Легочный альвеолярный протеиноз
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Болезнь, как правило, поражает людей в возрасте 20-60 лет.
В редких случаях ткань легких замещается грубой фиброзной тканью. Болезнь может прогрессировать, оставаться стабильной или проходить без лечения.
Когда альвеолы заполнены жидкостью, легкие не в состоянии снабжать кровь кислородом. Следовательно, большинство людей, страдающих этим заболеванием, испытывает одышку при физической нагрузке. У некоторых тяжелое затруднение дыхания развивается даже в покое. У большинства больных имеется кашель, обычно без мокроты, если они не курильщики.
При появлении одышки, быстрой утомляемости, кашля со скудной мокротой желтоватого цвета, цианоза кожных покровов, боли в груди.
Причины точно не известны. Существуют несколько теорий относительно происхождения альвеолярного протеиноза:
- Нарушение экзокринной функции альвеолоцитов с гиперпродукцией белково-липидного секрета.
- Снижение функциональной активности макрофагов и, как следствие, недостаточная эвакуация секрета.
- Гиперпродукция сурфактанта.
- Генетическая патология («болезнь накопления»).
- Экологические факторы.
- Профессиональные вредности.
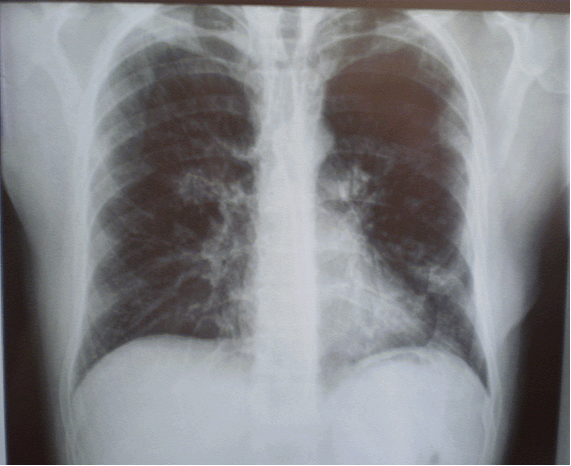
При рентгенологическом исследовании грудной клетки можно обнаружить очаговое затенение в обоих легких. Исследования функции легких показывают, что объем воздуха, который они могут вместить, снижен. Анализы крови выявляют низкий уровень кислорода в крови – сначала только во время физической нагрузки, а позже и в покое.
Для установления диагноза исследуют жидкость из альвеол. Чтобы получить образцы клеток из бронхов, врач с помощью бронхоскопа делает смыв с бронхов раствором соли. Иногда во время бронхоскопии производят биопсию (врач берет кусочек ткани легкого для микроскопического исследования). Если необходимо взять большее количество ткани, это можно сделать во время хирургической операции.
Когда симптомов нет или они незначительны, лечения не требуется. При появлении симптомов богатую белками жидкость из альвеол удаляют во время бронхоскопии путем промывания раствором соли. Иногда достаточно промывания лишь небольшой части легких, но, если симптомы тяжелые и уровень кислорода в крови низкий, дают наркоз, чтобы можно было промыть все легкое. Через 3-5 дней процедуру повторяют на другом легком, снова под наркозом. Для некоторых пациентов бывает достаточно одной подобной процедуры, в то время как другие нуждаются в них каждые 6-12 месяцев в течение многих лет.
Люди с легочным альвеолярным протеинозом часто страдают постоянной одышкой, но болезнь редко приводит к смерти, если указанные процедуры проводить регулярно. Эффективность других видов лечения, например использования йодида калия и ферментов, которые расщепляют белки, остается до конца не изученной. Кортикостероиды (гормональные препараты) не дают результатов и могут лишь увеличить опасность развития инфекционных осложнений.
Альвеолярный протеиноз
Альвеолярный протеиноз — заболевание неизвестной этиологии, характеризующееся умеренно прогрессирующей одышкой вследствие накопления в альвеолах продуктов обмена сурфактанта – поверхностно-активного вещества, предотвращающего повреждение альвеол. Болезнь была впервые описана S.H. Rosen и соавт. в 1958 г.
Альвеолярный протеиноз относится к редким заболеваниям; встречается в 1-4 случаях на 1 млн. Болеют преимущественно люди среднего возраста (20-50 лет), мужчины чаще, чем женщины (в соотношении 3:1).
![]()
Выделяют следующие формы:
- врождённая форма обусловлена мутациями генов, кодирующих структуру белков сурфактанта;
- приобретённая (или идиопатическая, т.е. с неустановленной причиной) форма (90% от общего числа заболевших, в 70% это курильщики);
- вторичный альвеолярный протеиноз (псевдопротеиноз) развивается как следствие вирусных инфекций; пневмоцистной пневмонии у больных гемобластозами; на фоне воздействия неорганической (в том числе асбестовой) пыли; токсических паров (особенно при производстве пластмасс); воздействия озона.
Патогенез
Ключевую роль в патогенезе заболевания играет нарушение обмена сурфактанта. Сурфактант — белково-липоидный комплекс, снижающий поверхностное натяжение в альвеолах. 80% его составляют фосфолипиды, 10% — холестерол и 10% — белки. Липиды и белки сурфактанта синтезируются альвеолоцитами. Инактивируется путём перехода в поверхностно-неактивные формы, основная часть которых (до 80%) реутилизируется альвеолоцитами. Остальная часть сурфактанта катаболизируется альвеолярными макрофагами. Нарушение регуляции процесса инактивации и утилизации составляющих сурфактанта ведёт к избыточному накоплению его в альвеолах, что приводит к патологии газообмена.
Клиническая картина
В течение длительного времени заболевание протекает бессимптомно. Его нередко случайно выявляют при профилактическом флюорографическом исследовании. Основной клинический признак болезни — медленно прогрессирующая одышка, которая может сопровождаться сухим или со скудной мокротой кашлем, субфебрильной температурой, болью в груди, похуданием, быстрой утомляемостью. Кровохарканье отмечается редко.
Течение болезни, чаще всего, хроническое. По мере прогрессирования усиливается цианоз, формируются «пальцы Гиппократа» (симптом барабанных палочек), похудание. Осложнения - присоединение бактериальной или грибковой инфекции, развитие лёгочной гипертензии и формирование лёгочного сердца. Туберкулёз осложняет течение альвеолярного протеиноза в 3-5% случаев.
Трудности диагностики обусловлены отсутствием специфических клинических признаков. От начала заболевания до установления диагноза нередко проходит около 3 лет. 40% больных первоначально ставят диагноз двусторонней пневмонии в отсутствии жалоб. Половине больных диагностируют туберкулёз и назначают противотуберкулёзную терапию.
Инструментальные исследования:
- рентгенография легких,
- компьютерная томография (МСКТ) грудной клетки,
- видеобронхоскопия,
- БАЛ - бронхоальвеолярный лаваж,
- микроскопия, иммуногистохимия жидкости, полученной при проведении БАЛ,
- лабораторная диагностика,
- для верификации диагноза показана биопсия лёгочной ткани.
Лечение и прогноз
При альвеолярном протеинозе назначение антибактериальных препаратов, глюкокортикостероидов, иммуносупрессантов нецелесообразно в связи с их неэффективностью. Единственный эффективный метод лечения больных — лечебный бронхоальвеолярный лаваж (БАЛ), применяемый с 1965 г. (промывание легкого стерильным изотоническим раствором с добавлением ферментов под наркозом). После лечения у пациентов уменьшается одышка, улучшаются показатели функции внешнего дыхания и газов крови, определяется положительная динамика изменений в лёгких на рентгенограммах и КТ. Прогноз благоприятный: течение болезни доброкачественное, медленно прогрессирующее. Спонтанная ремиссия наблюдается в 10-30% случаев. Летальные исходы обусловлены, прежде всего, прогрессированием дыхательной недостаточности.
Альвеолярный протеиноз легочный
Легочный альвеолярный протеиноз - это заболевание при котором в альвеолах легких накапливаются белково-липидные вещества. Классически, при компьютерной томографии альвеолярный протеиноз проявляется паттерном булыжной мостовой, однако, необходимо помнить, что протеиноз относится к редким заболеваниям при данном неспецифическом паттерне.
Эпидемиология
Легочный альвеолярный протеиноз редкое заболевание, встречается в молодых людей и лиц среднего возраста (в диапазоне 20-50 лет) [6, 7]. Имеется четкая связь с курением, в группе курильщиков у мужчин имеется большая предрасположенность (М:Ж ~ 2:1), у некурящих лиц данная тенденция отсутствует [4]. У пациентов до года заболевание сочетается с тимической алимфоплазией [6].
Клиническая картина
Клинические проявления как правило неспецифичны (диспноэ или минимально продуктивный кашель). Приблизительно треть пациентов не предъявляет жалоб. У детей проявления еще менее специфичны и включают диарею, рвоту, снижение массы тела, цианоз [6]. Симптоматика может быть обусловлена присоединением оппортунистической инфекции. Симптомы включают “барабанные палочки”, хрипы, цианоз.
Патология
Альвеолярные макрофаги являются ключевым звеном в поддержании гомеостаза сурфактанта, их дифференциация регулируются гранулоцитарно-макрофагальным колониестимулирующим фактором (GM-CSF). Приобретенный аутоиммунный ответ к данному полипептидному цитокину считается причиной развития легочного альвеолярного протеиноза у взрослых [4,12].
Маркеры
- повышение уровня воспалительных маркеров, напр. лактатдегидрогеназы (LDH) [4]
- (+) антитела против гранулоцитарно-макрофагального колониестимулирующего фактора (anti-GM-CSF) в бронхоальвеолярном лаваже или плазме [12]
Диагностика
Основное правило - радиологические изменения зачастую более выражены, чем тяжесть клинической картины [6].
Рентгенография
Изменений при рентгенографии не достаточно чтобы выставить диагноз [2]. Проявления варьируют и включают [4,5]:
- инфильтраты в виде крыла летучей мыши (бабочки):
- двусторонние симметричные инфальтраты с щажением верхушек и реберно-диафрагмальных углов
- напоминают по виду отек легких
- чаще встречается у взрослых
- напоминают милиарный паттерн
- чаще встречаются у детей
Плевральный выпот, кардиомегалия и лимфааденопатия не являются характерными признаками для неосложненного легочного альвеолярного протеиноза.
Компьютерная томография
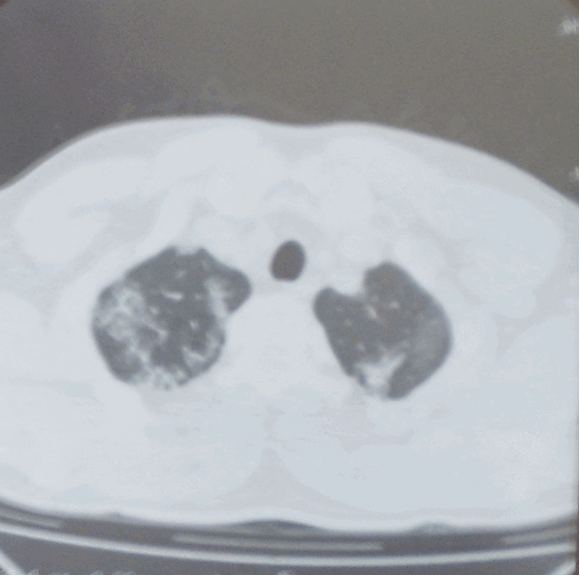
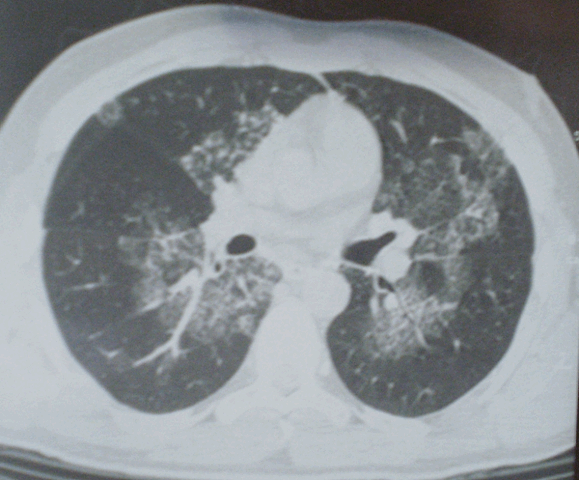
Два овновных признака альвеолярного протеиноза при ВРКТ:
Сочетание этих двух признаков называется паттерном булыжной мостовой, который является характерным, но не патогномоничным признаком. Дополнительно может встречаться консолидация легочной ткани. Изменения в легких носят пятнистое или географическое распределение, с некоторой предрасположенностью к локализации в нижних долях [2].
Признак матового стекла обычно разрешается после лечебного бронхоальвеолярного лаважа, однако утолщение перегородок может сохраняться [10]. Изменения подобные легочному фиброзу не типичны для легочного альвеолярного протеиноза [2], однако несколько чаще встречаются при развитии болезни в неонатальном периоде [12].
Осложнения
- суперинфекция различной этиологии [1] (встречается ≈30%)
Дифференциальный диагноз
Дифференциальный диагноз зависит от доминирующих проявлений:
- , плевральный выпот и кардиомегалия как правило отсутствуют при альвеолярном протеинозе
Перевод публикации - Dan J Bell, Yuranga Weerakkody et al. Pulmonary alveolar proteinosis [13].
Идиопатический альвеолярный протеиноз: клинический случай
Цель. В статье рассмотрены вопросы этиопатогенеза, клиники и диагностики альвеолярного легочного протеиноза на примере клинического случая пациента с альвеолярным протеинозом.
Особенности клинического случая. Представленный случай идиопатического альвеолярного протеиноза (АП) характеризуется минимальными клиническими проявлениями заболевания при выраженных изменениях в легочной ткани по данным лучевых методов исследования органов грудной клетки. Диагноз АП был подтвержден морфологически.
Заключение. Альвеолярный протеиноз представляет собой редкое заболевание легких интерстициальной природы, в основе которого лежит нарушение клиренса сурфактанта с накоплением в просвете альвеол патологического белково-липидного вещества. Выделяют три основных формы АП: врожденный, приобретенный (идиопатический) и вторичный АП. Основа диагностики АП – характерные изменения при мультиспиральной компьютерной томографии органов грудной клетки и данные морфологического исследования.
Введение. Альвеолярный протеиноз (АП) представляет собой редкое заболевание легких интерстициальной природы, в основе которого лежит нарушение клиренса сурфактанта с накоплением в просвете альвеол патологического белково-липидного вещества [1]. Впервые заболевание было описано в 1958 году Rosen et al. [2], и к настоящему времени в литературе опубликовано около 500 клинических случаев [3]. Встречается, как правило, в возрасте 30-50 лет, преимущественно у мужчин.
Этиология и патогенез АП. Выделяют три основных формы АП: врожденный, приобретенный (идиопатический) и вторичный АП [3]. Врожденный АП обусловлен мутациями генов, кодирующих синтез белков сурфактанта типа В или С, либо ?с-цепи рецептора к гранулоцитарно-макрофагальному колониестимулирующему фактору (ГМ-КСФ). Патогенез идиопатического АП связывают с наличием аутоиммунных антител к ГМ-КСФ, приводящих к повреждению либо функциональной недостаточности этого белка [4]; такая форма наиболее часто встречается у лиц молодого возраста. Вторичный АП развивается в результате воздействия этиологических факторов, приводящих к снижению числа и дисфункции альвеолярных макрофагов. К числу этиологических факторов вторичного АП относят: вирусные и бактериальные инфекции (микобактерии, грибы, пневмоцисты), контакт с аэрополлютантами (кремний, алюминий, пластмассы, тяжелые металлы), иммуносупрессивные состояния, в том числе лекарственно-обусловленные, гемобластозы (лейкозы, лимфомы) и др. [5].
Общепринятая концепция патогенеза АП заключается в следующем: под влиянием этиологических факторов в альвеолах появляется воспалительный экссудат, что приводит к активации альвеолярных макрофагов (АМ) и лимфатической системы с целью удаления белково-липидной фракции из альвеол. Одновременно происходит выработка альвеолоцитами II типа избыточного количества измененного сурфактанта, не обладающего поверхностно-активными свойствами. Большие количества сурфактанта поглощаются АМ, что вызывает развитие в них дегенеративных изменений и приводит к снижению их функциональной активности. Нагруженные липидной фракцией макрофаги - один из основных морфологических признаков АП. Вследствие указанных изменений в альвеолах происходит дальнейшее прогрессивное накопление белково-липидного вещества в их просвете. В свою очередь, это усиливает компенсаторную гиперфункцию альвеолоцитов II типа и приводит к еще большей продукции сурфактанта и усугубляет нарушение функции АМ. Таким образом происходит формирование патогенетического «порочного круга».
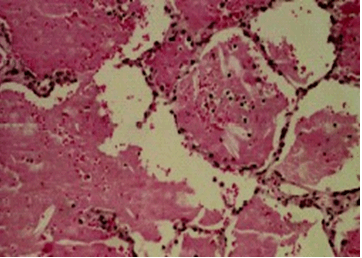
Патоморфологическая картина АП характеризуется следующими особенностями: преимущественное поражение базальных и задних отделов легких (плевра и средостение интактны); наличие на поверхности легких серовато-белых бугорков в виде зерен; наличие в альвеолах и бронхиолах больших количеств эозинофильного гранулярного ШИК-положительного белково-липидного вещества; наличие в просвете альвеол нагруженных липидной фракцией АМ («пенистые» макрофаги); гиперплазия и гипертрофия альвеолоцитов II типа [6].
Помимо вышеизложенного, одним из универсальных механизмов патогенеза всех форм АП является нарушение функции АМ, обусловленное дефицитом ключевого регулятора липидного обмена – рецептором активации пролиферации пероксисом ? (peroxisome proliferator-activated receptor ?) [7]. Недостаточность этого фактора приводит к повышению уровня холестерина и его метаболитов в бронхоальвеолярном лаваже [8]. Недостаточность этого фактора может быть скорректирована с помощью заместительной терапии ГМ-КСФ [9]. Интересно, что протеинозоподобные патогистологические изменения в ткани легких наблюдаются у мышей, в геноме которых отсутствует ген, кодирующий синтез ГМ-КСФ [9, 10], а ингаляционное введение этого биологически активного вещества уменьшает выраженность подобных изменений [11, 12].
При АП наибольшим изменениям подвергается система сурфактанта. Сурфактант продуцируется альвеолоцитами II типа и состоит на 90% из липидной фракции и на 10% - из белковой, а также из небольшого количества углеводных цепей. Приблизительно 80-90% липидной фракции представлено фосфолипидами, оставшуюся часть составляют холестерин, триацилглицерол и ненасыщенные жирные кислоты [13]. Изучение процессов синтеза, репарации и биодеградации сурфактанта в настоящее время является одним из приоритетных направлений пульмонологических исследований. Описано два основных пути катаболизма сурфактанта [14]: первый заключается в реализации полного цикла рециркуляции веществ, образующих сурфактант, в альвеолоцитах II типа; второй подразумевает обеспечение клиренса сурфактанта с помощью его фагоцитоза и биодеградации в альвеолярных макрофагах [15]. Регуляция этих путей осуществляется путем взаимодействия целого ряда цитокинов, в первую очередь, ГМ-КСФ, реализующего свои эффекты через соответствующие рецепторы [16]. Любые нарушения синтеза либо реализации действия этого фактора неизбежно приводят к развитию патологического процесса в альвеолах, обусловленного накоплением избыточного количества липидно-белковой субстанции, что лежит в основе развития и прогрессирования врожденного и идиопатического АП. Также немаловажную роль играют интерлейкины (ИЛ) (ИЛ-10, ИЛ-12 и ИЛ-18)[17], которые регулируют синтез ГМ-КСФ, механизмы локального фагоцитоза и синтеза антител и интерферонов [18]. Так формируется вторичный иммунологический дефект, обуславливающий присоединение вторичной инфекции и ухудшение прогноза у больных АП, что требует проведения первичной и вторичной антибактериальной и противовирусной профилактики.
Диагностика АП включает в себя сбор клинико-анамнестических данных (выявление семейного анамнеза, оценка синдрома дыхательной недостаточности), проведение компьютерной томографии органов грудной клетки и морфологическую верификацию [19]. Основным методом лечения АП в настоящее время является бронхоальвеолярный лаваж (БАЛ) [20]. Показано, что применение этого метода для симптоматической терапии АП дает стойкую ремиссию заболевания длительностью от нескольких месяцев до нескольких лет [20]. Также патогенетически обоснованными методиками терапии являются введение ГМ-КСФ (как парентерально, так и ингаляционно) и экстракорпоральные методы лечения, среди которых ведущее место занимают плазмаферез и применение мембранных оксигенаторов [21].
Клинический случай. Больной П., 32 лет, поступил в пульмонологическое отделение НУЗ «ЦКБ №1 ОАО «РЖД» в связи с выявлением при диспансерном обследовании изменений на флюорограмме в виде усиления и деформации легочного рисунка в прикорневых областях обоих легких. Амбулаторно была проведена рентгенография органов грудной клетки, на которой вышеуказанные изменения были подтверждены (рис. 1). Для дообследования и верификации диагноза направлен в стационар.
![]()
Рисунок 1. Рентгенограмма органов грудной клетки больного П.: легочный рисунок диффузно усилен и деформирован на всем протяжении за счет интерстициального компонента
При поступлении жалоб не предъявляет. Ранее проблем со стороны органов дыхания не отмечал. Около двух месяцев назад выявлены вышеописанные изменения на рентгенограмме ОГК, требующие уточнения характера интерстициальных изменений в легких. Клинически значимых хронических заболеваний в анамнезе нет. Наследственность не отягощена. Аллергологический анамнез: пыльца растений – риноконъюнктивит (симптоматическая терапия препаратами антигистаминового ряда). Проживает в районе с повышенной задымленностью (рядом с домом – промышленное предприятие). Профессия – помощник машиниста локомотива, проходил ежегодное медицинское освидетельствование. В анамнезе – табакокурение с индексом 15 пачка-лет. В детском и юношеском возрасте занимался легкой атлетикой, имеет спортивный разряд.
В объективном статусе клинически значимых отклонений от нормы не выявлено.
Общеклинические анализы крови и мочи, биохимический анализ крови – без патологии. По лабораторным данным отмечается повышение IgE до 189,6 Ме/мл (N до 100). АТ к ВИЧ (ИФА) - не обнаружены. HBs-антиген, анти-НСV (ИФА) - не обнаружены. RW- отр.
Исследование газового состава артериальной крови: рН – 7,394, рСО2 – 40,8 мм. рт. ст., рО2 – 86,3 мм. рт. ст. SaO2 – 98% (при дыхании атмосферным воздухом).
Цитологическое исследование биоптата слизистой бронхов: группы клеток цилиндрического эпителия, небольшое число лейкоцитов, макрофаги, слизь.
Данные инструментальных исследований: ЭКГ, ФВД – значимых отклонений от референсных значений не выявлено.
Фибробронхоскопия: картина двухстороннего эндобронхита.
Компьютерная томография органов грудной полости: на серии компьютерных томограмм получены изображения легких и средостения в аксиальной плоскости на уровне вдоха по стандартному протоколу. Легкие обычных размеров и формы, с дополнительной долей непарной вены в правом легком. Легочный рисунок резко деформирован по мелко- и среднепетлистому типу в прикорневых отделах с обеих сторон. Также отмечаются участки и зоны деформации легочного рисунка по мелко- и среднепетлистому типу и по типу «матового стекла» в плащевых отделах легких с обеих сторон – симптом «географической карты». Во всех отделах определяются множественные плевральные сращения. Корни легких структурны, с наличием множественных бронхопульмональных лимфатических узлов (0,2-0,5 см в диаметре) в структуре. Стенки трахеи и бронхов умеренно уплотнены, с наличием мелких бронхоэктазов в задне-базальных отделах слева. Сердце умеренно увеличено в размерах, преимущественно за счет левых отделов. Жидкости в плевральных полостях не обнаружено (рис. 2).
![]()
Рисунок 2а. Компьютерная томограмма органов грудной клетки: изменения легочного рисунка по типу «матового стекла». Множественные плевральные сращения
![]()
Рисунок 2б. Компьютерная томограмма органов грудной клетки: изменения легочного рисунка по типу «матового стекла». Множественные плевральные сращения
С целью морфологической верификации диагноза больному была произведена операция в объеме торакоскопии плевральной полости слева, миниторакотомии слева, краевой аппаратной резекции язычкового сегмента левого легкого.
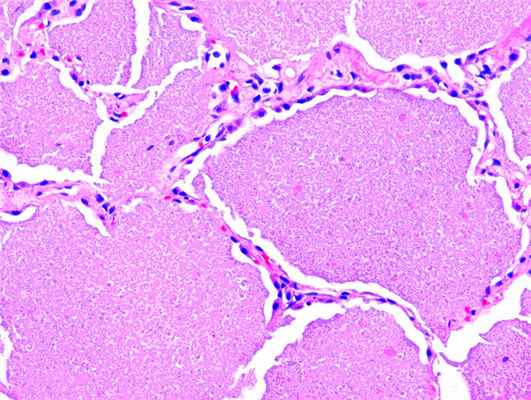
Проведено морфологическое исследование: в микропрепаратах ткань легкого, гистоархитектоника которой не нарушена. В просвете некоторых альвеол определяется эозинофильное гранулярное вещество с кристаллоидными включениями. Альвеолоциты II типа гиперплазированы. В отдельных полях зрения обнаруживается повышенное количество альвеолярных макрофагов. Часто обнаруживаются очаговые лимфоцитарные инфильтраты. Заключение: гистологическая картина характерна для легочного альвеолярного протеиноза (рис. 3).
![]()
Рисунок 3. Гиперплазия стенки бронхиол и скопление аморфного эозинофильного материала в просвете альвеол, мелкоточечные геморрагии (гематоксилин-эозин, х200)
Заключение. Таким образом, на основании данных компьютерной томографии органов грудной полости и данных морфологического исследования был сформулирован клинический диагноз: идиопатический легочный альвеолярный протеиноз, состояние после торакоскопии плевральной полости слева, миниторакотомии слева, краевой аппаратной резекции язычкового сегмента левого легкого. ДН 0 ст.
- отсутствие клинических проявлений заболевания (одышка, кашель, и др.);
- нетипичная для АП локализация поражения: по данным КТ ОГК отмечается поражение не только задне-базальных, но и верхних отделов легких (рис 2). Помимо этого, визуализируется поражение плевры в виде множественных плевральных сращений, что не характерно для АП.
- отсутствие показаний для проведения бронхоальвеолярного лаважа (в первую очередь, признаков прогрессирующей дыхательной недостаточности) обусловило дальнейшую тактику ведения пациента: рекомендовано наблюдение в динамике и проведение БАЛ при ухудшении клинического статуса.
Список литературы:
1. Kitamura T., Tanaka N., Watanabe J., Uchida K., Kanegasaki S., Yamada Y., Nakata K. Idiopathic pulmonary alveolar proteinosis as an autoimmune disease with neutralizing antibody against granulocyte/macrophage colony-stimulating factor. J Exp Med 1999; 190:875-80.
2. Rosen S.H., Castleman B., Liebow A.A. Название статьи? N Engl J Med 1958; 258: 1123-42.
3. Ceruti M., Rodi G., Stella G.M., Adami A., Bolongaro A., Baritussio A., Pozzi E., Luisetti M. Successful whole lung lavage in pulmonary alveolar proteinosis secondary to lysinuric protein intolerance: a case report. Orphanet Journal of Rare Diseases 2007;1750-7.
4. Seymour J.F., Presneill J.J. Pulmonary alveolar proteinosis (progress in the first 44 years). Am J Respir Crit Care Med 2002; 166:215-35.
5. Xie L., Zhao T., Wang Q., Chen L., Li A., Wang D., Qi F., Liu Y. Secondary pulmonary alveolar proteinosis associated with myelodisplastic syndrome. Clin Med J 2007; 120 (12):1114-6.
6. Vourlekis J.S., Greene K.E. Pulmonary alveolar proteinosis. Interstitial Lung Disease 2006; 865-76.
7. Bonfield T.L., Farver C.F., Barna B.P., Malur A., Abraham S., Raychaudhuri B., Kavuru M.S., Thomassen M.J. Peroxisome proliferator-activated receptor-gamma is deficient in alveolar macrophages from patients with alveolar proteinosis. Am J Respir Cell Mol Biol 2003; 29: 677-82.
8. Meaney S., Bonfield T.L., Hansson M., Babiker A., Kavuru M.S., Thomassen M.J. Serum cholestenoic acid as a potential marker of pulmonary cholesterol homeostasis: increased levels in patients with pulmonary alveolar proteinosis. J Lipid Res 2004; 45:2354-60.
9. Stanley E., Lieschke G.J., Grail D. еt al.. Granulocyte/macrophage colony-stimulating factor-deficient mice show no major perturbation of hematopoesis but develop a characteristic pulmonary pathology. Proc Natl Acad Sci USA 1994; 91:5592-6.
10. Dranoff G., Crawford A.D., Sadelain M. еt al. Involvement of granulocyte-macrophage colony-stimulating factor in pulmonary homeostasis. Science 1994; 264:713-6.
11. Huffman J.A., Hull W.M. Dranoff G., Mulligan R.C., Whitsett J.A. Pulmonary epithelial cell expression of GM-CSF corrects the alveolar proteinosis in GM-CSF-deficient mice. J Clin Invest 1996; 97:649-55.
12. Reed J.A., Ikegami M., Cianciolo E.R. еt al. Aerosolized GM-CSF ameliorates pulmonary alveolar proteinosis in GM-CSF-deficient mice. Am J Physiol 1999; 276: L556-L63.
13. Akino T. Lipid components of the surfactant system. In: Pulmonary surfactant. B. Robertson, L.M.G. van Golde and J.J. Batenburg, editors. 1992. Elsevier, Amsterdam. 19-31.
14. Hawgood S., Poutain F.R. The pulmonary collectins and surfactant metabolism. Annu Rev Physiol 2001; 63:495-519.
15. Trapnell B.C., Whitsett J.A., Nakata K. Pulmonary alveolar proteinosis. N Engl J Med 2003; 349:2527-39.
16. Trapnell B.C., Whitsett J.A. GM-CSF regulates pulmonary surfactant homeostasis and alveolar macrophage-mediated innate host defense. Annu Rev Physiol 2002; 64:775-802.
17. Yoshida M., Whitsett J.A. Interaction between pulmonary surfactant and alveolar macrophages in pathogenesis of lung disease. Cell Mol Biol 2004; 50:OL639 – OL648.
18. Wylam M.E., Ten R., Prakash U.B., Nadrous H.F., Clawson M.L., Anderson P.M. Aerosol
granulocyte-macrophage colony-stimulating factor for pulmonary alveolar proteinosis. Eur Respir J 2006; 27:585-93.
19. Shah P.L., Hansell D., Lawson P.R. et al. Pulmonary alveolar proteinosis: clinical aspects and current concept on pathogenesis. Thorax 2000; 55:67-77.
20. Dexter M.E., Cosgrove G.P., Douglas I.S. Managing a rare condition presenting with intractable hypoxemic respiratory failure. Chest 2007; 131:320-7.
21. Spock A. Long-term survival of pediatric patients with pulmonary alveolar proteinosis treated with lung lavage. Eur Respir J 2005; 25:1127.Легочный альвеолярный протеиноз
Легочный альвеолярный протеиноз представляет собой накопление сурфактанта в альвеолах. Этиология почти всегда неизвестна. Заболевание проявляется одышкой, недомоганием и усталостью. Диагноз ставится на основании результатов исследования промывных вод бронхоальвеолярного лаважа, хотя имеются характерные рентгенологические и лабораторные изменения. Лечение с помощью полного лаважа легких или, в некоторых случаях, рекомбинантный гранулоцитарный колониестимулирующий фактор макрофагов. На фоне лечение 5-летняя выживаемость составляет около 80%.
Этиология легочного альвеолярного протеиноза
Легочный альвеолярный протеиноз чаще всего является идиопатическим и встречается у здоровых мужчин и женщин в возрасте 30–50 лет. Редкие вторичные формы встречаются у больных острым силикозом Силикоз Причиной силикоза является вдыхание свободного кристаллического кремния, которое приводит к развитию узлового легочного фиброза. Хронический силикоз первоначально не вызывает никаких симптомов. Прочитайте дополнительные сведения Pneumocystis jiroveciiПневмония, обусловленная Пневмоциста Каринии Pneumocystis jirovecii – частая причина пневмонии у пациентов с иммунодефицитом, особенно у инфицированных вирусом иммунодефицита человека (ВИЧ) и получающих системные кортикостероиды. Прочитайте дополнительные сведенияСведения о сходстве или различии патофизиологических механизмов идиопатических и вторичных случаев отсутствуют.
Патофизиология легочного альвеолярного протеиноза
Считается, что развитие заболевания связано с нарушением переработки сурфактанта альвеолярными макрофагами вследствие патологического влияния гранулоцитарно-макрофагального колониестимулирующего фактора (ГМ-КСФ), возможно, в результате уменьшения или полного подавления функции общей бета-цепи рецептора ГМ-КСФ/интерлюкин ИЛ-13/ИЛ-5 мононуклеаров (выявляется только у некоторых детей; у взрослых отсутствует). Антитела к ГМ-КСФ были также найдены у большинства пациентов. Предполагается токсическое повреждение легких, но оно не доказано при вторичных ингаляционных причинах, таких как силикопротеиноз.
Альвеолы заполнены бесклеточным липопротеиновым ШИК-положительным сурфактантом. Клетки альвеол и интерстиция – без патологии. Поражаются преимущественно заднебазальные сегменты легкого. Плевра и средостение не вовлекаются.
Симптомы и признаки легочного альвеолярного протеиноза
Большинство пациентов с легочным альвеолярным протеинозом отмечает одышку при физической нагрузке, снижение массы тела, усталость, недомогание субфебрильную температуру. Также может наблюдаться, хотя и менее часто, кашель, иногда с вязкой мокротой. Утолщение концевых фаланг пальцев по типу «барабанных палочек» и цианоз наблюдаются редко. Инспираторные влажные хрипы выявляются редко, поскольку альвеолы заполнены жидкостью; их появление свидетельствует о развитии инфекции.
Диагностика легочного альвеолярного протеиноза
Подозрение на легочный альвеолярный протеиноз впервые возникает при выполнении рентгенографии органов грудной клетки по поводу неспецифических симптомов со стороны дыхательной системы. Рентгенологическое исследование выявляет двусторонние средне- и нижнедолевые затемнения в виде бабочки при нормальной структуре корней легких.
Для установления диагноза выполняют бронхоальвеолярный лаваж. Промывная жидкость молочного цвета или мутная и окрашивается положительно при периодическом окрашивании кислотой по Шиффу (ШИК-реакция). Промывные воды характеризуются наличием макрофагов, перегруженных сурфактантом, увеличением количества Т-клеток и высокой концентрацией апопротеина-А сурфактанта.
Торакоскопическая или открытая биопсия легких выполняется при наличии противопоказаний к бронхоскопии или при неинформативности исследования промывных вод. Исследования, как правило, назначаются до начала лечения
Исследование газового состава артериальной крови (ГАК)
КТ высокого разрешения (КТВР)
Исследование функции легких
При КТВР выявляются затемнения по типу «матового стекла», утолщение интралобулярных структур и междольковые перегородки типичной полигональной формы. Эти признаки не являются специфичными и могут выявляться при остром респираторном дистресс-синдроме, вирусной пневмонии, липоидной пневмонии, бронхоальвеолярном раке и пневмонии, вызванной Pneumocystis jirovecii.
Исследование газового состава артериальной крови позволяет диагностировать гипоксемию при легкой и умеренной физической нагрузке или в покое при более тяжелом течении заболевания.
Изменения в лабораторных показателях включают полицитемию, гипергаммаглобулинемию, увеличение активности лактатдегидрогеназы в сыворотке крови и увеличение сывороточных сурфактантных белков А и D. Изменения позволяют заподозрить заболевание, но не являются диагностическими.
Прогноз при легочном альвеолярном протеинозе
Без лечения легочный альвеолярный протеиноз разрешается самостоятельно у 10% пациентов. 40% пациентов выздоравливают после одной процедуры бронхоальвеолярного лаважа; другим пациентам требуется проведение лаважа каждые 6–12 месяцев в течение многих лет. 5-летняя выживаемость составляет около 80%; наиболее частая причина смерти – дыхательная недостаточность Обзор дыхательной недостаточности (Overview of Respiratory Failure) Острая дыхательная недостаточность – это угрожающее жизни пациента ухудшение оксигенации, вывода углекислого газа или, и того, и другого. Дыхательная недостаточность может быть вызвана нарушением. Прочитайте дополнительные сведения , типично развивающаяся в течение первого года после постановки диагноза. Инфекции требуют соответствующего лечения. Вторичные инфекции легких, вызванные бактериями (Mycobacteria, Nocardia) и другим организмами (например, Aspergillus, Cryptococcus и другими оппортунистическими грибами), развиваются часто и обусловлены нарушением функции макрофагов.
Лечение легочного альвеолярного протеиноза
Лечение не требуется пациентам с легочным альвеолярным протеинозом, не имеющим проявлений заболевания или при незначительной их выраженности.
У пациентов с беспокоящей одышкой, выполняют полный лаваж легких с помощью общей анестезии и двухпросветного эндотрахеального зонда. Промывание одного легкого проводится до 15 раз физиологическим раствором от 1 до 2 л, другое легкое при этом вентилируется. Затем процесс обращают.
Трансплантация легких не проводится, поскольку заболевание проявляется в трансплантированном легком.
Системные кортикостероиды не обладают лечебным эффектом и могут увеличивать риск вторичной инфекции. Роль ГМ-КСФ (при ингаляционном или подкожном введении) в лечении заболевания требует уточнения. Открытые исследования продемонстрировали клиническое выздоровление у 57% пациентов.
Основные положения
Рассмотреть легочной альвеолярный протеиноз у в остальном здоровых пациентов в возрасте от 30 до 50 лет, если рентген грудной клетки показывает двустороннего средне- и нижне-легочные поля затемнений, распределенные в форме бабочки с нормальным прикорневой областью.
Выполнить бронхоальвеолярный лаваж; если он противопоказан, или когда результаты не являются диагностическими, провести биопсию легких.
Если одышка является умеренной или тяжелой, лечить с помощью полного лаважа легких.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: