте менее 60 лет показано применение терапии индукции ремиссии острого миелобластно- го лейкоза.
11.3.2. Высокодозная химиотерапия с трансплантацией аллогенных гемопоэтических стволовых клеток показана всем пациентам с МДС в возрасте менее 60 лет при наличии HLA-идентичного родственного донора.
11.3.3. Критерии клинико-гематологического ответа при лечении первич
Читайте также: