Лучевые признаки точечной хондродисплазии у плода
Добавил пользователь Skiper Обновлено: 29.01.2026
Точечные хондродислазии – клинически и генетически гетерогенная группа заболеваний, при которых характерна точечная кальцификация костей скелета. Наиболее изученной является форма точечной хондродисплазии, описанная Эрихом Конради в 1914 г., а несколько позже – Карлом Хюнерманом. В 1977 г. Рудольф Хаппле предположил, что болезнь наследуется по доминантному, сцепленному с полом типу. Основными признаками считаются ассиметричное укорочение конечностей в местах точечного обызвествления эпифизов, контрактуры крупных суставов (30%), ранний сколиоз в связи с точечным обызвествлением позвонков, отставание в росте. Наблюдаются плоское лицо, запавшая переносица, гипоплазия скуловых костей и возникающий за счет этого антимонголоидный разрез глаз, катаракта (в 20% случаев). У новорожденных – эритема и толстые, трудно отделяемые чешуйки, у детей постарше – фолликулярная атрофодермия (расширение устьев фолликулов, из-за чего кожа становится «пористой», похожей на лимонную корку) и ихтиоз; волосы редкие и жесткие, в 25% случаев отмечается аллопеция. Эритематозные высыпания у новорожденных – пятнистые или линейные (по линиям Блашко), иногда наблюдается обширная эритродермия. Рентгенологически выявляется деформация тел позвонков, укорочение трубчатых костей (обычно одностороннее), точечная кальцификация, в первую очередь, появляющаяся в конечных отделах трубчатых, карпальных и тарзальных костей, отростках позвонков, седалищных и лонных костях.
Более чем в 95% случаев хондродисплазии Конради-Хюнермана болеют лица женского пола. Проявления заболевания у гетерозиготных носительниц мутаций в гене EBP очень варьируют и в первую очередь зависят от паттерна инактивации хромосом Х в тканях, подверженных изменениям у больных девочек. Описаны случаи гибели плода с множественными аномалиями развития, в то же время известны взрослые женщины с синдром Конради-Хюнермана, у которых признаки заболевания были выявлены только при прицельном обследовании врача. Проявление признаков синдрома варьирует у женщин из одной семьи и у носителей одинаковой мутации.
Долгое время считалось, что плоды мужского пола с хондродисплазией Конради-Хюнермана умирают еще во время внутриутробного развития. Однако сегодня известны несколько подтвержденных случаев заболевания у мальчиков с типичными признаками синдрома: один имел кариотип 47,XXY; четверо, оказалось, несли мозаичный генотип по гемизиготному носительству мутации в гене EBP. Также описаны фенотипы 11-ти мальчиков с гемизиготным генотипом по мутации в гене EBP: у всех наблюдалась умеренная или тяжелая задержка в развитии, гипотония, изменения в ЦНС (синдром Денди-Уокера, агенез мозолистого тела и др.), челюстно-лицевые и генитальные аномалии, катаракта, ихтиоз.
В Центре Молекулярной Генетики для диагностики хондродисплазии Конради-Хюнермана проводится поиск мутаций в гене EBP методом секвенирования кодирующей последовательности гена. При необходимости возможна оценка паттерна инактивации хромосом Х (лайонизации хромосом Х).
Хондродисплазия
Хондродисплазии — это общее название для группы наследственных заболеваний, вызванных нарушением образования или окостенения хрящевой ткани. Они возникают в результате генных мутаций, наследуются как по аутосомно-доминантному, так и по аутосомно-рецессивному типу. Патологии проявляются низкорослостью, разнообразными укорочениями и деформациями конечностей, поражением суставов. Для диагностики необходимы данные рентгенографии или компьютерной томографии, результаты генетического исследования. Лечение направлено на устранение или коррекцию существующих нарушений, для чего применяются ортопедические операции, ортезы, методы ЛФК и физиотерапии.
МКБ-10

Общие сведения
На сегодня известно более 100 вариантов хондродисплазии, многие из которых схожи между собой по клинической картине, поэтому постановка точного диагноза, как правило, затруднена. Заболевания не теряют своей актуальности, поскольку сопровождаются грубыми деформациями опорно-двигательного аппарата, что чревато инвалидностью, резким ухудшением качества жизни, некоторые имеют высокий показатель летальности в раннем возрасте. Достоверные статистические данные распространенности отсутствуют.
Причины хондродисплазии
Патология связана с точечными мутациями генов (COL2A1, SEDL, COMP и т.д.), отвечающих за образование белков хрящевого матрикса или некоторых регуляторных пептидов. Большинство типов хондродисплазии имеют аутосомно-доминантный тип наследования — при наличии генного дефекта у одного из родителей риск рождения больного ребенка равен 50%. Некоторые формы отличаются аутосомно-рецессивным механизмом передачи, для которого оба родителя должны быть носителями мутантного гена.
Патогенез
При большинстве типов хондродисплазий выявляется первичный дефект белков: разных типов коллагена, матрилина, олигомерного протеина. Все они в норме составляют хрящевую ткань, поэтому при их патологическом строении наблюдаются дефекты структуры, избыточное или недостаточное развитие хрящей. Нарушения также происходят на этапе образования регуляторных белков, к которым относят тирозинкиназный рецептор, факторы транскрипции, ферменты процессинга РНК.
Типичные деформации и укорочение конечностей часто сочетаются с расстройствами энхондрального окостенения, при котором определяется неструктурное беспорядочное расположение хондроцитов, вызванное дефектами окислительного фосфорилирования. Ряд форм болезни проявляются образованием костных выступов (экзостозов). Патогенетические механизмы многих типов хондродисплазий до сих пор точно не изучены.
Классификация
В современной генетике принят комплексный подход к систематизации хондродисплазий, учитывающий этиопатогенетические, клинико-рентгенологические особенности. Международная рабочая группа по хондродисплазиям представила вариант классификации из 17 разделов согласно типу генной мутации. В то же время в практической медицине актуально деление болезни с учетом характера поражения хрящевой ткани, которое включает 3 категории:
- Эпифизарные дисплазии. Сюда относят поражения суставного хряща, дисплазии хрящевой ткани собственно эпифиза. Основные представители группы: болезнь Волкова, спондилоэпифизарная дисплазия, псевдоахондроплазия, болезнь Фэрбанка.
- Физарные дисплазии. Характеризуются нарушениями формирования ростковой эпифизарной пластинки. Эта категория включает ахондроплазию (хондродистрофию), гипохондроплазию, экзостозную хондродисплазию.
- Метафизарные дисплазии. Эта группа патологий возникает вследствие задержки энхондрального роста на фоне неправильного окостенения хрящей метафизов. Они наименее изучены, описано лишь несколько вариантов: дисхондроплазия (болезнь Олье), хондродисплазии типа Янсена, Шмида, Мак-Кьюзика.
Симптомы хондродисплазии
Типичным признаком любой формы болезни является низкий рост (карликовость), связанный с нарушениями удлинения трубчатых скелетных костей. Рост взрослых пациентов составляет около 1,2-1,3 м. Карликовость сопровождается диспропорциями тела: при относительно нормальных размерах туловища у таких больных короткие конечности, крупный череп. При раннем закрытии черепных швов происходит деформация головы.
Хондродисплазии имеют множество симптомов, зависящих от конкретного варианта патологии. У страдающих ахондроплазией большой лоб, седловидный нос, выраженный прогиб в пояснице (лордоз), ноги приобретают саблевидную форму. Для дистрофической дисплазии характерно удлинение больших пальцев кистей рук, эквиноварусная косолапость. При эпифизарных дисплазиях отмечаются укорочение дистальных фаланг пальцев, вывихи тазобедренных суставов.
Помимо симптомов со стороны опорно-двигательной системы диагностируются множественные соматические признаки хондродисплазий. Изменения кожи и подкожной клетчатки проявляются пигментными пятнами, крупными липомами, гемангиомами, на ладонях и стопах зачастую формируются большие очаги гиперкератоза. У пациентов могут возникать гепатоспленомегалия, разнообразные поражения глаз: дистрофия хрусталика, астигматизм, миопия.
Осложнения
Часть хондродисплазий относят к летальным мутациям, при их наличии лишь немногие пациенты доживают до взрослого возраста. В эту категорию выделяют некоторые типы ахондрогенеза, танатофорную дисплазию, капомелическую дисплазию, ризомелическую форму точечной хондродисплазии. К нелетальным последствиям заболевания относят контрактуры суставов, деформации конечностей, грубые искривления позвоночника по типу кифосколиозов или лордозов.
Поскольку поражение костно-хрящевых структур обычно затрагивает позвоночник, у больных есть риск неврологических нарушений. Они обусловлены сдавлением спинного мозга при сужении позвоночного канала либо при стенозе большого затылочного отверстия. Осложнения включают нарушения чувствительности, снижение тонуса скелетной мускулатуры, тазовые расстройства. Вследствие деформации грудной клетки нередко нарастает дыхательная недостаточность.
Генные мутации могут затрагивать не только белки хрящевой ткани, но и компоненты соединительнотканных волокон другой локализации, с чем связано осложненное течение хондродисплазий. Нередко заболевания сопровождаются пороками развития сердца: наличием дополнительных хорд, патологиями клапанного аппарата, поражением крупных эластических сосудов. При вовлечении в процесс мышечной ткани сердца встречаются тяжелые формы кардиомиопатий.
Диагностика
Первичное обследование больных находится в компетенции ортопеда-травматолога, при необходимости к осмотру привлекают генетика, хирурга-ортопеда. Заподозрить наличие хондродисплазии удается по задержке роста в сочетании с множественными скелетными деформациями, однако выделить конкретную нозологическую единицу по физикальным данным затруднительно. План диагностики включает следующие методы исследования:
- Рентгенография костей. Рентгенологические признаки разнообразны: уплощение эпифизарных отделов трубчатых костей, деформация коротких трубчатых костей, неравномерные очаги окостенения. Патогномоничная картина наблюдается при ахондроплазии —на снимке поперечники трубчатых костей увеличены, корковое вещество утолщено, метафизы расширены, рельеф усилен.
- КТ костей скелета. Компьютерная томография имеет более высокую разрешающую способность, поэтому метод рекомендован при затруднениях в дифференциальной диагностике хондродисплазий. Также КТ информативна при мягких фенотипических вариантах, когда отсутствуют выраженные аномалии костного аппарата.
- Генетическое тестирование. На современном этапе медицины расшифрованы гены, отвечающие за большинство вариантов хондродисплазий. Для верификации диагноза используется секвенирование экзона, которое выполняется на базе специализированных генетических центров. По показаниям тестирование проводится ближайшим родственникам больного.
Лечение хондродисплазий
Специфическая терапия не разработана. Суть медицинской помощи заключается в максимально возможном восстановлении нормальных пропорций скелета, улучшении функциональности конечностей, предупреждения тяжелой инвалидности. Важную роль играет разъяснение пациентам недопустимости повышенных нагрузок на суставы, что может стать причиной резкого ухудшения состояния. Комплекс лечебных мероприятий при хондродисплазиях состоит из следующих направлений:
- Ортопедическая коррекция. Использование специальных ортезов, фиксаторов (например, при вывихе тазобедренного сустава) частично компенсирует имеющиеся анатомические аномалии. Чтобы улучшить походку, подбирается специальная ортопедическая обувь.
- Хирургические вмешательства. Хороший функциональный и косметический результат дают операции по удлинению конечностей, которые выполняются в несколько этапов. Также оперативная помощь показана для устранения крупных экзостозов, ликвидации суставных контрактур, коррекции стеноза спинномозгового канала.
- Реабилитация. Больным с хондродисплазиями назначаются индивидуальные курсы лечебной физкультуры, механотерапии, специального массажа. Для уменьшения мышечного напряжения полезны физиотерапевтические процедуры.
Прогноз и профилактика
Среди хондродисплазий есть летальные варианты, приводящие к гибели плода во внутриутробном периоде или ранней смерти ребенка, и мягкие формы, при которых возможна успешная реабилитация и социализация пациентов. Большое значение для улучшения прогноза имеет своевременная помощь хирургов-ортопедов, всесторонняя реабилитация. Учитывая генетические предпосылки заболевания, эффективные меры профилактики не разработаны.
2. Множественная эпифизарная хондродисплазия: особенности первичного эндопротезирования тазобедренного сустава// А.Ю. Милюков// Политравма. — 2017. — №3.
4. Экзостозная хондродисплазия взрослых (клиника, диагностика, хирургическое лечение)/ А.В. Балберкин. — 1994.
Гипохондроплазия
Гипохондроплазия – наследственное заболевание, относящееся к группе хондродисплазий, причина которого заключается в нарушении формирования хрящей и некоторых типов костей, что приводит к карликовости. Симптомами этой патологии являются укороченные конечности и пальцы, увеличенные относительные размеры кистей и стоп, небольшое ограничение движений в локтевом суставе и поясничный лордоз. Диагностика производится на основании осмотра пациента, данных рентгенологических исследований, молекулярно-генетического анализа. Лечения гипохондроплазии не существует, однако серьезных осложнений, значительно ухудшающих качество жизни пациента (кроме карликовости), также не наблюдается.
Гипохондроплазия – генетическая патология, которая во многом сходна с родственными ей хондродисплазиями – например, ахондроплазией и танатоформной дисплазией. Впервые это состояние было описано в 1913 году Равенной, который выявил наследственную природу этой формы карликовости и ее отличия от ахондроплазии. Встречаемость гипохондроплазии в настоящий момент точно неизвестна – некоторые врачи-генетики указывают цифры 1:100000, однако с этим согласны далеко не все исследователи.
Основное затруднение в вычислении встречаемости этого заболевания обусловлено тем, что более чем в 80% случаев оно возникает спонтанно по причине мутаций de novo. Остальная доля случаев гипохондроплазии (менее 20%) передается по аутосомно-доминантному механизму. Однако точно установлено, что данная патология встречается намного реже ахондроплазии (1:20000), хоть и имеет более легкое течение. Заболевание с равной степенью вероятности поражает как мужчин, так и женщин.
Причины гипохондроплазии
Большое сходство проявлений гипохондроплазии с ахондроплазией обусловлено тем, что причиной данных заболеваний являются мутации одного и того же гена - FGFR3, расположенного на 4-й хромосоме. Он кодирует последовательность фактора роста фибробластов-3, представляющего собой трансмембранный рецептор тирозинкиназы. В норме он несколько тормозит рост фибробластов и хондроцитов в ростовых пластинках эндохондральных костей, тем самым контролируя правильное развитие костной ткани. При мутациях гена FGFR3 полученный фактор роста фибробластов-3 имеет дефект и не может полноценно выполнять свои функции – в зависимости от характера генетического дефекта это может привести к танатоформной дисплазии, ахондроплазии или гипохондроплазии.
Выявлено несколько десятков мутаций гена FGFR3, результатом которых является развитие карликовости по типу гипохондроплазии. Наиболее распространенным является дефект, возникающий по причине замены азотистого основания (аденина или гуанина) в положении 1620 тринадцатого экзона гена. В результате в полученном от такого гена белке в положении 540 происходит замена аспараганина на лизин, что значительно изменяет тирозинкиназную чувствительность рецептора.
Кроме того, описано еще множество других замен азотистых оснований в гене и, как следствие, аминокислот в полученном протеине, которые приводят к развитию симптомов гипохондроплазии. Однако имеются указания, что у некоторых больных с клинической картиной этого заболевания при генетическом исследовании не было выявлено дефектов FGFR3. Это может говорить о том, что в развитии гипохондроплазии могут участвовать и другие гены.
Все мутации вышеуказанного гена наследуются по аутосомно-доминантному механизму, однако в подавляющем большинстве случаев они являются спонтанными и не выявляются у родителей или родственников больного. Подмечено, что у многих пациентов с гипохондроплазией возраст отцов превышает средний, что может указывать на герминативную природу появления мутаций гена FGFR3.
Симптомы гипохондроплазии
Как правило, в первые годы жизни ребенка гипохондроплазия ничем себя не проявляет – при рождении не наблюдается никаких отклонений от нормы, набор массы и психофизическое развитие происходят нормально. Первые признаки отставания в росте регистрируются в возрасте 3-4-х лет, становится заметной небольшая диспропорциональность тела (короткие руки и ноги, увеличенные ступни и кисти). Во многих случаях окружающие даже не сразу выявляют какие-либо необычные пропорции у больных – они просто выглядят невысокими коренастыми людьми. Форма черепа и черты лица при гипохондроплазии зачастую без особенностей, иногда может выявляться небольшая брахицефалия.
У больных иногда возникают незначительные сгибательные контрактуры локтевых суставов и очень редко – тазобедренных. Вальгусное искривление бедренных костей при гипохондроплазии не наблюдается, возможны аналогичные деформации голени. Примерно в половине случаев у больных возникает лордоз поясничного отдела позвоночника. Так как все эти проявления похожи на симптомы ахондроплазии, только выражены намного слабее, долгое время считалось, что это одна и та же патология, только отдельные исследователи относили ее к самостоятельной нозологической единице. Лишь современные исследования в области генетики однозначно доказали справедливость такого выделения гипохондроплазии.
Выявление гипохондроплазии производится на основании комплекса медицинских мер: консультации педиатра и ортопеда, рентгенологических исследований, молекулярно-генетического анализа. При осмотре больных старше 3-4-х лет обнаруживают характерные для патологии изменения – укороченные конечности, увеличенный относительный размер стоп и кистей, нарушения подвижности в локтевом и иногда в тазобедренном суставе, поясничный лордоз. Если гипохондроплазия имеет наследственный характер, то такие проявления всегда имеются у одного из родителей, так как это заболевание является аутосомно-доминантным. При здоровых родителях косвенным признаком, указывающим на эту патологию, может быть возраст отца на момент зачатия более 40-45 лет.
Намного больше информации о гипохондроплазии дают рентгенологические исследования костей, суставов и позвоночника. При обследовании последнего выявляют сужение спинномозгового канала в каудальном направлении, вогнутые контуры задней поверхности поясничных позвонков. Также рентгенологически наблюдается укорочение и уплотнение бедренных и плечевых костей, незначительное удлинение большеберцовой кости, эпифизы костей квадратной формы в области коленных суставов, уплощение вертлужной впадины. Примерно в двух третях случаев гипохондроплазии также диагностируется укорочение локтевой кости.
Генетическая диагностика гипохондроплазии включает в себя прямое секвенирование последовательности гена FGFR3 или отдельных его экзонов для выявления мутаций. Чаще всего производят исследование 13-го экзона гена, так как именно там располагаются дефекты в большинстве случаев заболевания. Возможна пренатальная диагностика посредством амниоцентеза или биопсии ворсин хориона.
Дифференциальный диагноз следует проводить с ахондроплазией и другими состояниями, которые сопровождаются укорочением конечностей. Различают эти патологии между собой при помощи рентгенологических и генетических методов исследования.
Лечение гипохондроплазии
Специфического лечения гипохондроплазии не существует, карликовость остается у человека на всю жизнь. Однако ряд других проявлений по типу контрактуры суставов и лордоза поясничного отдела позвоночника выражены умеренно. Поэтому прогноз гипохондроплазии относительно жизни и ее качества в большинстве случаев благоприятный. Лишь в некоторых случаях взрослым больным может потребоваться хирургическое вмешательство из-за сдавления поясничного отдела спинного мозга или его корешков (радикулит), которые возникают по причине лордоза и нарушения строения позвонков.
Профилактика
Профилактика заболевания сводится к пренатальной генетической диагностике, особенно это необходимо делать семейным парам, где один из родителей страдает от гипохондроплазии. Учитывая аутосомно-доминантный характер наследования этой патологии, вероятность рождения больного ребенка при здоровом втором родителе составляет 50%.
Лучевые признаки точечной хондродисплазии у плода
УЗИ, рентгенограмма при несовершенном остеогенезе у плода
а) Терминология:
1. Сокращения:
• Несовершенный остеогенез (НО)
2. Определения:
• Генетически и клинически гетерогенная группа заболеваний соединительной ткани, проявляющихся остеопорозом и переломами:
о 90% случаев связаны с патологией коллагена I типа (• Исторически выделяют 5 фенотипических групп НО (I-V) в зависимости от клинических проявлений
• В настоящее время известно 17 генетических причин НО
• В актуальной классификации НО подразделяются классические фенотипы:
о Фенотипы от легкой до средней степени тяжести (ранее - тип I, IV, V)
о Фенотипы с прогрессирующей деформацией и перинатальной летальностью (ранее - тип II, III)
б) Лучевая диагностика:
1. Общие сведения:
• Критерии диагностики:
о Отличить НО от других скелетных дисплазий позволяет наличие переломов
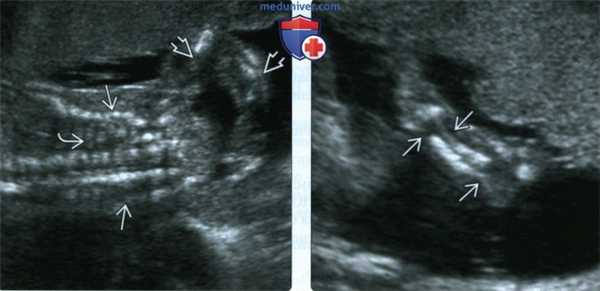
(Слева) УЗИ грудной клетки и верхней конечности плода во II триместре, фронтальная плоскость. Грудная клетка уменьшена, ребра неправильной формы с множеством переломов. Многочисленные переломы костей руки.
(Справа) Тот же случай, визуализация предплечья. Неоднородная оссификация локтевой и лучевой костей. Костные сегменты нередко смещены, за счет чего кость выглядит изогнутой или искривленной. В данном случае диагностирован перинатальнолетальный НО (II тип).
2. УЗИ при несовершенном остеогенезе у плода:
• Конечности:
о Укорочение и искривление трубчатых костей вследствие переломов
о Формирование ложных суставов
о Формирование мозолей придает костям «скомканный» вид
о Минерализация снижена
• Грудная клетка:
о При перинатально-летальном типе - множественные переломы ребер (в виде «рахитических четок»)
• Головной мозг:
о Визуализируется «излишне отчетливо», реверберационный артефакт отсутствует:
- Слабая минерализация черепа
о Череп деформируется при обычном нажатии датчиком
3. Рентгенография при несовершенном остеогенезе у плода:
• Генерализованная остеопения
• Задержка формирования черепа, определяется множество вормиевых костей
• Ребра истончены, вследствие переломов имеют вид «рахитических четок»
• Истончение компактного вещества и диафизов трубчатых костей
• В тяжелых случаях тела позвонков спадаются, присутствуют переломы ребер, трубчатые кости расширены и изогнуты вследствие компрессионных переломов
4. Рекомендации по лучевой диагностике:
• Предпочтительный метод исследования:
о УЗИ во II триместре
о При беременности высокого риска - ТВУЗИ в I триместре
• Советы по проведению исследования:
о Измерение всех трубчатых костей, поиск переломов:
- При перинатально-летальной форме укорочение значительное
о Соотносят окружности грудной клетки и живота:
- Грудная клетка уменьшена → повышен риск гипоплазии легких
о Отсутствие изменений по данным УЗИ не позволяет исключить НО у пациента группы высокого риска
о Менее тяжелые формы могут проявляться изолированным искривлением бедренных костей
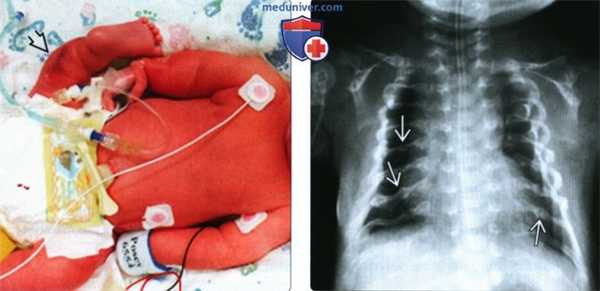
(Слева) Клиническая фотография новорожденного с НО. Клиническая форма -между перинатально-летальным (II тип) и прогрессирующим деформирующим (III тип) типами. Грудная клетка уменьшена, конечности укорочены. Множественные переломы конечностей, особенно ног, создают видимость их искривления.
(Справа) Тот же случай. Рентгенография грудной клетки. Грудная клетка незначительно уменьшена. Множественные переломы ребер. Проявления менее тяжелые, чем при перинатально-летальном НО.
в) Дифференциальная диагностика несовершенного остеогенеза у плода:
1. Танатофорная дисплазия:
• Оссификация костей, в том числе черепа, не нарушена
• Выраженное укорочение и искривление трубчатых костей
• При I типе бедренная кость изогнута в форме телефонной трубки; при II типе кости прямые
• При II типе - макроцефалия и череп в форме трилистника
2. Ахондрогенез:
• Гипоминерализация позвоночника, оссификация черепа вариабельна
• Часто - водянка плода, кистозная гигрома
• Выраженная микромелия
• При типе 1А - переломы ребер
3. Кампомелическая дисплазия:
• Гипоплазия лопатки
• Бедренная, большеберцовая или малоберцовая кости изогнуты под острым углом, что можно ошибочно принять за переломы
• Оссификация черепа не нарушена
• Часто - инверсия пола
4. Гипофосфатазия:
• Генерализованная гипоминерализация всех костей - кости «побиты молью»
• Переломы не характерны
• Микромелия
• Низкий уровень щелочной фосфатазы сыворотки крови у новорожденных и родителей

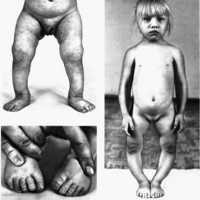
(Слева) Клиническая фотография матери и ребенка, страдающих НО IV типа. Темно-голубые склеры, лица треугольной формы определяется ложный сустав.
(Справа) Тот же случай. Явный ложный сустав вследствие многочисленных внутриутробных переломов. При сравнении длин стопы (в норме) и длинных костей (укорочение) выявляется микромелия.
г) Патологоанатомические особенности:
1. Общие сведения:
• Генетические факторы:
о 90% случаев - мутации генов COL1A1, COL1A2, кодирующих коллаген I типа
о Большинство типов НО наследуются по аутосомно-до-минантному типу
о 60% нетяжелых типов НО - мутации de novo
о Большинство повторных случаев II типа связано с гонадным мозаицизмом:
- Риск повторного возникновения - до 3%
о Сообщается о нескольких аутосомно-рецессивных генах
• Сопутствующие аномалии:
о Несовершенный дентиногенез
о Тугоухость
о Голубые или серые склеры
2. Стадирование, градация и классификация:
• Классификация van Dijk и Sillence (2014) основана на сочетании фенотипа и лежащих в основе генных мутаций
о Недеформирующий НО с голубыми склерами (I тип):
- Переломы у новорожденных находят редко
- Голубоватый оттенок склер
- Тип IA - зубы не изменены; тип IB - несовершенный дентиногенез (60%)
- Тугоухость (35-50%)
- Хрупкость костей уменьшается в подростковом возрасте; может рецидивировать с наступлением менопаузы
- При рентгенографии - вормиевы кости
- Гены COL1A1, COL1A2 наследуются по аутосомно-доминантному типу
о Перинатально-летальный НО (II тип):
- Темные серо-голубые склеры; трубчатые кости короткие и толстые
- Уменьшение грудной клетки, ребра в виде «рахитических четок»
- Выраженное укорочение конечностей, из-за переломов кости выглядят «скомканными»
- Деминерализация черепа
- Гены COL1A1, COL1A2 наследуются по аутосомно-доминантному типу
- Гены CRTAP, LEPRE1, PPIB наследуются по аутосомно-рецессивному типу
о Прогрессирующий деформирующий (III тип):
- У новорожденного - множественные переломы, прогрессирующая деформация конечностей, позвоночника, черепа
- Белые или серо-голубые склеры
- Треугольное лицо
- Выраженная карликовость
- Компрессия спинного мозга
- Дети обычно неходячие
- Аутосомно-доминантный тип наследования: COL1A1, COL1A2
- Аутосомно-рецессивный тип наследования: ВМР1, CRTAP, FKBP10, LEPRE1, PLOD2, PPIB, SERPINF1, SERPINH1, ТМЕМ38В, WNT1, CREB3L1
о Деформирующий НО с нормальными склерами (IV тип):
- Клинически и рентгенологически - между I и III типами
- Белые или серо-голубые склеры
- Карликовость
- Часто - несовершенный дентиногенез
- С возрастом развивается тугоухость
- Аутосомно-доминантный тип наследования: COL1A1, COL1A2, WNT1a
- Аутосомно-рецессивный тип наследования: CRTAP, PPIB, SP7
- Х-сцепленный тип наследования: PLS3
о НО с кальцификацией межкостных перепонок (V тип):
- Дефекты коллагена не I типа (1FITM5); аутосомно-доминантный тип наследования
- Классический фенотип напоминает спондилометафизарную дисплазию
2. Макроскопические изменения и исследование операционного материала:
• НО II типа (перинатально-летальный):
о Компактное вещество истончено, губчатое вещество рыхлое
о Повышение числа остеокластов/остеоцитов
д) Клинические особенности:
1. Клиническая картина:
• Самые частые субъективные и объективные симптомы:
о При УЗИ во II триместре определяются многочисленные переломы
о Описаны случаи НО III—IV типа с изолированным искривлением бедренной кости
• Другие субъективные и объективные симптомы:
о В I триместре - кистозная гигрома, утолщение воротникового пространства
о Диагностика возможна с 12-14 нед.
2. Демографические особенности:
• Возраст:
о При летальном НО - с увеличением возраста отца риск повышается
• Пол:
о Ж>М
• Эпидемиология:
о 1:10 000-20 000 живых новорожденных
о Заболеваемость НО у беременных 1:20 000-30 000
3. Естественное течение и прогноз:
• Различаются в зависимости от типа:
о I, IV типы: продолжительность жизни в норме или несколько снижена
о II тип: летальный в перинатальном периоде
о III тип: продолжительность жизни значительно снижена
• НО у беременной женщины:
о Склонность к атонии матки, образованию гематом, кровотечениям
о Ходьба затруднена, жалобы на боли в спине
о Преждевременные роды
о При карликовости - нарушение функции легких по рестриктивному типу
о Вероятность наследования НО в зависимости от типа составляет 25-50%
о Беременной показана ЭхоКГ с исследованием аорты
о КС дискутабельно:
- Переломы костей таза
- Описаны случаи разрыва матки при родах через естественные пути, что связывают с дефицитом коллагена в миометрии
• Другие осложнения:
о Артрит, сколиоз, разрывы сухожилий, боль в спине, дилатация корня аорты, базилярная инвагинация
4. Лечение несовершенного остеогенеза у плода:
• Пренатальное лечение плода отсутствует:
о НО матери - экспериментальная терапия бисфосфонатами (прегравидарный период)
• Во всех случаях - генетическое консультирование
• Биопсия ворсин хориона или амниоцентез для исследования коллагена:
о В некоторых случаях возможна молекулярная диагностика
о Сообщается о случаях успешной преимплантационной генетической диагностики
• Подозрение на летальный или тяжелый НО:
о Предлагается прерывание беременности
о Для генетического консультирования необходимо подтвердить диагноз
• Роды в условиях стационара, имеющего опыт работы с наследственной фетопатологией и скелетными дисплазиями
• КС не улучшает прогноз:
о Не увеличивает выживаемость при летальном НО
о Не снижает число перинатальных переломов при нелетальном НО
о Инструментальные роды противопоказаны
• При прерывании беременности или гибели плода показаны аутопсия и рентгенография:
о Биохимическое и молекулярно-генетическое исследование тканей
• В постнатальном периоде:
о Интрамедуллярные штифты для выпрямления и стабилизации трубчатых костей при тяжелом НО
о При тяжелом НО - циклическая терапия бисфосфонатами:
- Снижает скорость метаболизма костной ткани и повышает ее плотность
- Уменьшает частоту переломов и боль
о Физические упражнения, ортезирование
е) Список использованной литературы:
1. Cozzolino М et al: Management of osteogenesis imperfecta type I in pregnancy; a review of literature applied to clinical practice. Arch Gynecol Obstet. ePub, 2016
2. Reyes C et al: Risks and benefits of bisphosphonate therapies. J Cell Biochem. 117(1):20—8, 2016
3. Forlino A et al: Osteogenesis imperfecta. Lancet. ePub, 2015
4. Shaker JL et al: Recent developments in osteogenesis imperfecta. FlOOORes. 4(F1000 Faculty Rev):681, 2015
5. Van Dijk FS et al: Osteogenesis imperfecta: clinical diagnosis, nomenclature and severity assessment. Am J Med Genet A. 164A(6): 1470-81, 2014
Редактор: Искандер Милевски. Дата обновления публикации: 1.11.2021
Ахондроплазия
Ахондроплазия – это врожденное заболевание, при котором нарушается процесс роста костей скелета и основания черепа. Причиной развития патологии является генетическая мутация. Часть плодов гибнет внутриутробно. При рождении нарушения заметны с первых дней жизни: головка увеличена, конечности укорочены. В последующем наблюдается выраженное отставание в росте рук и ног при нормальном размере туловища, возникают вальгусные и варусные деформации конечностей, деформации позвоночника. Ахондроплазия диагностируется по данным осмотра, специальных измерений и рентгенографии. Лечение симптоматическое, направлено на предотвращение и устранение грубых деформаций.
Ахондроплазия (врожденная хондродистрофия, болезнь Парро-Мари, диафизарная аплазия) – генетическое заболевание, при котором наблюдается укорочение конечностей в сочетании с нормальной длиной туловища. Характерными особенностями являются низкий рост (130 и менее см.), изогнутый вперед позвоночник, седловидный нос и относительно большая голова с выступающими лобными буграми. По данным специалистов в сфере травматологии и ортопедии, ахондроплазия возникает у одного из 10 тысяч новорожденных, женщины страдают чаще мужчин. Способов полностью излечить ахондроплазию, восстановив рост и пропорции тела, в настоящее время не существует. Лечение направлено на минимизацию негативных последствий болезни.
Причины ахондроплазии
В основе патологии лежит нарушение развития костей вследствие генетически обусловленной дистрофии эпифизарных хрящей. Причиной развития заболевания является мутация гена FGFR3. В 20% случаев ахондроплазия передается по наследству, в 80% развивается в результате впервые возникшей мутации. Из-за хаотичного расположения клеток ростковой зоны происходит нарушение нормального процесса окостенения. В результате рост костей замедляется. При этом поражаются только кости, растущие по энхондральному типу: трубчатые кости, кости основания черепа и т. д. Кости свода черепа, растущие из соединительной ткани, достигают положенного размера, что приводит к несоответствию пропорций между головой и телом, становится причиной характерного изменения формы черепа.
Симптомы ахондроплазии
Нарушение анатомических пропорций заметно уже при рождении: ребенок имеет относительно большую голову, короткие ручки и ножки. Лоб выпуклый, мозговая часть черепа увеличена, затылочные и теменные бугры выпирают. В отдельных случаях возможна гидроцефалия. Отмечаются нарушения строения лицевого скелета, возникшие вследствие неправильного развития костей основания черепа. Глаза пациентов с ахондроплазией широко расставлены, находятся глубоко в орбитах, около внутренних углов глаз есть дополнительные складочки. Нос седловидный, сплющенный, с широкой верхней частью, лобные кости заметно выступают вперед, верхняя челюсть также значительно выступает вперед над нижней. Язык грубый, небо высокое.
Нижние и верхние конечности больных ахондроплазией равномерно укорочены, в основном – за счет проксимальных сегментов (бедер и плеч). Ручки новорожденного ребенка достают только до пупка. Все сегменты конечностей несколько искривлены. Стопы широкие и короткие. Ладони широкие, II-V пальцы короткие, практически одинаковой длины, I палец длиннее остальных. В первые месяцы жизни у пациентов с ахондроплазией на конечностях видны жировые подушки и кожные складки. Туловище нормально развито, грудная клетка не изменена, живот выпячен вперед, а таз наклонен кзади, поэтому ягодицы выступают сильнее, чем у здоровых детей.
У грудных детей с ахондроплазией чаще, чем у их здоровых сверстников, развивается внезапная смерть во сне. Предполагается, что причиной гибели в таких случаях является сдавление продолговатого мозга и верхней части спинного мозга из-за аномалии формы и размера затылочного отверстия. Кроме того, для детей страдающих ахондроплазией характерны нарушения дыхания из-за особенностей строения лица, больших миндалин и небольшой грудной клетки.
По мере взросления из-за извращения эпифизарного роста костей при нормальном периостальном росте кости все больше утолщаются, изгибаются, становятся бугристыми. Из-за повышенной эластичности эпифизарных и метафизарных отделов трубчатых костей возникают варусные деформации конечностей, быстро прогрессирующие при ранней нагрузке. Искривление еще больше усугубляется вследствие чрезмерной тяги хорошо развитых мышц и значительной массы нормально развивающегося туловища. Из-за нарушения нормальной оси конечностей у больных ахондроплазией формируются плосковальгусные стопы, коленные суставы становятся разболтанными.
Возникает ряд характерных для ахондроплазии деформаций. Бедренные кости искривляются и скручиваются внутрь в нижних отделах. Из-за неравномерного роста костей голени малоберцовая кость в верхнем отделе «выдвигается» вверх и перестает сочленяться с большеберцовой, а в нижнем «перекашивает» вилку голеностопного сустава. В результате голеностопный сустав разворачивается на 10-15 градусов внутрь, стопа уходит в положение супинации под углом 10-20 градусов. Верхние конечности также искривляются, особенно в области предплечий. Укорочение верхних конечностей сохраняется, однако у взрослых пациентов с ахондроплазией пальцы достают уже не до пупка, а до паховой складки.
У взрослых больных отмечается дефицит роста, обусловленный, в основном, укорочением нижних конечностей. Средний рост женщин составляет 124 см, мужчин – 131 см. Сохраняются и даже становятся более выраженными изменения головы и лицевого скелета: увеличенная мозговая часть черепа, выступающий и нависающий лоб, глубокая переносица, видимое нарушение прикуса. Возможно косоглазие.
Пациенты с ахондроплазией склонны к ожирению. Из-за зауженных носовых ходов у них часто развиваются средние отиты, формируется кондуктивная тугоухость. Из-за обструкции верхних дыхательных путей могут выявляться признаки дыхательной недостаточности. При ахондроплазии достаточно часто наблюдается сужение спинномозгового канала. Обычно оно возникает в поясничном, реже – в шейном или грудном отделе позвоночника. Может проявляться нарушениями чувствительности, парестезиями и болями в ногах. В тяжелых случаях возможно нарушение функции тазовых органов, парезы и параличи.
Постановка диагноза ахондроплазия производится детским ортопедом, не вызывает затруднений из-за характерного внешнего вида и пропорций тела пациента. Всех детей подробно осматривают, чтобы оценить степень отклонений от нормального развития скелета, данные заносят в таблицу. Эта таблица регулярно дополняется по мере роста ребенка, а внесенные в нее данные сравниваются со стандартной таблицей, специально составленной для больных с ахондроплазией.
Для оценки состояния различных органов и систем проводится комплексное обследование, назначаются консультации различных специалистов. Для исключения гидроцефалии новорожденных детей с ахондроплазией осматривает нейрохирург, при подозрении на гидроцефалию назначается МРТ головного мозга или более доступная компьютерная томография. Для изучения состояния носовых ходов и ЛОР-органов больных ахондроплазией направляют на консультацию к отоларингологу. Может также потребоваться консультация пульмонолога.
При рентгенографии черепа выявляется диспропорция между лицевой и мозговой частью. Затылочное отверстие уменьшено в размере, нижняя челюсть и кости свода черепа увеличены. Турецкое седло имеет характерную башмакообразную форму и плоское, удлиненное основание. Рентгенография грудной клетки при ахондроплазии обычно без изменений, в отдельных случаях грудина выдается вперед и несколько изогнута. Возможно утолщение ребер и их деформация в области перехода в хрящевые дуги. Иногда отсутствуют нормальные анатомические изгибы ключиц.
На снимках позвоночника больных ахондроплазией грубых изменений, как правило, также не выявляется, физиологические изгибы выражены слабее, чем у здоровых людей, при этом может выявляться поясничный гиперлордоз. Рентгенография таза свидетельствует об изменении размера и формы крыльев подвздошных костей – они имеют прямоугольную форму, развернуты и укорочены. Определяется также горизонтальное расположение крыши вертлужных впадин.
При рентгенографии трубчатых костей у пациентов с ахондроплазией выявляется укорочение и истончение диафизов, утолщение и бокаловидное расширение метафизов. Эпифизы погружены в метафизы по типу шарниров. На рентгенографии суставов видны деформация и неконгруэнтность суставных поверхностей, расширение суставных щелей и нарушение формы эпифизов. Рентгенограммы коленного сустава больных ахондроплазией свидетельствуют об удлинении малоберцовой кости, при рентгенографии голеностопного сустава определяется ротация и супинация. Достоверность клинического диагноза подтверждается с помощью генодиагностики.
Лечение ахондроплазии
Полное излечение пациентов силами современной ортопедии пока невозможно. Предпринимались попытки проводить лечение с использованием гормона роста, однако достоверных свидетельств эффективности этой методики при ахондроплазии получить не удалось. В раннем возрасте проводится консервативная терапия, направленная на укрепление мышц и профилактику деформации конечностей. Больным ахондроплазией назначают ЛФК, массаж, рекомендуют носить специальную ортопедическую обувь и т. д. Проводится профилактика ожирения.
Хирургические вмешательства при ахондроплазии показаны при выраженных деформациях конечностей и сужении спинномозгового канала. Для коррекции деформаций выполняется остеотомия, для устранения спинального стеноза – ламинэктомия. В ряде случаев также осуществляются операции для увеличения роста. Удлинение конечностей при ахондроплазии обычно проводится перекрестно, в два этапа: сначала удлиняется бедро с одной стороны и голень с другой, затем выполняются оперативные вмешательства на оставшихся сегментах.
Читайте также: