Лечение апластической анемии - пересадка костного мозга
Добавил пользователь Владимир З. Обновлено: 29.01.2026
2 ФГБОУ ВО «Красноярский государственный медицинский университет имени профессора В.Ф. Войно-Ясенецкого» Министерства здравоохранения РФ
В статье приводится случай успешного лечения больной со сверхтяжелой формой апластической анемии в сочетании с пароксизмальной ночной гемоглобинурией и хроническим гемолизом. Интерес клинического случая в том, что больной одновременно проводилось лечение выраженного внутрисосудистого гемолиза, обусловленного пароксизмальной ночной гемоглобинурией и сверхтяжелой формы апластической анемии. Учитывая выраженность внутрисосудистого гемолиза перед и после процедуры аллогенной трасплантации костного мозга, больной проведен короткий индукционный курс терапии препаратом Солирис (Экулизумаб) 600мг/сут в вену Д-8, Д-1, Д+10. Аллогенная трасплантация костного мозга больной проведена от сиблинга (родной сестры). В настоящее время функционирование трансплантата удовлетворительное, без потребности в ростовых факторах и заместительных гемотрансфузиях. Проявлений реакции «трансплантат против хозяина» не наблюдалось, была продолжена иммуносупрессивная терапия циклоспорином А.
1. Савченко В.Г. Клинические рекомендации по лечению апластической анемии /В.Г. Савченко, Е.Н. Паровичникова, Е.А. Михайлова, и др.; ред. В.Г. Савченко. – Москва, 2014. – 23с.
2. Кулагин А.Д. Апластическая анемия: иммунопатогенез, клиника, диагностика, лечение /А.Д. Кулагин, И.А. Лисуков, В.А. Козлов. – Новосибирск: Наука, 2008. – 236 с.
4. Кулагин А.Д., Лисуков И.А., Птушкин В.В. и др. Национальные клинические рекомендации по диагностике и лечению пароксизмальной ночной гемоглобинурии / А.Д. Кулагин, И.А. Лисуков, В.В. Птушкин // Онкогематология. – 2014. – № 2. – С. 3-11.
5. Пароксизмальная ночная гемоглобинурия: Информационно-обозревательная брошюра для врачей-гематологов / ред. Кулагина А.Д. – Научное общество медицинских инноваций. –Москва: Литература, 2015. – 29 с.
6. Kelly R. The pathophysiology of paroxysmal nocturnal hemoglobinuria and treatment with eculizumab / R. Kelly, S. Richards, P. Hillmen, A. Hill // Ther. Clin. Risk Manag. – 2009. – V.2009:5. – P. 911-921.
7. Parker C.J. Bone marrow failure syndromes: paroxysmal nocturnal hemoglobinuria / C.J. Parker // Hematol. Oncol. Clin. North Am. – 2009; 23: 333-46.
8. Hillmen P. Long-term safety and efficacy of sustained eculizumab treatment in patientswith paroxysmal nocturnal hemoglobinuria / P. Hillmen, P. Muus, A. Roth et al. // Br. J. Haemotol. – 2013. –162(1). – P. 62-73.
Это заболевание впервые было описано в 1888 г. Паулем Эрлихом. Название апластическая анемия был предложено в 1904 г. Чауфордом. Частота ежегодной встречаемости заболевания от 6 до 13 случаев на 1 000 000 населения. Апластическая анемия (АА) – заболевание крови, при котором в результате угнетения костномозгового кроветворения формируется панцитопения [1]. Иммунная агрессия, направленная на гемопоэтические клетки – предшественники за счет активности Т-лимфоцитов и киллеров, является основным механизмом нарушения кроветворения при апластической анемии. Отмечается гиперпродукция цитокинов, подавляющих гемопоэтические клетки и стимулирующих активацию Т-лимфоцитов [2]. В микропрепаратах костного мозга при апластической анемии отмечается полное опустошение костного мозга, присутствуют мелкие очаги гемопоэза [3]. Микроокружение костного мозга играет большую роль в развитии гемопоэтических клеток и в функционировании костного мозга, которое в свою очередь зависит от сети микроциркуляции мозга. Плотность сосудов костного мозга (плотность микроциркуляции) у больных с апластической анемией низкая. Это играет определенную роль в патофизиологии недостаточности мозга. Не исключено, что применение проангиогенных агентов в терапии апластической анемии сыграет определенную роль в восстановлении функции костного мозга.
Лечение апластической анемии [2]:
- Терапия, направленная на восстановление костного мозга.
- Заместительная терапия компонентами крови, лечение и профилактика инфекционных осложнений.
- Дополнительные методы лечения апластической анемии
- стероиды;
- спленэктомия;
- стимулирующие колонии факторы.
Пароксизмальная ночная гемоглобинурия (ПНГ) – Болезнь Гарлея, болезнь Маркиафавы – Микели, болезнь Штрюбинга – Маркиафавы – это приобретенное, прогрессирующее системное заболевание, при котором наблюдается внутрисосудистый гемолиз. Заболеваемость ПНГ достигает примерно 1 случай на 1000 000 жителей в год. Причиной является соматическая мутация в стволовой клетке, развивается тотальная цитопения с вовлечением в процесс тромбоцитов и лейкоцитов. Формируются тромбозы, нарушается деятельность многих органов, в том числе физиологическая и иммунная несостоятельность костного мозга [4]. Следует помнить, что следует проводить дифференциальную диагностику пароксизмальной ночной гемоглобинурией у пациентов с цитопенией. Поскольку приобретенную недостаточность клеток кроветворения можно дифференцировать между такими заболеваниями, как апластическая анемия, пороксизмальная ночная гемоглобинурия, миелодиспластический синдром, а также возрастная дегенерация костного мозга у совершенно здоровых людей, и синдромах с неопухолевым процессом, следует, по мере возможности, методом секвенирования уточнять патогенетическую мутацию при синдроме недостаточности кроветворения.
Называют три формы Пароксизмальной ночной гемоглобинурии:
- Классическая форма с признаками гемолиза.
- ПНГ у пациентов с апластической анемией.
Субклиническая форма заболевания у пациентов без клинических и лабораторных признаков гемолиза, но при наличии малого клона клеток с признаками Пароксизмальной ночной гемоглобинурии [4].
Лечение Пароксизмальной ночной гемоглобинурии, представленное ранее: трансфузии компонентов крови, терапия гемолиза, дефицита железа, стимуляторы развития молодых форм эритроцитов, противовоспалительные препараты. Главным образом, терапия носила симптоматический и полиативный характер [5].
Аллогенная трансплантация костного мозга (ТКМ) на данный момент является единственным радикальным методом лечения Пароксизмальной ночной гемоглобинурии. ТКМ сопряжена с высокой летальностью и развитием осложнений. Кроме того, в посттрансплантационный период иногда происходит восстановление ПНГ-клона и рецидив Пароксизмальной ночной гемоглобинурии [4].
В связи с высоким риском развития осложнений алло-ТКМ проводится при аплазии кроветворения (АА /ПНГ и АА/ субклиническая ПНГ), а также при злокачественной клональной трансформации Пароксизмальной ночной гемоглобинурии в Миелодиспластический синдром, острый лейкоз [6].
Лечение состоит из фазы индукции (600 мг в/венно 4 недели) и фазы поддерживающей терапии (900мг в/венно в течение 5-ой недели и далее каждые 14 дней). Солирис безопасен при длительной применении и позволяет значительно снизить частоту осложнений и смертность пациентов с Пароксизмальной ночной гемоглобинурией [7; 8]. В настоящее время экулизумаб применяется у пациентов с Пароксизмальной ночной гемоглобинурией при наличии тромботических осложнений, хронического гемолиза с нарушением функции органов и систем, трансфузионной зависимости вследствие хронического гемолиза, беременности у пациенток с Пароксизмальной ночной гемоглобинурией.
Апластическая анемия, по данным эпидемиологических исследований, встречается в Европе, Северной Америке, Дальнем и Ближнем Востоке. Довольно часто Апластическая анемия распространена в Корее. В Европейских странах распространенность апластической анемии составляет 2 случая на 1 млн населения в год при колебании этого показателя в зависимости от конкретной страны от 0,6 до 3 и более на 1 млн населения в год. Апластическая анемия часто сочетается с пароксизмальной ночной гемоглобинурией (ПНГ). Клон Пароксизмальной ночной гемоглобинурии у больных с апластической анемией находят в 50 % [5]. Апластическая анемия в сочетании с пароксизмальной ночной гемоглобинурией наблюдается в 2–4 случаях на 1 млн населения в год.
Приводим клинический случай диагностики и успешного лечения больной с апластической анемией сверхтяжелой формы в сочетании с пароксизмальной ночной гемоглобинурией.
Пациент М.Я.В., 1993 года рождения
Диагноз: Приобретенная идиопатическая апластическая анемия, сверхтяжелая форма. Курс комбинированной иммуносупрессивной терапии (тимоглобулин-22.01-26.01.16 г.) + циклоспорин А). Аллогенная родственная трансплантация костного мозга (14.06.16 г.).
Пароксизмальная ночная гемоглобинурия (12.2015 г.). Хронический внутрисосудистый гемолиз.
Осложнения: Транзиторная лекарственная нефротоксичность (ЦсА). Вторичный гемосидероз.
Дебют заболевания с августа 2015 г. – появление обильных длительных менструаций до 7 дней, подкожных кровоизлияний от незначительных травм, за медицинской помощью не обращалась. С ноября 2015 г. появление крови в кале.
08.12.2015 г. больная экстренно госпитализирована в гематологическое отделение КМКБ №7 г. Красноярска. В клинической картине выраженный анемический синдром (выраженная общая слабость, утомляемость, головокружение, одышка при ходьбе и физической нагрузке); геморрагический синдром (менаметроррагии, подкожные кровоизлияния, кровь в кале).
В гемограмме эритроциты 1,32*10 12 /л, ретикулоциты 72,5 %0, сфероцитоз, агрегация эритроцитов, гемоглобин 49 г/л, тромбоциты 16*10 9 /л, лейкоциты 2,66*10 9 /л, палочкоядерные 1 %, сегментоядерные 4 %, лимфоциты 87 %, моноциты 7 %, СОЭ 52 мм/ч. Миелограмма: пунктат значительно сниженной клеточности. Мегакариоциты в пунктате не найдены. Бластов нет. Проба Кумбса (прямая, непрямая) – отрицательная. В биохимическом анализе крови: Лактатдегидрогеназа- 1194,0 Ед/л (N до 450 Ед/л), билирубин общий 22,2 ммоль/л (прямой — 5,6 ммоль/л, непрямой 16,6 ммоль/л). Трепанобиопсия: изменения отражают гипоплазию костномозгового кроветворения. ПНГ-клон выявлен: среди гранулоцитов (FLAER-CD24) – 52,72 %, среди моноцитов (FLAER-CD14) – 58 %, на эритроцитах 11 %.
Таким образом, на основании гистологического, цитологического, иммуноферментного типирования – исследований выставлен диагноз: Приобретенная идиопатическая апластическая анемия, сверхтяжелая форма/ пароксизмальная ночная гемоглобинурия, впервые выявленная.
Проводилась заместительная – гемокомпонентная терапия (трансфузии эритроцитарной взвеси № 4, тромбоконцентрата № 40), симптоматическая терапия.
В декабре 2015 г. пациент заочно консультирован профессором НИИ детской онкологии, гематологии и транспланталогии (ДОГиТ) им. Р.М. Горбачевой (Санкт-Петербург) д.м.н. А.Д. Кулагиным: рекомендовано HLA-типирование пациента и сиблинга (родная сестра). Но на тот период времени сиблинг (сестра) находилась на 7-м месяце беременности. По результатам типирования пациент и ее сестра полностью совместимы.
В январе, марте 2016 г. повторные заочные консультации в НИИ ДОГиТ им. Р.М. Горбачевой г. Санкт-Петербурга: показано проведение аллогенной родственной трансплантации костного мозга после родоразрешения и прекращения грудного вскармливания у родственного донора. С учетом выраженности внутрисосудистого гемолиза для снижения рисков процедуры аллогенной трансплантации костного мозга показано проведение короткого индукционного курса терапии препаратом экулизумаб.
До момента аллогенной трансплантации костного мозга пациент неоднократно проходил стационарное лечение в гематологическим отделении КМКБ № 7 г. Красноярска, где проводилась гемокомпонентная терапия (трансфузии эритроцитарной взвеси, тромбоконцентрата).
Во время очередной госпитализации в январе 2016 г. проведен курс АТГ (тимоглобулин 800мг/курс)+ГКС. Терапию больная перенесла удовлетворительно, признаков сывороточной болезни не было. С марта 2016 г. начат прием циклоспорина А в дозе 400 мг/сут, переносимость удовлетворительная.
Госпитализация в отделение трансплантации костного мозга ПСПбГМУ им. академика И.П. Павлова НИИ ДОГиТ им. Р.М. Горбачевой – 06.06.16 г. Донор: сестра 1988 г.р., полностью совместима по HLA-системе с большой и малой несовместимостью по АВО-АII>ВIII).
Режим кондиционирования: флударабин 240 мг в/венное, бусульфан 520 мг per os, тимоглобулин 326 мг в/венное. Профилактика трансплантат против хозяина (РТПХ): циклоспорин 1560 мг в/венное, метотрексат 60 мг в/венное. Профилактика хронического внутрисосудистого гемолиза — экулизумаб 600 мг/сут в/венное Д-8, Д-1, Д+10. 14.06.16г. проведена операция Аллогенная трансплантация костного мозга. Введение препаратов режима кондиционирования, трансфузию трансплантата перенесла без осложнений. В течение раннего посттрансплантационного периода наблюдались осложнения: токсический гепатит 2 ст., на фоне введения метотрексата, проводилась терапия гепатопротекторами с положительной динамикой; фебрильная нейтропения, ответ на тиенам.
Восстановление периферической крови: лейкоциты >1х10 9 /л/Д+24; нейтрофилы >0,5х10 9 /л/Д+19, тромбоциты >50х10 9 /л/Д+20. Пункция костного мозга: Д+28 — нормоклеточный костный мозг, все ростки представлены, кариотип 46,ХХ, донорский химеризм 90-97%; Д+43/Д+62 – в миелограмме все ростки представлены, химеризм полный донорский (97 %). Исследование ПНГ-клона: Д+24 – сохраняется минорный клон среди гранулоцитов (FLAER-CD24) – 0,01 %, среди моноцитов (FLAER-CD14) – 0,01 %; лабораторных и клинических проявлений внутрисосудистого гемолиза нет. Д+43 – ПНГ клон среди моноцитов и гранулоцитов не детектируется, среди эритроцитов – минорный (CD59 – 0,55 %). Д+65 – среди моноцитов и гранулоцитов не детектируется, среди эритроцитов – минорный (CD59 – 0,30 %).
С Д+23 переведена на пероральный прием циклоспорина А 200 – 250 мг/сут. Признаков острой реакции «трансплантат против хозяина» не было. Амбулаторно продолжался прием противомикробных, антибактериальных, противогрибковых препаратов, с целью профилактики инфекционных осложнений, в назначенной дозе. Еженедельно больная наблюдалась врачами-гематологами г. Красноярска, проводился контроль анализов крови и биохимических показателей, а также коррекция дозы циклоспорина А.
В сентябре 2016 г. пациент находился на плановом обследовании и лечении в отделении ТКМ для взрослых ИДГиТ им. Р.М. Горбачевой г. Санкт-Петербурга. Пункция костного мозга (Д+97): нормоклеточный костный мозг, все ростки представлены, химеризм полный донорский (90-97%). Гемограмма: эритроциты 3,31*10 9 /л, гемоглобин 105 г/л, ретикулоциты 1,24%0, тромбоциты 165*10 9 /л, лейкоциты 2,5*10 9 /л, нейтрофилы 71,8%. Исследование ПНГ-клона (Д+98): ПНГ-клон не детектируется.
Функционирование трансплантата удовлетворительное, без потребности в ростовых факторах, заместительных гемотрансфузиях. Проявлений реакции трансплантат против хозяина нет, продолжена иммуносупрессивная терапия циклоспорином А (125мг/сут), доза редуцирована в связи с органической токсичностью (гиперкреатининемия).
Повторный плановый осмотр с ноября 2016 г. в отделении трансплантации костного мозга для взрослых ИДГиТ им. Р.М. Горбачевой г. Санкт-Петербурга. Пункция костного мозга (Д+153): умеренной клеточности костный мозг, все ростки представлены. Гемограмма: эритроциты 3,58*10 9 /л, гемоглобин 116 г/л, ретикулоциты 2,31%0, тромбоциты 162*10 9 /л, лейкоциты 3,3*10 9 /л, нейтрофилы 65,9 %. Исследование ПНГ-клона (Д+157): ПНГ-клон не детектируется. Функционирование трансплантата удовлетворительное, без потребности в ростовых факторах и заместительных гемотрансфузиях. Группа крови донорская. Проявлений реакции трансплантат против хозяина нет, продолжается иммуносупрессивная терапия циклоспорином А. Больная продолжает наблюдаться у гематологов в отделении трансплантации костного мозга для взрослых ИДГиТ им. Р.М. Горбачевой г. Санкт-Петербурга и гематологов города Красноярска.
Таким образом, своевременная высококвалифицированная диагностика, современный подход в терапии данного заболевания, обеспечили удовлетворительный исход процесса у пациента, с улучшением клинического состояния и благоприятным прогнозом для здоровья.
Лечение апластической анемии - пересадка костного мозга
При апластической анемии терапией намечаются следующие цели (Берчану):
1) устранение обусловившей заболевание причины;
2) достижение наиболее полной ремиссии путем:
а) замещающего лечения, чтобы обеспечить необходимое время для восстановления костного мозга;
б) лечения инфекционных осложнений;
3) стимулирование кроветворения.
Отдельные гигиенические мероприятия представляются обязательными, в этой связи больным указывать на необходимость избегать кожные инфекции — применением антисептических веществ, инфекции дыхательных путей — избегая места большого скопления людей. Рекомендуется также избегать физическую травму вызывающую кровотечение.
Замещающее лечение при апластической анемии. Наиболее целесообразным представляется переливание крови. Всеми рекомендуется этот способ лечения когда показатель гемоглобина понижается до менее 8 г/100 мл. Перфузия тромбоцитной массой показана при тяжелом геморрагическом синдроме, в то время как переливание концентрата гранулоцитов необходимо при осложнении болезни инфекцией неподдающейся обычной антибиотикотерапии.
Терапия инфекций при апластической анемии. Рекомендуются культуры крови, мочи, желчи, кожных поражений, мокроты и пр. в целях выявления возбудителя. Однако в срочных случаях до получения результата от культур и антибиограммы, целесообразно назначать антибиотик широкого диапазона действия, например ампициллин. Полезно не забывать, что у таких больных довольно редкие инфекции как, например, гистоплазмой, кандидой, аспергиллом — частое явление.
Страдающих тяжелой формой агранулоцитоза или нейтропении (менее 500/мм3) изолировать, преимущественно в стерильные комнаты.
Лечение кортикостероидными гормонами при апластической анемии. Такое мероприятие оправдывает себя во многих случаях апластической анемии (для стимулирования кроветворения, при геморрагическом синдроме, сопутствующем расплавлении крови и пр.). Большинством авторов рекомендуется 1—2 -месячный курс лечения умеренными дозами (от 30 до 50 мг. в сутки). Если за этот срок не отмечаются положительные результаты, прекратить лечение.
Лечение, стимулирующее костный мозг при апластической анемии. До настоящего времени, в частности у больных детского возраста, наиболее эффективным представляется применение андрогенных гормонов.
Видимо мужские гормоны обусловливают выделение эритропоэтина и сенсибилизацию кроветворной ткани к этому веществу. С особым успехом, в частности детям, применяется синтетический тестостерон (оксиметолон), причем медикамент назначается внутрь, дозами от 2 до 4 мг на кг. веса тела в сутки. Когда этот медикамент отсутствует, может оказаться полезным и тестостерон-пропионат принимаемый также внутрь из расчета 0,25 мг/кг веса тела/сутки. Лечение проводить не менее 3 месяцев.
Отдельные авторы назначают это лечение на сроки, достигающие 9 месяцев. Разные статистические материалы и данные разведочных исследований не выявляют какой-либо существенной разницы между леченными андрогенами и иными способами.
Многочисленные эксперименты доказали роль карбоната лития в поощрении продукции моноцитами фактора, стимулирующего колонии. До настоящего времени не проводились разведочные работы, однако авторы, сделавшие попытки в этом направлении рекомендуют назначение карбоната лития из расчета 450—600 мг/сутки в течении 3—4 недель, при условии наличия в костном мозге гранулоцитных предшественников и периодической проверки уровня лития в крови. Отсутствие положительного результата вскрывает нецелесообразность дальнейшего применения этого лечения (Williams, Ward).

Пересадка костного мозга при апластической анемии
При костномозговой аплазии после облучения смертельными дозами Mathe применял пересадку костного мозга с весьма хорошими результатами. После успешного завершения этого эксперимента и до настоящего времени в технике пересадки органов — — вообще, и костного мозга — в частности, достигнуты существенные прогрессы.
В данный момент пересадка составляет терапевтический метод выбора в случаях заболевания молодых людей тяжелой апластической анемией (Geary).
Критерии для постановки диагноза тяжелой апластической анемии следующие: анемия с наличием 1% ретикулоцитов, 500/мм3 гранулоцитов, 20 000/мм3 тромбоцитов и в результате биоптического исследования пораженного гипоплазией костного мозга — 25% нормальной клеточной массы.
Статистические данные центров (Seattle), применяющих эту терапию, указывают на полную ремиссию у 50% больных, в противоположность 10% — леченных условными способами (Thomas D., Sterb).
При этом возникают следующие проблемы:
— отбор доноров костного мозга путем стандартизации по системе "Human Leucocytes Antigen" (HLA) и осуществеления смешанных культур лимфоцитов. По статистическим материалам в системе HLA, у половины какой-либо данной группы больных отмечено сходство с их родственниками, в то же время в пределах семьи шансы совместимости оцениваются до 25%;
— даже при строгой совместимости — отдача трансплантата наблюдается в 26% случаев.
Существует ряд способов годготовки больных в целях облегчения пересадки и избежания явления отдачи.
Применение циклофосфамида составляет стандартный метод лечения, но больным, в отношении которых тест лимфоцитотоксичности показывает предварительную сенсибилизацию к костному мозгу донора, назначаются прокарбазин, глобулин антитимоцит и общее облучение. Недавно в Seattle начало применяться циклофосфамид в сочетании с периферическими одноядерными клетками донора для дополнения клеток-штамм, при этом полученные результаты — обнадеживающие.
Известно, что у примерно 80% больных с предварительной сенсибилизацией наблюдается явление отдачи трансплантата.
После пересадки назначается метотрексат в целях предупреждения явления «трансплантат против хозяина», которое, в течение ближайших трех месяцев после операции, составляет причину 20% смертных исходов.
Однако тяжелые инфекции составляют причину остальных, быть может даже большинства, смертных исходов.
Отмечается также развитие интерстициальной пневмонии у 68% больных и заболевание вызываемое цитомегалическим вирусом или Pneumocystis carinii (Silver).
До настоящего времени результаты обнадеживающие. В этой связи отмечаются большая продолжительность выживаемости страдающих костномозговой аплазией с хорощо приживленным трансплантатом, чем у леченных условными способами (Storb и Thomas) и, как уже было сказано, более высокий процент полной ремиссии.
Иммуносупрессорная терапия (циклофосфамидом и пр. средствами) испробована при генуинной апластической анемии, поскольку были выявлены некоторые иммунологические механизмы (Ascensao). Недавно была сделана попытка лечения этого заболевания либо только антилимфоцитным глубилином, либо сочетая его с костномозговой пересадкой (Geary). Статистическими данными еще не подтверждена эффективность такого лечения, однако прогрессы, достигнутые в деле выявления иммунологических механизмов при апластической анемии сделают возможным отбор больных для иммуносупрессорной терапии.
С той же иммуносупрессорной целью проводилась спленэктомия, но лишь с частично положительными результатами (Берчану).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Показания для пересадки костного мозга - техника
Целью пересадки костного мозга является замена поврежденных или отсутствующих гематопоэтических клеток получателя, клетками донора в норме. Непрерывное возобновление миелоидного и лимфоидного клеточных рядов обеспечивается клетками костного мозга, ряда полипотентного штамма. Следовательно, по существу, пересадка костного мозга это перевивка клеток штамм.
При выборе донора отдавать предпочтение, в первую очередь, однояйцевым близнецам, одинаковых с точки зрения тканевой совместимости. Но так как подобная возможность представляется исключительно редко, наиболее частые пересадки используют костный мозг брата или сестры, при условии их идентичности с получателем по наиболее важному комплексу гистосовместимости (идентичный HLA — А и В, также смешанный отрицательный лимфоцитный тест). К отбору костного мозга от нероднящихся лиц, даже при соответственной гистосовместимости, прибегать лишь в случаях крайней необходимости.
В случае пересадки от однояйцевого близнеца или при комбинированном иммуном недостатке, получателя не следует подвергать подготовке. Во всех остальных случаях проводить иммуносупрессорную терапию в целях развития переносимости получателя к перевиваемым ему клеткам. У больных с апластической анемией для индуцирования такой переносимости применять циклофосфамид в количестве 50 мг/кг веса тела, в течение последовательных 4 дней. По истечении 36 часов от последней дозы, пересадить костный мозг.
При злокачественных заболеваниях, в частности острой лейкемии, подготовка получателя преследует также цель устранения злокачественных клеток.
Предпочтение отдается сочетанию общего облучения дозой 900—1000 рад. и циклофосфамида. Избегать переливания крови вообще, а от будущего донора костного мозга — в частности, для предотвращения иммунизации получателя к антигенам гистосовместимости, находящимся на лейкоцитах и тромбоцитах.
Техника пересадки костного мозга относительно несложна. Костный мозг отбирать в условиях общей анестезии путем ряда пункций в грудину и подвздошную кость. Отбирать 300—500 мл костномозговой массы, что у получателя должно соответствовать 10 8 —10 9 ядерных клеток/кг веса тела. После фильтрации костномозговая масса вводится больному капельным внутривенным путем. По иной технике внутривенное введение костного мозга проводится непосредственно после отбора, с помощью специального шприца (Mathe и сотр.).
Иммуносупрессия неизменно ведет к костномозговой аплазии, продолжающейся 10—20 дней после пересадки костного мозга. В течение этого периода больной нуждается в тромбоцитных концентратах, для сохранения числа тромбоцитов на уровне более 20 000/мм 3 , равно как и гранулоцитных концентратов. В качестве донора гранулоцитов отдавать предпочтение членам семьи, по возможности донору костного мозга. Вопреки трудностям, с которыми сопряжено получение гранулоцитов их введение показано даже когда у больного не наблюдается явное бактериальное или грибковое заражение.
Больной должен получать их до момента, когда он станет способным вырабатывать большее количество гранулоцитов, чем получаемое путем переливания. В целях предупреждения инфекции подобных больных определять в больницу и помещать в асептическую изоляционную палату.
Когда в результате действия трансплантата больной преодолевает стадию костномозговой аплазии, следующей угрозой является воздействие, вырабатываемых перевитыми полипотентными клетками-штамм, лимфатических клеток на ткани и органы получателя. Эта реакция трансплантата против получателя весьма тяжелая, она развивается у примерно 70% больных, получивших костный мозг от братьев или сестер с одинаковыми первостепенными гистосовместимыми системами.
В таких случаях поражены кожа, печень, кишечник, повышается температура, уменьшается вес тела, развиваются понос и желтуха. При тяжелой форме смертность составляет 85%. Лечения метотрексатом, эндоксаном и преднизоном мало эффективны.
Бактериальная, вирусная и грибковая инфекции составляют наиболее частую причину смертельного ихсода за счет агранулоцитоза, иммуной недостаточности и реакции трансплантата против получателя. Легочные инфекции, обусловливаемые цитомегалическим вирусом или Pneumocistis carinii считаются наиболее опасными.
Показания к пересадке костного мозга касаются, в первую очередь, апластической анемии, независимо от этиологии. В таких случаях преследовать повторное заселение костного мозга больного нормальными клетками-штамм.
Несколько больных живут уже более 10 лет после пересадки костного мозга от однояйцевых близнецов. В условиях тяжелой апластической анемии, при которой смерть составляет 80—90%, испытана аллогеническая пересадка от гистосовместимых братьев и сестер. Выживают примерно 50% больных.
Недавно отмечен прогресс в отношении выживания больных, страдающих апластической анемией после пересадки, именно в тех случаях, когда пересадка делается до получения больным переливаня крови или клеточных концентратов. В этой группе выживаемость составила более 70 % больных. У большинства погибших была доказана функциональность пересаженных клеток; смертные исходы были отнесены за счет инфекции или реакции трансплантата против получателя. Пересадка костного мозга предоставила лишний аргумент по вопросу патогенеза за счет дефекта клеток-штамм при апластической анемии.
При врожденной иммуной недостаточности (иммуный недостаток в сочетании с синдромом di George) предложена пересадка костного мозга для заселения вновь центральных лимфатических органов клетками-предшественниками нормальных лимфоцитов. Больные не нуждаются в иммуносупрессии, при этом число необходимых клеток-штамм значительно меньше. Возможно назначение концентратов клеток-штамм малыми повторными дозами.
К сожалению выживают лишь больные, получившие костный мозг от однояйцевого близнеца; в остальных случаях реакция трансплантата против получателя, полностью лишенного иммуной защиты, крайне тяжелая.
В отношении страдающих острой лейкемией для которых донором является однояйцевой близнец, вопрос о назначении пересадки костного мозга может быть принят во внимание с появлением болезни. Неудачные случаи отнесены за счет возвратного лейкемического процесса примерно спустя 7 недель после облучения и пересадки. В нескольких случаях удалось обнаружить злокачественное преобразование клеток донора.
Добавление химиотерапии, до облучения, и иммунотерапии лимфоцитами донора и инактивированными лейкемическими аутологическими клетками после пересадки, улучшило результаты. Отдельные больные, леченные этим способом, продолжают жить более 4 лет. Результаты слабее, когда донор костного мозга не однояйцевой близнец. Однако из 100 больных, леченных бригадой Е.Д. Томаса, 13 живут уже более 3 лет на стадии полной ремиссии, без какого-либо поддерживающего лечения.
Одинаковый способ применяли страдающим твердой опухолью (нейтробластома, лимфома), однако подобных случаев мало и результаты нечеткие. Следует особо отметить случаи аутологической пересадки костного мозга страдающим подобными опухолями. Отбор костного мозга у данного больного и его хранение при температуре —80° или —180° делают терапию более активной. Костномозговая аплазия, обусловленная терапией устраняется применением законсервированных аутологических костномозговых клеток.
Опыт радиобиологической клиники Больницы Фундень насчитывает 22 случая остеосаркомы, леченных крупными дозами циклофосфамида (9,5—11,1 г. в течение 10—11 дней). Асептическое изолирование и применение костного мозга от однояйцевого близнеца, в случаях тяжелой костномозговой аплазии и инфекционного синдрома, привели к ремиссии аплазии и сделали возможным подход к радиотерапевтическому и/или хирургическому лечению больного. Пересадка аутологического костного мозга может оказаться полезной и при бластическом припадке в условиях хронической гранулоцитной лейкемии.
При этом костномозговые клетки, законсервированные в хронический период болезни, впрыскиваются при появлении костномозговой аплазии после усиленных химио- и радиотерапии.
При случайной острой лучевой болезни пересадка костного мозга составляет единственную возможность для выживания пострадавшего. Летальная доза облучения у человека составляет более 600 рад. — когда облучение носит общий характер. Назначать однозначные костномозговые клетки спустя 6—10 дней после общего летального облучения. Возможно осуществление постоянного трансплантата, со всеми соответствующими последствиями, или временную пересадку до восстановления структур, способствующих возобновлению собственного кроветворения.
В случае облучения, при котором определенная часть костного мозга остается неповрежденной, можно расчитывать на повторное обсеменение отмершего костного мозга клетками-штамм, выходящими из оставшейся нормальной территории.
Когда же доза облучения превышает 1000 рад. степень поражения костномозговой стромы делает невозможной колонизацию гомологичными клетками-штамм.
Апластическая анемия ( Гипопластическая анемия )
Апластическая анемия – угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
МКБ-10
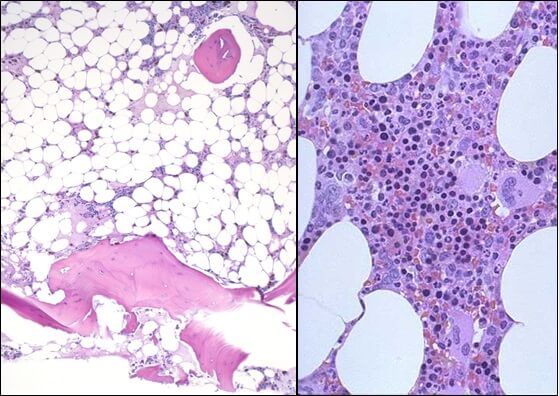
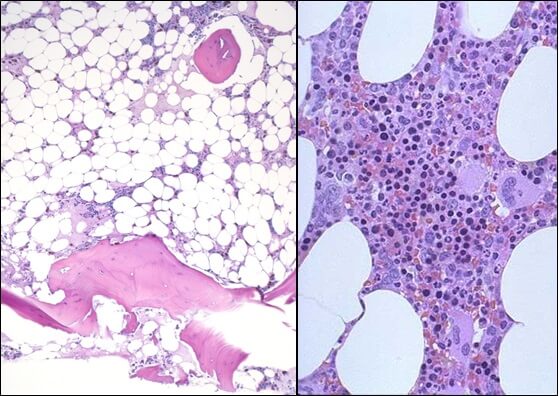
Общие сведения
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
Причины
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
Патогенез
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Классификация
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Осложнения
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений – кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Диагностика
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз и профилактика
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.
Миелодиспластический синдром
Миелодиспластический синдром – группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение – переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Миелодиспластический синдром – группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома – курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие – при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким – около 8 месяцев. Вероятность отторжения костного мозга после трансплантации – около 10%.
Читайте также:
- КТ, МРТ при синоназальной доброкачественной смешанной опухоли
- Подагра - кристаллический синовит. Диагностика и лечение
- Возбудители отогенных внутричерепных осложнений. Внутричерепные осложнения средних отитов
- Причины устойчивости опухолевых клеток к химиотерапии
- Проба с физической нагрузкой в кардиологии. Физиология физических нагрузок