Лечение гипопроконвертинемии - схема
Добавил пользователь Алексей Ф. Обновлено: 30.01.2026
Гипопроконвертинемия, или дефицит фактора VII (лабильный фактор, болезнь Александера), была впервые обнаружена в 1951 году. Считается самым распространенным из редких расстройств свертывания крови (на 500 тысяч человек около 1,3 тысячи больных). Передается по наследству обоими родителями-носителями дефектного гена. Одинаково распространена как среди мужчин, так и среди женщин.
Фактор VII — это белок, который при связывании с тканевым фактором способствует протеканию процесса свертывания, приводя к образованию тромбов. Если фактор VII не вырабатывается в необходимом количестве, организм не может сформировать фибриновый сгусток должным образом.
Препараты, увеличивающие малое производство фактора VII
- антибиотики;
- разжижители крови, например, варфарин;
- лекарственные препараты от рака, например, интерлейкин-2;
- антитимоцитарный глобулин.
Заболевания, которые способствуют гипопроконвертинемии:
- заболевания печени;
- миелома;
- сепсис;
- апластическая анемия;
- дефицит витамина К.
Симптомы гипопроконвертинемии
Отсутствуют при слабой и средней форме заболевания. Дети, рожденные с гипопроконвертинемией, в течение первого полугода жизни подвержены внутричерепным кровоизлияниям, кровотечениям желудочно-кишечного тракта.
У взрослых людей с этим расстройством свертывания крови могут присутствовать такие симптомы, как появление кровоподтеков по телу, кровоизлияние в суставы и мышцы, кровотечения после операции, образование гематом в носу, на поверхности кожи и даже в мочеполовой системе. У женщин часто наблюдается меноррагия – затяжные менструации, болезненные и длительные (более 7 дней).
Диагностика заболевания проводится при помощи анализа крови на тромбопластиновое и протромбиновое время. Чаще всего это заболевание появляется у людей с заболеваниями печени и дефицитом витамина К, а также у больных, принимающих оральные антикоагулянты.
Лечение гипопроконвертинемии
Основное лечение: употребление рекомбинантного фактора VII. В качестве альтернативы могут быть использованы концентраты протромбина (протромбиновые комплексы), свежезамороженная плазма.
Болезнь Оврена (парагемофилия)
Болезнь Оврена (парагемофилия), или дефицит фактора V, — редкое расстройство кроветворения. Тяжелая степень расстройства проявляется у больных уже в раннем возрасте.
Самые распространенные симптомы парагемофилии это: обильные и длительные носовые кровотечения, кровоподтеки на лице и теле, гемартроз. Больные отличаются высоким риском кровотечений, в том числе внутричерепных, легочных, желудочных, кишечных.
Частота заболевания - примерно 1 человек на миллион. Регионы, где распространена болезнь Оврена: Индия, Иран.
Недостаточность фактора V обычно вызвана мутациями в гене F5. Он регулирует активность белка свертывания крови В. Очень редко заболевание провоцируется специальными антителами, распознающими фактор свертывания как чужеродный элемент в крови, таким образом, организм как бы отторгает полезный компонент крови.
Лечение болезни Оврена
Заключается в приеме специальной формы фактора V. Профилактика этого заболевания подразумевает избегание тяжелых физических нагрузок, ограничение активности, отказ от экстремальных видов спорта, связанных с падениями и ударами.
Геморрагические нарушения, обусловленные антикоагулянтами в крови
Оборотные антикоагулянты — это антитела, которые нейтрализуют специфические факторы свертывания в естественных условиях (например, аутоантитела против фактора VIII или фактора V) или ингибируют определенные белки крови. Иногда эти вещества вызывают кровотечения путем связывания протромбина.
Если у человека присутствует обильное кровотечение, которое невозможно остановить, врачи предполагают наличие оборотных антикоагулянтов.
Антифосфолипидные антитела обычно вызывают тромбоз. Тем не менее, у некоторых пациентов антитела связываются с протромбиново-фосфолипидным комплексом и провоцируют гипопротромбинемию и кровотечение. Подобные геморрагические нарушения возникают у женщин в послеродовом периоде или у пожилых пациентов без острых симптомов какого-либо заболевания.
Симптомы циркуляции антикоагулятнов в крови
Как и при любых нарушениях свертываемости крови, при циркуляции антикоагулятнов в крови процесс свертывания нарушается, возникают спонтанные кровотечения, кровоподтеки, болезненные менструации у женщин.
Лечение циркуляции антикоагулятнов в крови
Показана терапия с применением циклофосфамида, ритуксимаба. Они подавляют продукцию аутоантител у больных без гемофилии. Управление острой кровопотерей у больных с гемофилией, осуществляется за счет фактора VIII или фактора VII.
Гипергепаринемия
Гипергепаринемия — это состояние, при котором в плазме крови повышена концентрация фермента гепарина. Причина этого заболевания — наследственная, передается оно в том случае, если оба родителя являются носителями дефектного гена. Гепарин отвечает за прекращение процесса свертывания крови.
При гепаринемии время свертывания крови после получения травм и от момента начала кровотечения увеличивается, соответственно, при гипергепаринемии кровь перестает свертываться вообще. Это серьезное заболевание встречается крайне редко, и может быть спровоцировано как наследственностью, так и факторами среды.
Гипергепаринемия изучена мало, в медицинской литературе, как отечественной, так и зарубежной, сведений по данному термину крайне мало. Большинство случаев подобных заболеваний можно назвать скорее гепаринемией обычной, нежели гипергепаринемией. Без лечения данное заболевание приводит к смерти от потери крови.
Симптомы гипергепаринемии
Гипергепаринемия выражается в следующих симптомах:
- отсутствие свертывания крови;
- неврологические симптомы (усталость, вялость, бессонница, повышенная утомляемость);
- высокая вероятность мозговых, внутримышечных, суставных и черепных кровотечений.
Лечение гепаринемии
Прием факторов свертывания крови в зависимости от дефицита определенного фактора.
Лечение гипопроконвертинемии - схема
Клиника гипопроконвертинемии - лабораторная диагностика
Гипопроконвертинемия имеет очень раннее начало. Оно происходит при самом рождении, проявляясь (чаще) либо пупочными геморрагиями, либо (реже) менингеальными геморрагиями. Обе формы являются особенно суровыми, часто с летальным исходом.
В течение жизни больного, геморрагические явления могут возникать (реже) спонтанно и (чаще) вследствие провокации. Наружные формы (эпистаксис, ротовые геморрагии, покровные геморрагии через раны, диггестивные геморрагии, мено-метроррагии) встречаются наиболее часто; они возникают в тех же этиологических условиях как и при недостатке Ф. V. Следует подчеркнуть, что метроррагии бывают иногда такими тяжелыми, что нуждаются в произведении терапевтической гистеректомии in extremis. Что касается внутренних геморрагии, они бывают реже и принимают вид экхимозов, гематом и гемартрозов. Последние, даже если и случаются намного реже, чем при гемофилии, являются не менее опасными и ведут к инвалидизирующим последствиям. Из всех недостатков плазматических факторов, недостаток Ф. VII вызывает чаще всего геморрагии на уровне ЦНС. (у детей).
Возникновение кровотечений при гипопроконвертинемии редко бывает спонтанным. Обычно они посттравматические и их суровость зависит непосредственно от тяжести травмы. Хирургические вмешательства вызывают у этих больных особенно тяжелые геморрагии, которые ставят их жизнь под угрозу.
Эволюция и осложнения гипопроконвертинемии. Эволюция парагемофилии Александера менее благоприятная, чем при болезни Оврена. Бывают экзитусы по поводу тяжелых геморрагии при рождении (отсечение пуповины или менингеальные кровотечения во время схваток), частота которых достигает около 20%. У взрослого смертность по поводу церебральных геморрагии — редчайшее явление; зато гемартрозы оставляют больным такую степень инвалидности, которая если и не настолько суровая как при гемофилии, все же является опасным осложнением.
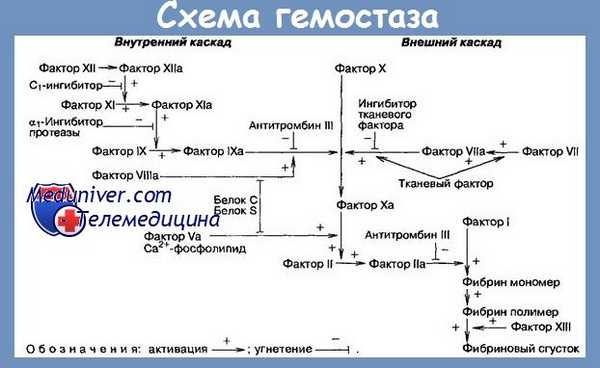
Лабораторное исследование при гипопроконвертинемии показывает:
а) Тесты с аномалийными результатами:
1) TQ значительно продлен. Мы замечаем, что сокращение Ф. VII в плазме влияет на результат TQ в гораздо большей мере, чем сокращение Ф. V; поэтому цифры TQ в случаях гипопроконвертинемии больше, чем цифры при гипопроакцелеринемии. Коррекция TQ может производиться с нормальной сывороткой (24-часовой), но не с нормальной плазмой адсорбированной на SО4Ba.
2) Тест ТФ VII + ТФ X дает очень удлиненный результат. Это специфический тест для этого заболевания (но только в сопоставлении с Т. Stypven-цефалина, который дает нормальный результат). Тест указывает и плазматический уровень Ф. VII, который при этом заболевании падает ниже 5% у больных (гомозиготов) и находится между 30 и 50% у здоровых носителей порока (гетерозиготов).
б) Тесты с нормальными результатами: число тромбоцитов, В.К., капиллярная резистентность, ретракция сгустка, ТС, ТН, РТТ, РТТК, толерантность к гепарину, Т.Ф. II, Т.Ф. V, Т. Stypevn-цефалина, ТСР, TGT (во всех трех вариантах), Т. тромбина и TEG — все дают нормальные результаты.
Нормализация in vitro тестов с патологическими результатами получается путем коррекции с PPSB или с сывороткой нормального лица.
Клинический положительный диагноз гипопроконвертинемии является предположительным и основывается на данных анамнеза и симптоматологии. Лабораторный диагноз достоверный, причем ключевым тестом является Т.Ф. VII + X.
Дифференциальная диагностика производится по отношению тех же заболеваний, как и при недостатке Ф. II.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Гипопроакцелеринемия - история изучения, причины
Гипопроакцелеринемия — геморрагический синдром, возникающий благодаря недостатку Ф.V., который характеризуется в клиническом отношении суровыми провоцированными геморрагическими явлениями. Этот синдром, выделенный из группы геморрагических диатезов с удлиненным TQ, обособил в 1947 г. Owren, который дал ему название парагемофилии, благодаря сходству клинической картины с той, которая наблюдается при гемофилии.
Различаются две формы гипопроакцелеринемии: врожденная и приобретенная. Форма, которую описал Owren (носящая его имя) — врожденная форма. Это редкое заболевание, число опубликованных до сих пор случаев достигает 80. Частота заболеваемости — 0,05/100 000. Приобретенная дефицитность менее серьезная, но зато встречается более часто. Она может быть единичной или ассоциированной с недостатком других плазматических факторов коагуляции: Ф.II, VII, VIII, IX, X.
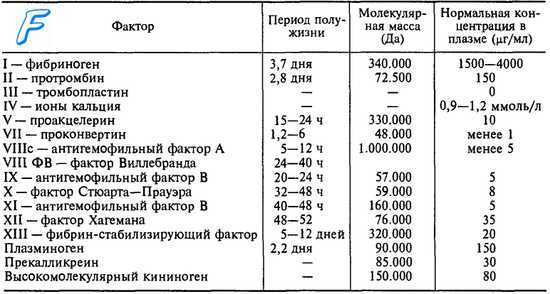
Патофизиология гипопроакцелеринемии. Основное расстройство врожденной формы — недостаток синтеза Ф.V. У нормального лица этот синтез имеет место в печени и индуцируется парой генов, находящихся на паре соматических хромозомов. У больных дефект состоит в ингибиции этих генов, что имеет своим результатом чрезвычайно слабый синтез Ф.V. (5%), ниже минимального уровня обеспечивающего гемостаз (20%); в результате происходит весьма суровый геморрагический диатез.
Генетическая автосомально-рецессивная передача, как и ее фенотипичные проявления, повидимому идентичны тем, которые встречаются при врожденной гипопротромбинемии (ограниченная казуистика не позволяет более категоричное утверждение). Существуют и случаи гомозиготизма (выражающиеся клинически в виде болезни) и случаи гетерозиготизма (здоровые носители порока более многочисленные чем первые, с Ф. VII между 20 и 50%). Порок бывает обычно семейным; как девочки, так и мальчики затронуты в одинаковой мере.
Приобретенная форма встречается в следующих ситуациях:
а) У оперированных больных, в течение первой постоперационной недели, с максимальной интенсивностью на 3—5 дни. Она происходит благодаря, с одной стороны, потере Ф. V в течение внутриоперационного кровотечения, а с другой стороны, расходу Ф. V в течение осуществления гемостаза.
б) У облученных больных (особенно тех, которым назначается Р32), имея в виду, что синтез Ф. V заторможен ионизирующей радиацией, которая к тому же увеличивает и расход Ф. V в циркуляции. Из всех плазматических факторов коагуляции, Ф. V самый чувствительный к этой агрессии.
в) У больных с геморрагической формой скарлатины.
г) В ассоциации с недостатком Ф. II, Ф. VII, Ф. IX, Ф. X, мы встречаем приобретенную форму при заболеваниях печеночной паренхимы, острых лейкемиях, в конечной стадии карцином, суровых питательных анемиях, спруэ.
д) В ассоциации с недостатком Ф. VIII, мы встречаем эту форму при острых стадиях ДВС. и при остром фибринолотическом синдроме.
ГИПОПРОКОНВЕРТИНЕМИЯ
ГИПОПРОКОНВЕРТИНЕМИЯ (греч, hypo- +проконвертин +греч. haima кровь) — семейно-наследственная форма геморрагического диатеза, обусловленная дефицитом проконвертина (фактора VII).
Фактор VII — белок, мигрирующий при электрофорезе между альфа- и бета-глобулинами. Стабилен при хранении, не утилизируется в процессе свертывания крови, а поэтому обнаруживается и в плазме, и в сыворотке. Синтезируется в печени. Период полураспада in vivo составляет 4—6 час. (см. Проконвертин).
Заболевание впервые описал Александер (В. Alexander, 1951). Оврен (P. A. Owren, 1952) доказал его семейно-наследственный характер.
Статистика болезни не разработана. Однако, судя по данным литературы, заболевание встречается относительно редко. Болеют лица обоего пола.
Содержание
Этиология
Заболевание связывают с патол, геном, расположенным на аутосоме и ответственным за недостаточный синтез проконвертина. Наследование дефицита проконвертина является рецессивным и не связано с полом.
Описаны геморрагические диатезы, обусловленные наличием патологического фактора VII, со структурными изменениями его молекулы.
Г. может быть и вторичной. Она возникает при заболеваниях, протекающих с поражением печени, сопровождающихся снижением ее функций, в частности при нарушении синтеза факторов свертывания (тяжелые формы острых гепатитов, циррозы, лейкозы). При этих состояниях, как правило, определяется недостаточность и других факторов свертывания (фибриногена, протромбина, Ас-глобулина и т. д.). Низкий уровень проконвертина наблюдается при лечении антикоагулянтами непрямого действия — дикумарином и его производными.
Патогенез
При Г. любой этиологии нарушается вторая фаза свертывания крови — образование тромбина, поскольку фактор VII принимает участие во внешнем механизме свертывания крови, способствуя (вместе с тканевым тромбопластином, фактором V и ионами кальция) активации фактора X, превращающего протромбин в тромбин (см. Свертывающая система крови).
Патологическая анатомия
Морфол. изменения обусловлены явлениями постгеморрагической анемии: малокровие внутренних органов и тканей.
Клиническая картина
Клиническая картина проявляется только у гомозиготных носителей патол, гена, у гетерозиготных носителей Г. выявляется лишь при лабораторном исследовании крови (нек-рым удлинением протромбинового времени и уменьшением содержания VII фактора).
Заболевание начинается обычно с первых дней или недель жизни, реже — в подростковом периоде. П. Шеваллье и Ж. Бернар различают соответственно две формы Г.— раннюю и позднюю. Ранняя Г. характеризуется более тяжелым течением, начинаясь пупочным кровотечением, поздняя Г. протекает более спокойно, нередко проявляясь при наступлении первых менструаций, переходящих в профузные маточные кровотечения. При достижении больными Г. зрелого возраста кровоточивость исчезает (но дефицит фактора VII сохраняется) .
Клинически Г. выражается симптомами повышенной кровоточивости: кровоизлияния в кожу, слизистые оболочки, суставы, мышцы, носовые и жел.-киш. кровотечения, меноррагии. При очень низком содержании в крови фактора VII описаны смертельные кровоизлияния в мозг. Однако проявления кровоточивости не всегда адекватны содержанию проконвертина в крови. У некоторых больных при выраженной Г. даже большие хирургические вмешательства не осложняются кровотечением в послеоперационном периоде.
Клин, проявления геморрагического диатеза, обусловленного наличием патол, фактора, не отличаются от симптомов Г.
Клиника вторичной Г. характеризуется появлением кровоточивости на фоне проявлений основного заболевания; Г. при лечении антикоагулянтами непрямого действия проявляется возникновением геморрагий, первым признаком которых может быть гематурия.
Осложнения связаны с кровоизлияниями в суставы. Возникающие гемартрозы (см.) способствуют деформации суставов и развитию тугоподвижности в них. В отдельных случаях образуются обширные гематомы со сдавлением нервных стволов и крупных сосудов. Иногда гематомы нагнаиваются с развитием симптомов интоксикации и сепсиса. При обширных кровоизлияниях в брюшную полость может развиться картина острого живота (см.).
Диагноз
Диагноз основывается на данных анамнеза: признаки повышенной кровоточивости у других членов семьи (как мужского, так и женского пола); клин, признаках болезни и данных лабораторных исследований.
Для выявления дефицитного фактора проводят: определение протромбинового времени по Квику; определение протромбинового времени при добавлении 0,1 объема «старой» плазмы или сыворотки; определение потребления протромбина; исследование парциального тромбопластинового времени.
При Г. удлинено протромбиновое время, к-рое нормализуется после добавления «старой» плазмы и сыворотки. Потребление протромбина и парциальное тромбопластиновое время нормальны.
При подозрении на Г., обусловленную наличием патол, фактора VII, необходимо дополнительно исследовать содержание фактора VII иммунол. методом.
При вторичных Г. одновременно выявляется недостаточность и других факторов свертывания.
Дифференциальный диагноз должен проводиться с геморрагическими диатезами, обусловленными дефицитом других факторов протромбинового комплекса (факторы II, V и X). С этой целью проводят тесты коррекции протромбинового времени: к плазме больного добавляется 0,1 объема специально приготовленных сред (нормальная плазма; плазма, адсорбированная BaSO4; плазма длительных сроков хранения; сыворотка). Определяют также потребление протромбина, тест генерации тромбопластина и парциальное тромбопластиновое время.
При дефиците фактора II (протромбина) протромбиновое время нормализуется после добавления нормальной и «старой» плазмы и не нормализуется после добавления сыворотки, т. к. фактор II стабилен при хранении и потребляется во время свертывания. Потребление протромбина и тест генерации тромбопластина не нарушены. Парциальное тромбопластиновое время удлинено, оно нормализуется добавлением нормальной плазмы.
При недостаточности фактора V (Ас-глобулина) нормализация протромбинового времени достигается добавлением нормальной плазмы и плазмы, адсорбированной BaSO4. Старая плазма и сыворотка не восполняют дефицита, к. фактор V быстро разрушается при хранении и потребляется в процессе свертывания. Парциальное тромбопластиновое время удлинено, оно нормализуется добавлением нормальной плазмы и плазмы, адсорбированной BaSO4.
При снижении уровня фактора X одновременно с удлинением протромбинового времени, к-рое нормализуется при исследовании со свежей и «старой» плазмой и сывороткой, выявляются нарушения в I фазе свертывания крови (потребление протромбина и тест генерации тромбопластина). Последние корригируются нормальной сывороткой. Парциальное тромбопластиновое время удлинено, нормализуется добавлением нормальной плазмы и сыворотки.
Лечение
Лечение сводится к остановке возникших кровотечений путем переливания сред, содержащих фактор VII (кровь, плазма, сыворотка, PPSB и др.), до повышения уровня фактора VII до 5—15% нормы. Переливания, учитывая короткий период полураспада дефицитного фактора, должны производиться каждые 4— 8 час. до прекращения кровотечений. При умеренных кровотечениях и при небольших хирургических вмешательствах производят трансфузии крови или плазмы. При необходимости массивных трансфузий (при обильных жел.-киш. кровотечениях, больших хирургических вмешательствах) применяют концентраты, содержащие факторы II, VII, IX и X (напр., PPSB).
Клинический эффект от переливания держится обычно в течение трех недель, хотя лабораторно дефицит фактора вновь обнаруживается через несколько часов.
Применение витамина В12 при наследственной Г. не повышает уровня проконвертина, использование этого препарата в комплексе леч. мероприятий не усиливает их эффективности.
Кортикостероидные гормоны также не способствуют купированию проявлений кровоточивости при этом заболевании.
Ингибиторы фибринолиза (ε-АКК, РАМВА и др.) могут повышать действие переливаемых сред, особенно при применении их после хирургических вмешательств.
Местные средства — тампоны с фибринной губкой, тромбином, змеиным ядом Расселла — эффективны только в сочетании с трансфузиями плазмы и крови.
Лечение вторичной Г. сводится к применению средств, положительно влияющих на основной процесс, а в случае развития геморрагий — к проведению активной трансфузионной терапии.
При возникновении Г. на фоне лечения антикоагулянтами непрямого действия следует немедленно прекратить введение этих препаратов, назначить витамин К и рутин (до 0,1 г в сутки).
Прогноз
Поскольку Г. имеет наследственный характер, полного выздоровления не наступает. Трудоспособность больных в значительной мере зависит от частоты, длительности и локализации геморрагий. Прогноз ухудшается при кровоизлияниях в головной мозг с выраженными неврол, симптомами. Прогноз вторичной Г. зависит прежде всего от течения основного заболевания.
Профилактика
Профилактика сводится к созданию охранительного режима с максимальным исключением внешних воздействий (резких физ. напряжений, ушибов и других травм).
Целесообразны профилактические трансфузии, которые показаны при тяжелом течении Г. с частыми и интенсивными случаями кровоточивости или при обострении.
Большое значение имеют медикогенетические консультации, ориентирующие супругов из семей с наследственной недостаточностью фактора VII в отношении возможности рождения больного Г. ребенка.
Библиография: Кассирский И. А. и: Алексеев Г. А. Клиническая гематология, М., 1970; ЛориеЮ.И.иОрло-в а Л. Д. Геморрагический диатез, вызванный дефицитом VII фактора (проконвертина), Пробл, гематол. и перелив, крови, т. 6, № 3, с. 22, 1961, библиогр.; Alexander а. о. Congenital SPGA deficiency, J. clin. Invest., v. 30, p. 59§, 1951; Falter M. J. a. K a u f m a n M. F. Congenital factor VII deficiency, J. Pediat., y. 79, p. 298, 1971; Goodnight S. H. a. o. Factor VII antibody — neutralizing material in hereditary and acquired factor VII deficiency, Blood, v. 38, p. 1, 1971; Hall C. A. a. o. A clinical and family study of hereditary proconvertin (factor VII) deficiency, Amer. J. Med., v. 37, p. 172, 1964, bibliogr.; Quick A. J. Hemorrhagic; diseases and thrombosis, Philadelphia, 1966; Seeler R. A. Congenital hypo-prothrombinemias, deficiency of factors II „ VII and X, Med. Clin. N. Amer., v. 56, p. 127, 1972, bibliogr.; Strauss H. S. Surgery in patients with congenital factor VII deficiency (congenital hypoproconver-tinemia), Blood, v. 25, p. 325, 1965, bibliogr.
Гипопроконвертинемия - история изучения, причины
Гипопроконвертинемия — геморрагический синдром, возникающий по поводу недостатка синтеза Ф. VII, характеризующийся в клиническом отношении тяжелыми провоцированными геморрагиями. Существуют врожденная форма и приобретенная форма.
Обособленная с нозологической точки зрения в рамках группы геморрагических диатезов с удлиненным TQ, врожденная форма считалась весьма частым заболеванием; в дальнейшем было установлено, что под названием гипопроконвертинемии существовало в дейтсвительности два порока, которые разделились на: недостаток Ф. VII и недостаток Ф. X.
После переоценки случаев в свете вышеуказанного, констатировалось, что это заболевание возникает намного реже, причем частота его равняется 0,1/100 000. Среди животных это заболевание встречается у собак породы Бигль ("Beagle").
Первый случай описал и изучил Alexander в 1951 г., откуда данному заболеванию было присвоено имя «парагемофилия Александера»; до 1979 г. в литературе опубликовано 85 случаев.
Чистая приобретенная форма до сих пор не упоминалась в литературе по специальности. Сообщается лишь о случаях приобретенного недостатка Ф. VII в ассоциации с недостатком Ф. II, IX и X, в обстоятельствах упомянутых при недостатке Ф. II (авитаминоз К) и недостатков Ф. II, V, IX, X при заболеваниях печеночной паренхимы.
Патофизиология гипопроконвертинемии
Основное расстройство гипопроконвертинемии — недостаток синтеза Ф. VII, который у нормального лица выполняется гепатоцитами и регулируется парой генов, находящихся на соматических хромозомах. У больных, ингибиция этих генов имеет своим результатом недостаточный синтез Ф. VII, ниже необходимого уровня для гемостаза (25%), что порождает геморрагический диатез.
Генетическая передача автосомально-рецессивного характера и ее фенотипические выражения подобны тем, которые существуют при недостатке Ф. V. Этот недостаток обычно семейный; он затрагивает в одинаковой мере и мальчиков и девочек.
Гомозиготы, благодаря затронутости обоих генов (Ф. VII = 0—5%) представляют клинические признаки болезни; гетерозиготы, благодаря затронутости только одного гена (Ф. VII = 30—50%) являются здоровыми носителями порока и выявляются лишь по случаю лабораторного исследования.
Приобретенная форма, в сочетании с недостатками других плазматических факторов коагуляции, встречается в этиопатогенетических обстоятельствах, подобных тем, которые мы наблюдаем при недостатке Ф. П.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Читайте также:
- Избегающее/ограничивающее расстройство потребления пищи
- Профилактика отравлений ядохимикатами в сельском хозяйстве
- Сужение левого предсердно-желудочкового отверстия. Митральный стеноз
- Диагностика бронхолегочного аспергиллеза. Лечение бронхолегочного аспергиллеза
- Лучевая диагностика химического ожога пищевода (эзофагита от химического воздействия)