Механизмы развития апластической анемии - патогенез
Добавил пользователь Дмитрий К. Обновлено: 29.01.2026
Механизмы развития апластической анемии - патогенез
Результаты последних исследований кроветворной клетки-штамм, развитие техники костномозговой культуры в пробирке на полутвердой среде, равно как и накопление данных о пересадке костного мозга вызвали необходимость пересмотра проблемы патогенетических механизмов при апластической анемии.
Обсуждаются следующие 3 основных механизма:
1) сокращение популяции клеток-штамм;
2) неполноценность кроветворной микросреды;
3) иммунологические механизмы.
1. Сокращение численности клеток-штамм доказано при генуинных апластических анемиях и анемиях, развивающихся под воздействием токсических веществ, ионизирующего облучения и пр. Так, в костном мозге соответствующих больных отмечена небольшая концентрация CFU-c и CFU-E, что, косвенным образом, свидетельствует о сокращении численности кроветворных клеток-штамм.
Другую аномалию этих клеток составляет неспособность к репликации в связи с чем восстановление костного мозга более не осуществляется (Geary, Kern).
Статистика результатов пересадки костного мозга подтверждает эту гипотезу, поскольку у 48% из 110 больных, страдающих апластической анемией, отмечено полное восстановление, а это говорит о повторном заселении костного мозга нормальными кроветворными клеткамиштамм.
В природе эта гипотеза подтверждена существованием мышей W.WW, у которых отмечены наличие врожденной апластической анемии и неспособность к образованию селезеночных колоний (CFU-S), следовательно количественное сокращение кроветворных клеток-штамм, состояние, на которое можно положительно воздействовать пересадкой костного мозга от мышей w/w в норме.
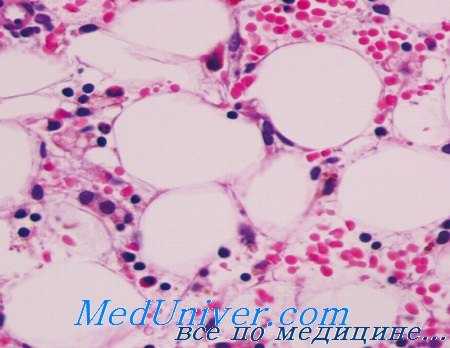
2. Неполноценность кроветворной микросреды обнаружена Knospe и Crosby при экспериментах на облученных животных. Это явление можно проследить и в естественных условиях, у мышей Sl/Sld, в костномозговой строме которых не происходит кроветворение и которые отличаются наличием врожденной апластической анемии. На их анемию можно воздействовать селезеночной тканью мышей, с которой, в организм животного, поступает необходимая кроветворная строма; вместе с тем костномозговые клетки мышей в норме не оказывают одинаковое влияние, поскольку перевиваемые клетки-штамм лишены необходимой развитию микросреды.
Отсутствуют убедительные данные в отношении подобного механизма у человека, вопреки ряду исследований, результаты которых выявили поражение сосудисто-соединительнотканной стромы при апластической анемии.
3. Иммунологические механизмы апластической анемии. Недавно проведенные исключительно тщательные исследования установили наличие этих механизмов в отношении порождающих клеток.
Различаются следующие 2 иммуносупрессорных механизма — один опосредованный антителами против кроветворных предшественников и другой, опосредованный цитотоксическими лимфоцитами.
Так, в ряде заболеваний нейтропенией были выявлены антитела вида иммуноглобулинов Г или М, обусловливающие супрессию CFU-c (Kelton), в то время как во многих случаях эритробластопении отмечены антитела вида иммуноглобулина Г, которые угнетали образование CFU-E (тип А) или действовали как антиэритропоэтин (тип В) (Krantz).
Также описаны нейтропении и эритробластопении, обусловленные супрессорным механизмом, опосредованным чрезмерной активностью супрессорных лимфоцитов Т (Hoffmann, Kagan).
В кроветворной клетке-штамм предполагается (по материалам пересадок костного мозга болезнь «трансплантат против хозяина») наличие антител, действующих против поверхностных антигенов клеток-штамм, однако такое высказывание еще не подтверждено средствами современной техники (Silver, Geary).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Фолиеводефицитная анемия
Фолиеводефицитная анемия - мегалобластическая анемия, обусловленная нарушением костномозгового кроветворения вследствие дефицита фолиевой кислоты (витамина B9). При фолиеводефицитной анемии развиваются все признаки анемического синдрома (бледность, слабость, тахикардия, снижение АД), незначительная желтуха, увеличение селезенки, анорексия, неустойчивый стул. С целью подтверждения диагноза проводится исследование гемограммы, костного мозга, определение уровня фолиевой кислоты в эритроцитах и сыворотке крови. Лечение B9-дефицитной анемии требует проведения заместительной терапии фолиевой кислотой.
МКБ-10

Общие сведения
Фолиеводефицитная анемия - комплекс клинико-гематологических проявлений, вызванных недостаточностью фолиевой кислоты вследствие ее ограниченного поступления, снижения абсорбции или повышенного расходования. Фолиеводефицитная анемия встречается реже, чем В12-дефицитная, однако часто сочетается с последней, а также нередко сопутствует железодефицитной анемии. Особую опасность фолиеводефицитная анемия представляет для беременных, поскольку может приводить к формированию пороков развития нервной системы плода, повышать риск отслойки плаценты, преждевременных родов и рождения недоношенного ребенка. Это обусловливает актуальность проблемы не только в рамках гематологии, но также педиатрии, акушерства и гинекологии.

Причины
Фолиевая кислота (витамин Вс, В9) поступает в организм в составе соединений - фолатов, присутствующих в овощах (бобовых, брокколи, шпинате, салате, спарже), печени, мясе, шоколаде, дрожжах и др. Запасы фолатов в организме составляют 5-10 мг, а минимальная ежесуточная потребность в них - 50 мкг (в период беременности в 2-3 раза выше). После прекращения поступления фолиевой кислоты извне, истощение ее эндогенных запасов наступает через 3-5 месяцев. Клинические проявления фолиеводефицитной анемии развиваются при снижении уровня витамина В9 в сыворотке крови менее 4 нг/мл.
Этиология фолиеводефицитной анемии может быть связана с недостаточным экзогенным поступлением витамина В9, нарушением его всасывания в кишечнике или повышенным выведением из организма. Чаще всего дефицит фолиевой кислоты носит алиментарный характер; он может быть обусловлен несбалансированным или недостаточным питанием, длительной термической обработкой пищи, вскармливанием детей козьим молоком и т. п.
Повышенную потребность в фолиевой кислоте испытывают беременные и кормящие женщины, недоношенные дети, подростки, онкобольные, пациенты с гемолитической анемией, гемобластозами, эксфолиативным дерматитом, псориазом. Эти категории лиц находятся в группе риска по развитию фолиеводефицитной анемии.
Нарушение абсорбции фолиевой кислоты в ЖКТ наблюдается при хроническом алкоголизме, состоянии после обширной резекции тонкого кишечника, целиакии, лимфоме кишечника, болезни Крона, спру, недостатке витамина В12, приеме антагонистов фолатов (противосудорожных препаратов, барбитуратов, оральных контрацептивов и др.). Чрезмерному выведению фолиевой кислоты из организма могут способствовать цирроз печени, гемодиализ, сердечная недостаточность.
Патогенез
Всасывание фолиевой кислоты происходит в двенадцатиперстной и начальном отделе тощей кишки. После поступления в кровоток она связывается с белками плазмы и транспортируется в печень, где значительная часть фолиевой кислоты откладывается в депо, остальная часть выводится с мочой.
В организме фолиевая кислота присутствует в виде коферментной формы - тетрагидрофолиевой кислоты, активно участвующей в синтезе глутаминовой кислоты, пиримидиновых и пуриновых оснований, а также тимидинмонофосфата – компонента ДНК. При фолиеводефицитной анемии, в первую очередь, нарушается синтез нуклеиновых кислот активно делящихся кроветворных клеток, в результате чего нормобластическое кроветворение сменяется на мегалобластическое. Следствием неэффективного гемопоэза служит развитие анемии, сочетающейся с лейкопенией и тромбоцитопенией.
Симптомы фолиеводефицитной анемии
Фолиеводефицитная анемия чаще развивается у молодых пациентов и беременных женщин. Среди клинических проявлений преобладают признаки анемического синдрома: бледность кожи с оттенком субиктеричности, слабость, тахикардия, артериальная гипотония, головокружения.
В отличие от пернициозной анемии, при дефиците фолиевой кислоты не развиваются неврологические нарушения (фуникулярный миелоз), а расстройства функции желудочно-кишечного тракта выражены незначительно. В числе последних иногда отмечается анорексия, неустойчивость стула, атрофический гастрит, глоссит, незначительная спленомегалия. При углубленном обследовании может выявляться миокардиодистрофия.
У пациентов с эпилепсией, шизофренией, психическими нарушениями фолиеводефицитная анемия усугубляет течение основного заболевания. Дефицит фолиевой кислоты в период беременности является фактором риска формирования дефектов нервной трубки (анэнцефалии, гидроцефалии, менингоцеле), врожденных пороков сердца, расщепления губы и нёба («заячьей губы» и «волчьей пасти»), гипотрофии плода. Кроме этого, у беременной повышается вероятность выкидыша, кровотечения, преждевременного родоразрешения.
После родов женщины с фолиеводефицитной анемией в большей степени подвержены развитию послеродовой депрессии, а дети раннего возраста - задержке психомоторного развития, нарушению работы кишечника, снижению иммунитета.
Диагностика
В общем анализе крови при фолиеводефицитной анемии отмечается гиперхромия, макроцитоз, лейкопения, тромбоцитопения, снижение количества ретикулоцитов. Подтверждению диагноза способствует определение снижения фолиевой кислоты в сыворотке крови (норма 6-20 нг/мл) и эритроцитах (норма – 100-450 нг/л). При исследовании миелограммы выявляется гиперплазия красного ростка, мегалобластный тип кроветворения.
При фолиеводефицитной анемии проба с гистидином, принятым внутрь, оказывается положительной: экскреция формиминглутаминовой кислоты с мочой значительно увеличивается (>18 мг). При миокардиодистрофии, по данным ЭКГ, имеет место нарушение реполяризации миокарда левого желудочка. С помощью УЗИ органов брюшной полости определяется увеличение селезенки. Фолиеводефицитную анемию приходится дифференцировать с В12-дефицитной анемией, острым эритромиелозом, миелодиспластическим синдромом, пароксизмальной ночной гемоглобинурией, гипопластической и аутоиммунной гемолитической анемией.
Лечение фолиеводефицитной анемии
Лечение фолиеводефицитной анемии требует нормализации питания, устранения провоцирующих факторов, проведения заместительной терапии. Пациентам назначается прием фолиевой кислоты внутрь в дозе 1-5 мг в сутки в течение 4-6 недель под контролем лабораторных показателей крови. При терапии антагонистами витамина В9 назначается его парентеральное введение. Пациенты с нарушением всасывание фолиевой кислоты, должны находиться под наблюдением гематолога и пожизненно получать заместительную терапию.
Профилактика
В профилактическом приеме фолиевой кислоты нуждаются беременные, больные с талассемией, гемолитической анемией. В целях предупреждения патологии плода и акушерских осложнений прием фолиевой кислоты по 0,4 мг/сутки необходимо начинать еще в рамках прегравидарной подготовки (за несколько месяцев до зачатия) и продолжать на протяжении всей беременности и грудного вскармливания. Известно, что профилактический прием фолиевой кислоты, начатый еще до наступления беременности, позволяет снизить частоту рождения детей с врожденными пороками ЦНС в 3,5 раза.
Гемолитическая анемия
Гемолитическая анемия – патология эритроцитов, отличительным признаком которой является ускоренное разрушение красных кровяных телец с высвобождением повышенного количества непрямого билирубина. Для данной группы заболеваний типично сочетание анемического синдрома, желтухи и увеличения размеров селезенки. В процессе диагностики исследуется общий анализ крови, уровень билирубина, анализ кала и мочи, УЗИ органов брюшной полости; проводится биопсия костного мозга, иммунологические исследования. В качестве методов лечения используется медикаментозная, гемотрансфузионная терапия; при гиперспленизме показана спленэктомия.
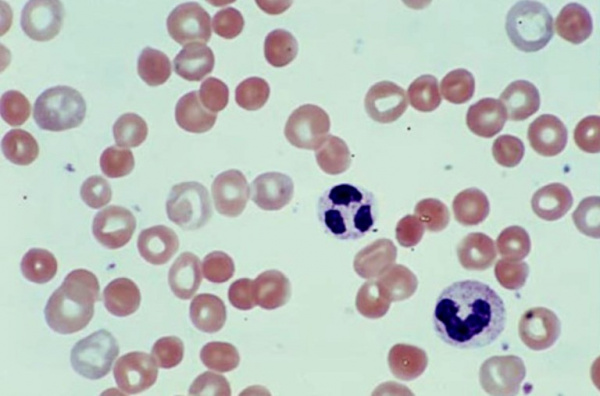
Гемолитическая анемия (ГА) - малокровие, обусловленное нарушением жизненного цикла эритроцитов, а именно преобладанием процессов их разрушения (эритроцитолиза) над образованием и созреванием (эритропоэзом). Данная группа анемий очень обширна. Их распространенность неодинакова в различных географических широтах и возрастных когортах; в среднем патология встречается у 1% населения. Среди прочих видов анемий на долю гемолитических приходится 11%. Патология характеризуется укорочением жизненного цикла эритроцитов и их распадом (гемолизом) раньше времени (через 14-21 день вместо 100-120 суток в норме). При этом разрушение эритроцитов может происходить непосредственно в сосудистом русле (внутрисосудистый гемолиз) или в селезенке, печени, костном мозге (внесосудистый гемолиз).
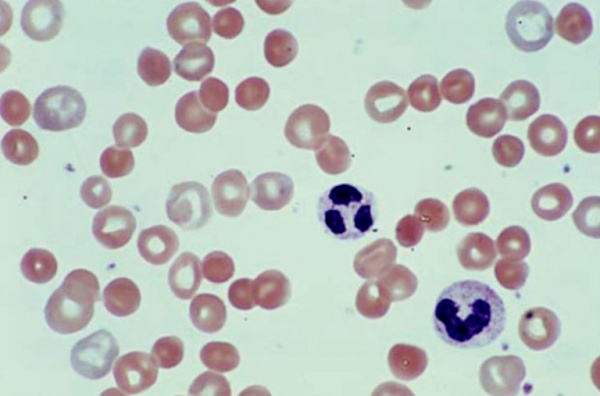
Этиопатогенетическую основу наследственных гемолитических синдромов составляют генетические дефекты мембран эритроцитов, их ферментных систем либо структуры гемоглобина. Данные предпосылки обусловливают морфофункциональную неполноценность эритроцитов и их повышенное разрушение. Гемолиз эритроцитов при приобретенных анемиях наступает под влиянием внутренних факторов или факторов окружающей среды, среди которых:
- Аутоиммунные процессы. Образование антител, агглютинирующих эритроциты, возможно при гемобластозах (остром лейкозе, хроническом лимфолейкозе, лимфогранулематозе), аутоиммунной патологии (СКВ, неспецифическом язвенном колите), инфекционных заболеваниях (инфекционном мононуклеозе, токсоплазмозе, сифилисе, вирусной пневмонии). Развитию иммунных гемолитических анемий могут способствовать посттрансфузионные реакции, профилактическая вакцинация, гемолитическая болезнь плода.
- Токсическое действие на эритроциты. В ряде случаев острому внутрисосудистому гемолизу предшествует отравление мышьяковистыми соединениями, тяжелыми металлами, уксусной кислотой, грибными ядами, алкоголем и др. Вызывать разрушение клеток крови может прием определенных лекарств (противомалярийных препаратов, сульфаниламидов, производных нитрофуранового ряда, анальгетиков).
- Механическое повреждение эритроцитов. Гемолиз эритроцитов может наблюдаться при тяжелых физических нагрузках (длительной ходьбе, беге, лыжном переходе), при ДВС-синдроме, малярии, злокачественной артериальной гипертензии, протезировании клапанов сердца и сосудов, проведении гипербарической оксигенации, сепсисе, обширных ожогах. В этих случаях под действием тех или иных факторов происходит травматизация и разрыв мембран изначально полноценных эритроцитов.
Центральным звеном патогенеза ГА является повышенное разрушение эритроцитов в органах ретикулоэндотелиальной системы (селезенке, печени, костном мозге, лимфатических узлах) или непосредственно в сосудистом русле. При аутоиммунном механизме анемии происходит образование антиэритроцитарных АТ (тепловых, холодовых), которые вызывают ферментативный лизис мембраны эритроцитов. Токсические вещества, являясь сильнейшими окислителями, разрушают эритроцит за счет развития метаболических, функциональных и морфологических изменений оболочки и стромы красных кровяных телец. Механические факторы оказывают прямое воздействие на клеточную мембрану. Под влиянием этих механизмов из эритроцитов выходят ионы калия и фосфора, а внутрь поступают ионы натрия. Клетка разбухает, при критическом увеличении ее объема наступает гемолиз. Распад эритроцитов сопровождаются развитием анемического и желтушного синдромов (так называемой «бледной желтухой»). Возможно интенсивное окрашивание кала и мочи, увеличение селезенки и печени.
Классификация
В гематологии гемолитические анемии подразделяются на две большие группы: врожденные (наследственные) и приобретенные. Наследственные ГА включают следующие формы:
- эритроцитарные мембранопатии (микросфероцитоз – болезнь Минковского-Шоффара, овалоцитоз, акантоцитоз) – анемии, обусловлены структурными аномалиями мембран эритроцитов
- ферментопении (энзимопении) – анемии, вызванные дефицитом тех или иных ферментов (глюкозо-6-фосфатдегидрогеназы, пируваткиназы и др.)
- гемоглобинопатии- анемии, связанные с качественными нарушениями структуры гемоглобина или изменением соотношения его нормальных форм (талассемия, серповидно-клеточная анемия).
Приобретенные ГА подразделяются на:
- мембранопатии приобретенные (пароксизмальная ночная гемоглобинурия – б-нь Маркиафавы-Микели, шпороклеточная анемия)
- иммунные (ауто- и изоиммунные) – обусловлены воздействием антител
- токсические – анемии, обусловленные воздействием химических веществ, биологических ядов, бактериальных токсинов
- механические - анемии, вызванные механическим повреждением структуры эритроцитов (тромбоцитопеническая пурпура, маршевая гемоглобинурия)
Симптомы
Наследственные мембранопатии, ферментопении и гемоглобинопатии
Наиболее распространенной формой данной группы анемий является микросфероцитоз, или болезнь Минковского-Шоффара. Наследуется по аутосомно-доминантному типу; обычно прослеживается у нескольких представителей семьи. Дефектность эритроцитов обусловлена дефицитом в мембране актомиозиноподобного белка и липидов, что приводит к изменению формы и диаметра эритроцитов, их массивному и преждевременному гемолизу в селезенке. Манифестация микросфероцитарной ГА возможна в любом возрасте (в младенчестве, юношестве, старости), однако обычно проявления возникают у детей старшего возраста и подростков. Тяжесть заболевания варьирует от субклинического течения до тяжелых форм, характеризующихся часто повторяющимися гемолитическими кризами. В момент криза нарастает температура тела, головокружение, слабость; возникают боли в животе и рвота.
Основным признаком микросфероцитарной гемолитической анемии служит желтуха различной степени интенсивности. Вследствие высокого содержания стеркобилина кал становится интенсивно окрашенным в темно-коричневый цвет. У пациентов с болезнь Минковского-Шоффара наблюдается склонность к образованию камней в желчном пузыре, поэтому часто развиваются признаки обострения калькулезного холецистита, возникают приступы желчной колики, а при закупорке холедоха конкрементом - обтурационная желтуха. При микросфероцитозе во всех случаях увеличена селезенка, а у половины пациентов – еще и печень. Кроме наследственной микросфероцитарной анемии, у детей часто встречаются другие врожденные дисплазии: башенный череп, косоглазие, седловидная деформация носа, аномалии прикуса, готическое нёбо, полидактилия или брадидактилия и пр. Пациенты среднего и пожилого возраста страдают трофическими язвами голени, которые возникают в результате гемолиза эритроцитов в капиллярах конечностей и плохо поддаются лечению.
Энзимопенические анемии связаны с недостатком определенных ферментов эритроцитов (чаще - Г-6-ФД, глутатион-зависимых ферментов, пируваткиназы и др). Гемолитическая анемия может впервые заявлять о себе после перенесенного интеркуррентного заболевания или приема медикаментов (салицилатов, сульфаниламидов, нитрофуранов). Обычно заболевание имеет ровное течение; типична «бледная желтуха», умеренная гепатоспленомегалия, сердечные шумы. В тяжелых случаях развивается ярко выраженная картина гемолитического криза (слабость, рвота, одышка, сердцебиение, коллаптоидное состояние). В связи с внутрисосудистым гемолизом эритроцитов и выделением гемосидерина с мочой последняя приобретает темный (иногда черный) цвет. Особенностям клинического течения гемоглобинопатий - талассемии и серповидно-клеточной анемии посвящены самостоятельные обзоры.
Приобретенные гемолитические анемии
Среди различных приобретенных вариантов чаще других встречаются аутоиммунные анемии. Для них общим пусковым фактором выступает образование антител к антигенам собственных эритроцитов. Гемолиз эритроцитов может носить как внутрисосудистый, так и внутриклеточный характер. Гемолитический криз при аутоиммунной анемии развивается остро и внезапно. Он протекает с лихорадкой, резкой слабостью, головокружением, сердцебиением, одышкой, болями в эпигастрии и пояснице. Иногда острым проявлениям предшествуют предвестники в виде субфебрилитета и артралгий. В период криза стремительно нарастает желтуха, не сопровождающаяся кожным зудом, увеличивается печень и селезенка. При некоторых формах аутоиммунных анемий больные плохо переносят холод; в условиях низких температур у них может развиваться синдром Рейно, крапивница, гемоглобинурия. Вследствие недостаточности кровообращения в мелких сосудах возможны осложнения в виде гангрены пальцев ног и рук.
Токсические анемии протекают с прогрессирующей слабостью, болями в правом подреберье и поясничной области, рвотой, гемоглобинурией, высокой температурой тела. Со 2-3 суток присоединяется желтуха и билирубинемия; на 3-5 сутки возникает печеночная и почечная недостаточность, признаками которых служат гепатомегалия, ферментемия, азотемия, анурия. Отдельные виды приобретенных гемолитических анемий рассмотрены в соответствующих статьях: «Гемоглобинурия» и «Тромбоцитопеническая пурпура», «Гемолитическая болезнь плода».
Осложнения
Каждый вид ГА имеет свои специфические осложнения: например, ЖКБ – при микросфероцитозе, печеночная недостаточность – при токсических формах и т.д. К числу общих осложнений относятся гемолитические кризы, которые могут провоцироваться инфекциями, стрессами, родами у женщин. При остром массивном гемолизе возможно развитие гемолитической комы, характеризующейся коллапсом, спутанным сознанием, олигурией, усилением желтухи. Угрозу жизни больного несут ДВС-синдром, инфаркт селезенки или спонтанный разрыв органа. Неотложной медицинской помощи требуют острая сердечно-сосудистая и почечная недостаточность.
Определение формы ГА на основе анализа причин, симптоматики и объективных данных относится к компетенции гематолога. При первичной беседе выясняется семейный анамнез, частота и тяжесть протекания гемолитических кризов. В процессе осмотра оценивается окраска кожных покровов, склер и видимых слизистых, производится пальпация живота для оценки величины печени и селезенки. Сплено- и гепатомегалия подтверждается при проведении УЗИ печени и селезенки. Лабораторный диагностический комплекс включает:
- Исследование крови. Изменения в гемограмме характеризуются нормо- или гипохромной анемией, лейкопенией, тромбоцитопенией, ретикулоцитозом, ускорением СОЭ. В биохимических пробах крови определяется гипербилирубинемия (увеличение фракции непрямого билирубина), увеличение активности лактатдегидрогеназы. При аутоиммунных анемиях большое диагностическое значение имеет положительная проба Кумбса.
- Анализы мочи и кала. Исследование мочи выявляет протеинурию, уробилинурию, гемосидеринурию, гемоглобинурию. В копрограмме повышено содержание стеркобилина.
- Миелограмму. Для цитологического подтверждения выполняется стернальная пункция. Исследование пунктата костного мозга обнаруживает гиперплазию эритроидного ростка.
В процессе дифференциальной диагностики исключаются гепатиты, цирроз печени, портальная гипертензия, гепатолиенальный синдром, порфирии, гемобластозы. Пациента консультируют гастроэнтеролог, клинический фармаколог, инфекционист и другие специалисты.
Лечение
Различные формы ГА имеют свои особенности и подходы к лечению. При всех вариантах приобретенной гемолитической анемии необходимо позаботиться об устранении влияния гемолизирующих факторов. Во время гемолитических кризов больным необходимы инфузии растворов, плазмы крови; витаминотерапия, по необходимости – гормоно- и антибиотикотерапия. При микросфероцитозе единственно эффективным методом, приводящим к 100 % прекращению гемолиза, является спленэктомия.
При аутоиммунной анемии показана терапия глюкокортикоидными гормонами (преднизолоном), сокращающая или прекращающая гемолиз. В некоторых случаях требуемый эффект достигается назначением иммунодепрессантов (азатиоприна, 6-меркаптопурина, хлорамбуцила), противомалярийных препаратов (хлорохина). При резистентных к медикаментозной терапии формах аутоиммунной анемии выполняется спленэктомия. Лечение гемоглобинурии предполагает переливание отмытых эритроцитов, плазмозаменителей, назначение антикоагулянтов и антиагрегантов. Развитие токсической гемолитической анемии диктует необходимость проведения интенсивной терапии: дезинтоксикации, форсированного диуреза, гемодиализа, по показаниям – введение антидотов.
Прогноз и профилактика
Течение и исход зависят от вида анемии, тяжести протекания кризов, полноты патогенетической терапии. При многих приобретенных вариантах устранение причин и полноценное лечение приводит к полному выздоровлению. Излечения врожденных анемий добиться нельзя, однако возможно достижение длительной ремиссии. При развитии почечной недостаточности и других фатальных осложнений прогноз неблагоприятен. Предупредить развитие ГА позволяет профилактика острых инфекционных заболеваний, интоксикаций, отравлений. Запрещается бесконтрольное самостоятельное использование лекарственных препаратов. Необходимо тщательная подготовка пациентов к гемотрансфузиям, вакцинации с проведением всего комплекса необходимых обследований.
4. Клинические рекомендации по диагностике и лечению аутоиммунных гемолитический анемий/ Цветаева Н.В., Никулина О.Ф. - 2014.
Апластическая анемия ( Гипопластическая анемия )
Апластическая анемия – угнетение функции кроветворения красного костного мозга (эритроцитопоэза, лейкопоэза и тромбоцитопоэза), приводящее к пангемоцитопении. К основным клиническим проявлениям гематологического синдрома принадлежат головокружение, слабость, обмороки, одышка, покалывание в груди, кожные геморрагии, кровотечения, склонность к развитию инфекционно-воспалительных и гнойных процессов. Заболевание диагностируется на основании характерных изменений гемограммы, миелограммы и гистологического исследования трепанобиоптата. Лечение патологии включает проведение гемотрансфузий, иммуносупрессивной терапии, миелотрансплантации.
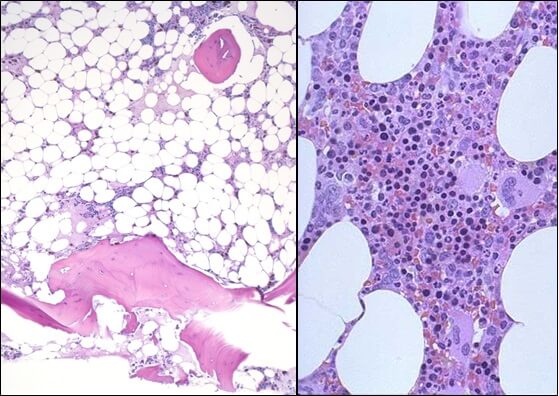
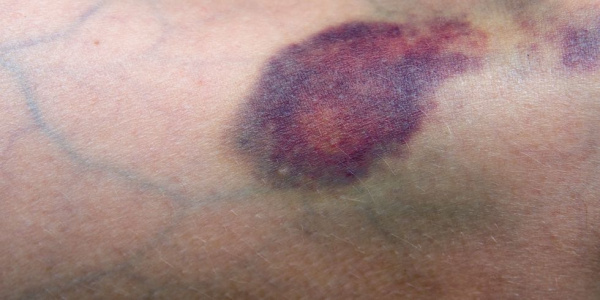
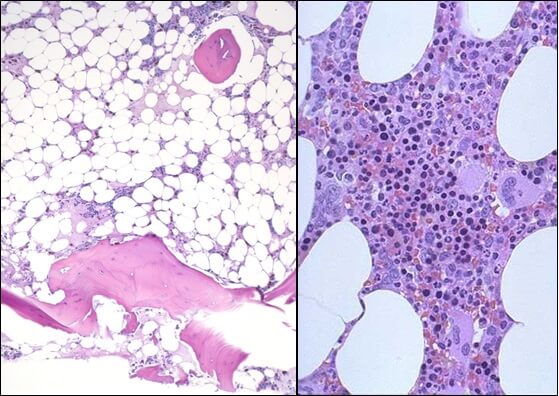
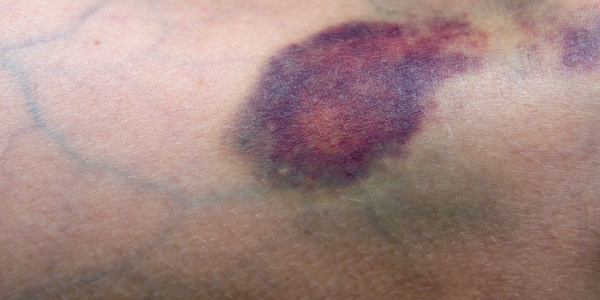
Апластическая (гипопластическая) анемия – тяжелое расстройство гемопоэза (чаще всех его звеньев), сопровождающееся развитием анемического, геморрагического синдромов и инфекционных осложнений. Развивается в среднем у 2 человек на 1 млн. населения в год. Приблизительно с одинаковой частотой патология поражает мужчин и женщин. Возрастные пики заболеваемости приходятся на возраст 10–25 и старше 50 лет. При данной патологии в костном мозге чаще нарушается образование всех трех типов клеточных элементов крови (эритроцитов, лейкоцитов и тромбоцитов), иногда - только одних эритроцитов; в зависимости от этого различают истинную и парциальную апластическую анемию. В гематологии данный вид анемии относится к числу потенциально фатальных заболеваний, приводящих к гибели 2/3 заболевших.
По происхождению апластическая анемия может быть врожденной (связанной с хромосомными аберрациями) и приобретенной (развившейся в течение жизни). Принято считать, что угнетение миелопоэза связано с появлением в красном костном мозге и крови цитотоксических T-лимфоцитов, производящих фактор некроза опухолей и γ-интерферон, которые в свою очередь подавляют ростки кроветворения. Запускать этот механизм могут различные внешнесредовые (химические соединения, физические явления, лекарственные вещества), а также эндогенные факторы (вирусы, аутоиммунные реакции). К числу наиболее значимых причин относят:
- Прием миелотоксических препаратов. Достоверно установлена связь анемии с приемом некоторых противоопухолевых, противосудорожных, антибактериальных, антитиреоидных, противомалярийных препаратов, транквилизаторов, препаратов золота и др., обладающих потенциальным миелотоксическим эффектом. Лекарственные вещества могут вызывать как прямое повреждение стволовых кроветворных клеток, так и опосредованное - через аутоиммунные реакции. Анемии, связанные с таким механизмом развития, называются лекарственными.
- Контакт с химическими и физическими агентами. Супрессию костного мозга может вызывать взаимодействие с органическими растворителями, соединениями мышьяка, бензольными соединениями, пестицидами, облучение всего тела. В некоторых случаях недостаточность гемопоэза является временной и обратимой - главными факторами здесь являются концентрация/доза вещества и время контакта. супрессию костного мозга.
- Вирусные инфекции. Из вирусных агентов наибольшее значение уделяется возбудителям гепатитов В, С и D. В этом случае гипопластическая анемия обычно развивается в течение полугода после перенесенного вирусного гепатита. При изучении патогенеза было замечено, что репликация вируса происходит в мононуклеарах крови и костного мозга, а также в иммунных клетках. Предполагается, что подавление миелопоэза в этом случае является своеобразным иммунным ответом, возникающим против клеток, несущих на своей поверхности вирусные антигены. Такой вид анемии выделяется в отдельную форму – постгепатитную. Среди других вирусных инфекций называются ЦМВ, инфекционный мононуклеоз, грипп.
Также описаны случаи панцитопении, вызванные инфицированием туберкулезом, интоксикацией, лучевой болезнью, лимфопролиферативными заболеваниями (тимомой, лимфомой, хроническим лимфобластным лейкозом), беременностью. Почти в половине наблюдений причину анемии выявить не удается - такие случаи относят к идиопатической форме.
В основе апластической анемии может лежать либо первичное повреждение гемопоэтических стволовых клеток, либо нарушение их эффективной дифференцировки. При наследственных анемиях недостаточность гемопоэза опосредована кариотипическими аберрациями, приводящими к нарушению репарации ДНК и невозможности репликации стволовых клеток костного мозга. В случае приобретенной анемии под влиянием этиофакторов наблюдается активация Т-клеток, которые начинают продуцировать цитокины (интерферон-гамма, ФНО), поражающие клетки-предшественники гемопоэза. В стволовых клетках костного мозга повышается экспрессия генов, отвечающих за апоптоз и активизацию клеточной гибели. Основные клинические проявления обусловлены пангемоцитопенией – снижением в составе крови всех ее форменных элементов (эритроцитов, лейкоцитов, тромбоцитов).
Кроме различных этиологических вариантов (лекарственного, постгепатитного, идиопатического), различают острую (до 1 мес. течения), подострую (от 1 до 6 мес.) и хроническую (более 6 мес.) форму заболевания. Анемию, протекающую с избирательным угнетением эритропоэза, называют парциальной красноклеточной аплазией. На основании выраженности тромбо- и гранулоцитопении данная форма анемии подразделяется на 3 степени тяжести:
- очень тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,2х109/л)
- тяжелую (тромбоцитов менее 20,0х109/л; гранулоцитов менее 0,5х109/л), по данным трепанобиопсии – низкая клеточность костного мозга (менее 30% от нормы)
- умеренную (тромбоцитов более 20,0х109/л; гранулоцитов более 0,5х109/л)
Симптомы апластической анемии
Поражение трех гемопоэтических ростков (эритро-, тромбоцито- и лейкопоэза) обусловливает развитие анемического и геморрагического синдромов, инфекционных осложнений. Дебют апластической анемии обычно происходит остро. Анемический синдром сопровождается общей слабостью и утомляемостью, бледностью кожи и видимых слизистых, шумом в ушах, головокружением, покалыванием в груди, одышкой при нагрузке.
Основным проявлением тромбоцитопении выступает геморрагический синдром. Больные отмечают появление петехий и экхимозов на коже, повышенную кровоточивость десен, спонтанные носовые кровотечения, меноррагии. Возможно возникновение гематурии, маточных и желудочно-кишечных кровотечений. Следствием лейкопении и агранулоцитоза служит частое развитие инфекционных процессов – стоматитов, пневмоний, инфекций кожи и мочевыводящих путей. Для апластической анемий нехарактерны похудание, лимфаденопатия, гепато- и спленомегалия – при этих признаках следует искать другую причину пангемоцитопении.
Врожденная апластическая анемия (синдром Фанкони) обычно развивается у детей в возрасте до 10 лет и кроме аплазии костного мозга характеризуется другими нарушениями: микроцефалией, гипоплазией почек, низкорослостью, аномалиями развития верхних конечностей (гипоплазией первой пястной и лучевой кости), гипоспадией, гиперпигментацией кожи, крайней степенью тугоухости и др. При наследственной анемии Эстрена-Дамешека отмечается тотальное поражение кроветворения и панцитопения при отсутствии врожденных аномалий развития. Для анемии Даймонда-Блекфена или парциальной красноклеточной аплазии характерно только снижение количества эритроцитов.
Летальный исход может быть обусловлен кровоизлияниями во внутренние органы, массивными кровотечениями, инфекционными осложнениями, анемической комой. Наиболее грозное из геморрагических осложнений – кровоизлияние в головной мозг (геморрагический инсульт). Больные склонны к частым и тяжело протекающим вирусным и бактериальным инфекциям респираторного тракта. Значительное или стремительное снижение уровня красных кровяных телец может привести к анемической коме. При молниеносной форме крайне быстро развиваются тяжелейшая анемия, иммунодефицит, коагулопатии, имеющие фатальные последствия.
Оценка гематологического статуса включает внимательный клинический осмотр и проведение тщательной лабораторной диагностики. При физикальном обследовании выявляется выраженная бледность или желтушность кожи, артериальная гипотония, тахикардия. Основу диагностического алгоритма составляет проведение общего и биохимического анализа крови, стернальной пункции, трепанобиопсии:
- Исследования крови. Для гемограммы при гипопластической анемии типичны эритро-, лейкоцито- и тромбоцитопения, нейтропения и относительный лимфоцитоз. Оценка биохимических показателей (печеночных проб, нефрологического комплекса, сывороточного железа, билирубина) информативна для исключения других анемий.
- Исследованиепунктата костного мозга. В миелограмме обнаруживается уменьшение количества миелокариоцитов и мегакариоцитов, снижение клеточности. В трепанобиоптате определяется замещение красного костного мозга жировым (желтым).
В рамках диагностического поиска апластическую анемию необходимо дифференцировать с мегабластными (В12-дефицитными, фолиеводефицитными) анемиями, идиопатической тромбоцитопенической пурпурой, пароксизмальной ночной гемоглобинурией, острым лейкозом.
Лечение апластической анемии
Больные с апластической анемией госпитализируются в специализированные отделения. Им обеспечиваются полная изоляция и асептические условия для предупреждения возможных инфекционных осложнений. Проведение эффективного лечения является сложной проблемой практической гематологии. В зависимости от уровня цитопении используются следующие лечебные подходы:
- Иммуносупрессиная терапия. При умеренной цитопении назначается фармакотерапия, включающая комбинацию антитимоцитарного иммуноглобулина и циклоспорина А. Поддерживающая терапия проводится анаболическими стероидами или их сочетанием с циклоспоринами.
- Гемотрансфузии. В комплексе с курсом иммуносупрессивной терапии при низких показателях красной крови показано проведение заместительной гемотрансфузионной терапии (переливание тромбоцитов и эритроцитарной массы), плазмафереза. Данная мера не оказывает воздействия на патогенетическое звено заболевания, но позволяет восполнить дефицит кровяных телец, не вырабатываемых костным мозгом.
- Трансплантация КМ и СК. Наиболее благоприятные прогнозы на долгосрочную выживаемость оказывает выполнение аллогенной трансплантации костного мозга. Однако ввиду сложности подбора иммунологически совместимого донора процедура используется ограниченно. В качестве экспериментальных подходов рассматриваются аутологичные трансплантации, пересадка стволовых клеток периферической крови. Больным с нетяжелой формой анемии может быть показано проведение спленэктомии, эндоваскулярной окклюзии селезеночной артерии.
Прогноз определяется этиологической формой, тяжестью и остротой течения анемии. Критериями неблагоприятного исхода служат быстрое прогрессирование заболевания, тяжелый геморрагический синдром и инфекционные осложнения. После трансплантации костного мозга ремиссии удается достичь у 75–90% пациентов. Первичная профилактика данной разновидности анемии предполагает исключение влияния неблагоприятных внешнесредовых факторов, необоснованного применения лекарственных препаратов, предупреждение инфекционной заболеваемости и др. Пациентам с уже развившимся заболеванием требуется диспансерное наблюдение гематолога, систематическое обследование и длительная поддерживающая терапия.
2. Комплексная программа диагностики апластической анемии с определением прогностически значимых патогенетических особенностей заболевания. Методические рекомендации. - 2015.
4. Апластическая анемия: современные представления о патогенезе и терапии/ Айсариева Б. К., Раймжанов А. Р., Айтбаев К.// Молодой ученый. - 2011 - №9.
Причины апластической анемии - конституционная апластическая анемия Фанкони
Этиология апластической анемии весьма разнообразна. Основные причины заболевания и их классификация приведены в ниже.
Этиологическая классификация апластических анемий:
I. Генуинные
II. Конституционные
III. Вызываемые физическими или химическими факторами
IV. Вирусные заболевания (гепатит), бактериальные инфекции (туберкулез).
V. Иммунологические заболевания (диссеминированная красная волчанка, аллергия и пр.).
VI. Ночная пароксизмальная гемоглобинурйя.
VII. Прочие: беременность, эндокринные болезни (Симмондса), хронический панкреатит и пр.
Не забывать, что тот же возбудитель обусловливает различные аспекты костного мозга вплоть до очень тяжелой формы аплазии.

Генуинная форма апластической анемии
К этой группе заболеваний относятся все те случаи, при которых не обнаруживается какой-либо причинный фактор (50%).
Однако, за последние десятилетия отмечается рост показателя заболеваемости этой формой болезни, что, видимо, следует отнести за счет загрязненности среды (ионизирующие излучения), питания, чрезмерного потребления медикаментов, вирусных заболеваний.
Привлекает внимание очень тяжелое течение этой формы у детей, картина носит острый характер и нередко смертельный исход наступает быстро. Гистологическое исследование выявило различную степень поражения костного мозга — от нормопластического аспекта вплоть до весьма тяжелой аплазии.
Необходимо отметить также формы с минимальной бластической реакцией в строении костного мозга или с лимфоидной реакцией, которые нередко трудно различить от острой лейкемии с небольшим процентом » или от апластической формы хронической лимфатической лейкемии (Bryon, Dreyfus и Bessis).
Впрочем в литературе описаны случаи так называемой генуинной апластической анемии, которые, по существу, оказывались «предлейкемическим состоянием» (Dreyfus и Bessis).

Конституционная или семейная генуинная анемия - апластическая анемия Фанкони
Первый случай был описан Фанкони в 1927 г. К этой патегории относятся случаи семейного характера, развивающиеся у детей с костномозговой аплазией и дефектами, как, например, синдактилия, правое сердце, стрельчатое небо, микроцефалия, умственная отсталость.
Гематологическая картина отражает наличие периферической панцитопении и нормоцитной или умеренно макроцитной анемий; показатель плодного гемоглобина бывает завышенным. При этом костный мозг представляется гипопластическим, жирным, иной раз нормо- или гипоцеллюлярным (Fanconi, Rohr Williams). Синдром видимо определяет рецессивный ген, в то же время цитогенетические исследования выявили большое разнообразие структурнохромосомных сдвигов (Bloom и сотр.). Описаны случаи, преобразовавшиеся в дальнейшем в острую лейкемию (Wintrobe).
Отмечается также другой вид семейной панцитопении, когда этому заболеванию сопутствует недостаточность поджелудочной железы по причине ее кистовидного фиброзирования (Williams).
Читайте также: