Миоклоническая эпилепсия с атаксией и глухотой
Добавил пользователь Skiper Обновлено: 27.01.2026
При деменции отмечают выраженные органические поражения большого и промежуточного мозга. В некоторых случаях, таких как сочетание болезни Альцгеймера и сенильной деменции, а также болезнь Пика, основой является дегенеративный процесс и гибель нервных клеток в ассоциативных полях коры головного мозга со вторичными изменениями подкоркового белого вещества.
К деменции может также приводить ограниченная дегенерация нейронов зрительного бугра. При других заболеваниях, таких как хорея Гентингтона, спиноцеребральная дегенерация и некоторые виды дегенеративных изменений в подкорковых ядрах и больших полушариях, деменция сопровождается дегенерацией нейронов скорлупы и хвостатого ядра.
Артериосклеротическое поражение может привести к множественным инфарктам в зрительном бугре, базальных ядрах, стволе мозга и больших полушариях, которые сопровождаются деменцией. Могут поражаться двигательные, чувствительные и зрительные проекционные поля, а также ассоциативные поля.
В тех случаях, когда в клинической картине преобладает умственная деградация, это состояние называют мультиинфарктной деменцией.
К ступору, коме или деменции приводят травмы, вызывающие ушиб извилин головного мозга, особенно нижних и передних отделов лобной и височной долей, а также некрозы и кровоизлияния в области среднего мозга. Большинство заболеваний, приводящих к деменции, поражают довольно обширную область; лобные и теменные доли страдают чаще, чем другие отделы головного мозга.
Б некоторых случаях могут действовать и другие механизмы. Так, например, снижение умственной деятельности и нарушение походки происходят в результате длительного повышения внутричерепного давления и хронической гидроцефалии (при значительном расширении желудочков мозга давление может не превышать 180 мм рт. ст.). Основным фактором является компрессия подкоркового белого вещества.
К выраженному расстройству корковых функций может приводить сдавление одного или обоих полушарий головного мозга хронической субдуральной гематомой. При сифилисе и некоторых вирусных инфекциях, например герпетическом энцефалите, одной из причин развития деменции является диффузный воспалительный процесс.
Некоторые токсикозы и болезни обмена веществ, обсуждаемые в гл. 349 и 350, могут приводить к нарушению деятельности нервной системы и создавать клиническую картину, сходную, если не идентичную, таковой при деменции. Следует помнить, что биохимические расстройства также вызывают нарушение деятельности нервной системы.
Клиническая классификация деменций
I. Болезни, при которых деменция является единственным проявлением невро¬логического или терапевтического заболевания
А. Болезнь Альцгеймера и сенильная деменция
II. Болезни, при которых деменция сопровождается другими неврологическими проявлениями, но нет явного наличия другого заболевания
А. Всегда в сочетании с другими неврологическими симптомами:
1. Хорея Гентингтона (хореоатетоз)
2. Лейкодистрофии: болезнь Шильдера, адренолейкодистрофия, мета-хроматическая лейкодистрофия и сходные демиелинизирующие забо¬левания (спастический парез, псевдобульбарный паралич, слепота, глухота)
3. Липофусциноз и другие болезни накопления липидов (миоклонические приступы, слепота, спастичность, мозжечковая атаксия)
4. Миоклонус-эпилепсия (множественные миоклонии, генерализованные судорожные припадки, мозжечковая атаксия)
5. Болезнь Крейтцфельда — Якоба (множественные миоклонии и моз¬жечковая атаксия)
6. Дегенеративные поражения мозжечка и большого мозга (мозжечко-вые атаксии, оливопонтоцеребеллярная и другие)
7. Дегенерации, при которых происходят изменения в базальных ядрах и больших полушариях (апраксия и ригидность) и надъядерный пара¬лич (паралич вертикального взора, кривошея)
8. Деменция со спастической параплегией
9. Кальцификация базальных ядер (идиопатическая и гиперпаратиреоз)
10. Болезнь Геллервордена — Шпатца (пирамидные и экстрапирамидные знаки)
11. Деменция при болезни Паркинсона (тремор, ригидность, брадикинезии)
Б. Часто в сочетании с другими неврологическими симптомами:
1. Церебральный артериосклероз и ишемический инфаркт
2. Опухоль головного мозга, особенно глиомы лобной и теменной долей, а также мозолистого тела
3. Травма головного мозга (ушиб мозга, кровоизлияния в средний мозг, хроническая субдуральная гематома)
4. Болезнь Маркьяфавы — Биньями (апраксия и другая лобная симпто¬матика)
5. Нормотензивная гидроцефалия (почти всегда с атаксией походки и часто с нарушением функции тазовых органов)
6. Хронические инфекционные поражения ЦНС: криптококкоз, токсоплазмоз, деменция при синдроме приобретенного иммунодефицита (СПИД)
III. Болезни, при которых деменция обычно сочетается с клиническими и лабо¬раторными признаками других заболеваний
Б. Болезнь Кушинга
В. Недостаточность питания, например пеллагра, синдром Вернике —
Корсакова, подострая сочетанная дегенерация спинного и головного мозга (недостаточность витамина Bi2) Г. Нейросифилис: прогрессивный паралич, менинговаскулярный сифилис
Д. Гепатолентикулярная дегенерация наследственная и приобретенная
Е. Хроническая лекарственная интоксикация (барбитураты и другие седативные препараты)
Не все из перечисленных заболеваний имеют одинаковое значение в качестве причин деменций. Некоторые из них встречаются редко. Из 84 больных с подтвержденной пресенильной деменцией (возраст 65 лет), обратившихся в Британский неврологический центр, у 58% выявили атрофию головного мозга, по-видимому альцгеймеровского типа, у 10% мультиинфарктную деменцию. У 30% больных обнаружили нормотензивную гидроцефалию, алкогольную деменцию, болезнь Крейтцфельда — Якоба, хорею Гентингтона, посттравматические остаточные явления или алкоголизм. У больных в возрасте старше 65 лет атрофию мозга выявили в 80% случаев, мультиинфарктную деменцию — в 15% случаев (Tomlinson). Вероятно, статистические данные, собранные Wells при осмотре 417 больных с деменцией, обращавшихся в университетскую клинику, являются наиболее репрезентативными. Эти данные представлены в табл. 23.3.
Миоклоническая эпилепсия с атаксией и глухотой
Миоклоническая эпилепсия с атаксией и глухотой
В 1968 г. May и White сообщили о семье, в которой у 6 больных в четырех поколениях был обнаружен синдром, включающий миоклонус, мозжечковую атаксию и пейросепсорную глухоту.
Клинические данные. Нервная система. У 4 из 6 больных в анамнезе имелись указания на большие судорожные припадки и у 2 из этих 4 наблюдался миоклонус. Миоклонические эпизоды появлялись в возрасте между 12 и 20 годами и заключались в сокращениях мускулатуры головы, туловища и конечностей. Некоторое время спустя, обычно в третьем десятилетии жизни, начинались генерализованные припадки. У 2 из 6 больных больших судорожных припадков не было. В юности появлялись нарушения координации в руках и ногах.
Они наблюдались у 3 из 6 больных. Атаксия, когда она имелась, прогрессировала медленно. Неврологическое исследование 3 больных выявило неустойчивую походку на широко расставленных ногах, легкий или умеренный интенционный тремор, тремор при пальценосовой или пяточно-коленной пробах, атаксию туловища. Нистагма, патологии черепно-мозговых нервов, потери чувствительности и мышечных атрофии не выявлено, хотя речь у больных с атаксией была дизартричной. Психических расстройств не отмечалось.
Орган слуха. Нарушения слуха впервые были замечены в детстве или в молодости. Они медленно прогрессировали, приводя в результате к резко выраженной глухоте в поздние годы жизни. На аудиограмме одного больного 32 лет была обнаружена двусторонняя нейросенсорная глухота (около 40 дБ). У другого больного в 50-летнем возрасте отмечалась глубокая потеря звуковоеприятия. Он слышал только громкие звуки, направленные в ухо.
Вестибулярная система. Результаты вестибулярных проб не описаны.
Лабораторные данные. У 3 обследованных больных на электроэнцефалограмме обнаружена нормальная активность в покое. У 2 больных с эпилептическими припадками в анамнезе световая стимуляция индуцировала продолжительные генерализованные спайки. В одном случае наблюдалось сочетание мноклонических сокращений в двуглавой мышце с корковыми спайками.
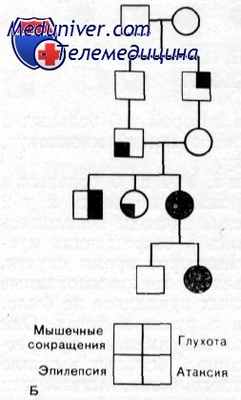
Наследственность. Проявления синдрома у 6 больных умеренно варьировали. У пробанда и у его матери был полный синдром, включающий глухоту, миоклонус, эпилепсию и атаксию. У дяди по линии матери отмечались глухота и атаксия без эпилепсии. У тетки и у деда по матери была только эпилепсия, а у прадеда по матери — только глухота. Никто из этих трех последних лиц обследован не был. Создается впечатление, что синдром наследуется по аутосомно-доминантному типу с варьирующей экспрессивностью.
Диагноз. Миоклонус характерен для нескольких разных заболеваний. Доброкачественный эссенциальный миоклонус наследуется по аутосомно-доминантному типу, но он не сочетается с атаксией или глухотой. Миоклоническая эпилепсия Лафора наследуется также по аутосомно-доминантному типу, но она характеризуется быстро нарастающим распадом психики, развивающимся в середине подросткового возраста после выявления эпилепсии.
При аутосомно-рецессивно наследующейся миоклонической эпилепсии Унверрихта, после выявления симптомов болезни, в юности развивается только легкое слабоумие. Миоклоническая эпилепсия наблюдается при гаргоилизме, который наследуется по аутосомно-рецессивному типу, выявляется в первые несколько лет жизни и приводит к смерти в 10-летнем возрасте. Миоклоническая эпилепсия Рамсея Ханта (Ramsey Hunt) наследуется также по аутосомно-рецсссивпому типу и включает миоклонус с атаксией, но без глухоты.
Лечение. При мноклопических сокращениях наиболее эффективен диазепам (диазепам, валиум). Большие судорожные припадки эффективно лечатся фенитоином (фенитоин, дифенилгидантоин, дилантин) и фенобарбиталом. При дефектах слуха можно использовать слуховые аппараты.
Прогноз. Заболевание медленно прогрессирует и ведет к расстройствам походки и слуха.
Выводы. Характеристика данного синдрома включает: 1) аутосомно-доминантное наследование с варьирующей экспрессивностью; 2) миоклоническую и grand mal эпилепсию, начинающуюся между 12 и 30 годами и встречающуюся примерно в половине случаев; 3) медленно прогрессирующую мозжечковую атаксию, начинающуюся в юности; 4) медленно прогрессирующую нейросенсорную глухоту, начинающуюся в детстве.
Новое в эпилептологии
Ведущий рубрики: Владимир Игоревич Харитонов — невролог-эпилептолог Украинского медицинского центра реабилитации детей с органическим поражением нервной системы МЗ Украины, действительный член Европейской академии эпилептологии (EUREPA) и Международной ассоциации детских неврологов (ICNA)
Синдром Альперса – Гуттенлохера – трагический синдром раннего детства, характеризующийся задержкой психомоторного развития различной степени тяжести, резистентной эпилепсией раннего начала (до 4-летнего возраста), эпизодами психомоторного регресса, дисфункцией печени либо печеночной недостаточностью, которая возникает вследствие назначения антиконвульсантов. Этот синдром является результатом мутации гена POLG1. Авторы приводят описание двух случаев, когда у детей с данным синдромом миоклонические припадки манифестировали на 20-й и 60-й день с момента рождения.
В первом случае ЭЭГ-исследование показывало низкоамплитудные полиспайки и комплексы полиспайк – волна, а также высокоамплитудные медленные волны, которые перемежались с паттерном вспышка – подавление.
Во втором случае наблюдались низкоамплитудные полиспайки, комплексы полиспайк – волна с периодическими десинхронизациями основной активности. В первом случае кортикальная слепота и печеночная недостаточность развились на фоне приема вальпроатов. Авторы упоминают единичный случай данного синдрома, когда резистентный эпилептический статус был установлен только после проведения функциональной гемисферэктомии.
MIRAS может возникать в детском возрасте как энцефалопатия с гепатопатией, ювенильной рефрактерной эпилепсией, мигренеподобными головными болями либо как группа атаксий – нейропатий, характерная для взрослого возраста. Данный синдром клинически проявляется как рецессивная атаксия, координаторные нарушения, дизартрия, хореоатетоз, личностные изменения, когнитивные нарушения, гемианопсия, нейропатия или эпилепсия и возникает чаще всего вследствие мутации гена POLG1 (мутации p.748W>S, p.1143E>G).
Синдром Лея развивается после мутации митохондриальной либо нуклеотидной ДНК. Митохондриальная форма встречается намного реже и имеет материнский путь наследования. Это вид наиболее часто возникает вследствие мутации m.8993T>G гена ATP6. Эпилепсия является типичным состоянием, сопровождающим синдром Лея. Наиболее часто у пациентов появляются генерализованные тонико-клонические припадки, однако изредка – эпилепсия парциальная непрерывная или синдром Веста. Недавно проведенные исследования показали, что появление синдрома Веста связано с ранней манифестацией синдрома Лея, но сам синдром Веста не влияет на прогноз последнего, и наоборот. В группе из пяти пациентов с синдромом Лея у трех наблюдались парциальные припадки, у одного – генерализованные тонические и у одного – миоклонические. У одного больного с ювенильным синдромом Лея вследствие мутации m.14487T>C эпилепсия была одним из ведущих симптомов совместно с атрофией зрительного нерва, атаксией, дистонией. Эпилепсия также наблюдалась у 17-месячного ребенка в состоянии сонливости, ано-рексии, дыхательной недостаточности, задержки моторного развития, несвоевременного выделения антидиуретического гормона, симметричного поражения шейного отдела спинного мозга и нижних отделов ствола головного мозга. У пациентов с синдромом Лея, унаследованным от матерей, эпилепсия проявляется относительно редко.
Синдром MELAS клинически характеризуется инсультоподобными эпизодами, энцефалопатией, эпилепсией и лактоацидозом. В 80% случаев он возникает вследствие перехода m.3243A>G в гене tRNA(Leu). Синдром может сочетаться с парциальными, генерализованными припадками, включая эпилептический статус. В исследовании при участии пяти пациентов с данным синдромом эпилепсия развивалась в возрасте 14-39 лет. Перед появлением эпиприступов у больных наблюдались инсультоподобные эпизоды (n = 3), мигрень (n = 2), диабет (n = 2), задержка психомоторного развития (n = 2). У четырех пациентов появились парциальные припадки и у одного – тонико-клонические. ЭЭГ-запись у больных показала диффузное (n = 4), одностороннее замедление (n = 1), фокальные острые волны в задних отделах (n = 4), симметричные острые волны (n = 2), фотосенситивность (n = 2). У лиц с мутацией m.3243A>G, манифестирующей с диабета и глухоты, эпилепсия наблюдалась редко.
Синдром MEMSA наиболее часто возникает вследствие мутации гена POLG1. Церебеллярная атаксия – это, как правило, первый симптом, развивающийся в подростковом возрасте. Миопатия – следующий симптом – может быть дистальной, проксимальной или же проявляться только при физических нагрузках. Миоклоническая эпилепсия является обязательной составной час-тью данного синдрома. Она развивается относительно поздно и проявляется судорогами в руках с вторичной генерализацией. Припадки бывают устойчивы к противоэпилептической терапии и даже анестезии и, как правило, сопровождаются прогрессированием энцефалопатии.
Основные клинические симптомы синдрома MERRF – атаксия, миопатия и миоклоническая эпилепсия. Мутация, наиболее часто вызывающая этот синдром, – переход m.8344A>G. Мио-клоническая эпилепсия также наблюдалась вследствие мутации m.8363G>A, передаваемой по женской линии. В ис- следовании, включавшем 5 пациентов с данным синдромом, начало эпилепсии варьировало в возрасте 7-50 лет. Припадки были классифицированы как миоклонические (n = 5) и тонико-клонические (n = 1). ЭЭГ показала диффузное замедление (n = 2), фокальные затылочные острые волны (n = 1), комплексы полиспайк – волна (n = 3), симметричные острые волны с фотосенситив-ностью (n = 1).
Митохондриальные нарушения с нечасто развивающимися судорогами представлены IOSCA, синдромом Керн-са – Сейра, LBSL, LHON, NARP.
IOSCA – состояние, возникающее вследствие мутации гена POLG1, проявляющееся развитием спиноцеребеллярной атаксии в грудничковый период и иногда сопровождаемое эпилепсией. Последняя, как правило, развивается на поздних этапах развития заболевания, в частности, проявляется в виде миоклонических или парциальных припадков.
Синдром Кернса – Сейра характеризуется развитием хронической прог-рессивной наружной офтальмоплегии, пигментной ретинопатии, нарушениями кардиальной проводимости, а также дополнительными фенотипическими манифестациями. Заболевание возникает вследствие одиночной обширной делеции mtDNA. Эпилепсия не является классическим проявлением при этом синдроме, однако иногда наблюдается у отдельных пациентов. ЭЭГ обычно показывает только замедление фоновой активности.
LBSL клинически проявляется медленной прогрессивной дисфункцией пирамидальных, церебеллярных путей, дорсальной колонны, что приводит к снижению ощущений вибрации и положения тела. Дополнительные симптомы включают легкие психические нарушения, медленно прогрессирующую атаксию, спастичность и нейропатию. Данный синдром возникает вследствие мутации гена DARS2, который кодирует митохондриальную аспартил-тРНК-синтетазу. Синд-ром, развивающийся в грудничковый или ювенильный период, приводит к потере возможности ходить в подростковом возрасте или после 20 лет. Эпилепсия появляется в единичных случаях.
LHON клинически характеризуется атрофией зрительного нерва с обширным выпадением центрального зрения, наиболее часто поражает молодых мужчин. Синдром возникает вследствие наличия одной из трех гомоплазми-ческих LHON мутаций m.3460G>A, m.11778G>A, m.14484T>C генов ND1, ND4, ND6 соответственно. Иногда у пациентов могут возникать экстраокулярные симптомы, такие как мигрень, умственная отсталость или эпилепсия. У некоторых больных эпилепсия даже может предшествовать появлению глазных симптомов. Иногда эпилепсия может быть доминирующим проявлением синдрома, а также может быть устойчивой к традиционной противоэпилептической терапии.
NARP представляет собой синдром, который клинически характеризуется развитием нейропатии, атаксии, пигментного ретинита. Наиболее частой причиной его возникновения является мутация m.8993T>G гена ATP6. Эпилепсия с тяжелым течением наблюдается в формах раннего начала, но является редким феноменом в формах, дебютирующих в подростковом или взрослом возрасте. Некоторые пациенты могут не страдать припадками и в то же время иметь эпилептиформные изменения на ЭЭГ. Эпилепсия у таких больных может возникнуть по мере прогрессирования заболевания.
При систематизации особенностей течения эпилепсии интересно отметить, что практически все миоклонические формы в исследовании, приведенном авторами, относились к синдрому MERRF с классической мутацией m.8344A>G. Фотопароксизмальные реакции наблюдались у пациентов с синдромами MERRF, MELAS и Лея.
Лечение эпилепсии при митохонд-риальных нарушениях не особенно отличается от терапии таковых иного происхождения. Однако необходимо обращать внимание на некоторые моменты, например, не назначать этим пациентам антиконвульсанты с токсическими для митохондрий побочными эффектами, поскольку они могут вызвать тяжелые, иногда стремительные (возможно смертельные) нежелательные реакции, участить припадки и сделать эпилепсию резистентной. Наиболее известный антиконвульсант с токсическим действием на митохондрии – вальпроевая кислота, особенно у лиц с мутацией гена POLG1, может вызвать появление печеночной недостаточности. Поэтому назначение вальпроатов пациентам с выявленными мутациями гена POLG1 является абсолютным противопоказанием. Альтернативным видом лечения считается кетогенная диета. У лиц с синдромом Лея из-за мутации PDH гена E1a генерализованные тонико-клонические припадки могут исчезать при назначении пирувата натрия в дозе 0,5 г/кг/сут перорально. Так-же проводились попытки стимуля-ции блуждающего нерва у больных митохондриальными заболеваниями с миоклоническими и парциальными припадками, однако были неэффективны.
Панель "Нейродегенеративные заболевания"
Нейродегенеративные заболевания представляют собой широкий спектр различных по своей природе болезней, обусловленных постепенной гибелью отдельных групп нервных клеток и характеризующихся неуклонно прогрессирующим неврологическим дефицитом, включая двигательные расстройства, психоэмоциональные и когнитивные (вплоть до деменции) нарушения и эпилептические приступы. Необходимо отметить, что нейродегенеративные заболевания могут манифестировать как во взрослом, так и в детском возрасте.
Эпидемиология
Более 40 млн людей по всему миру страдают различными нейродегенеративными заболеваниями, из них порядка 10 млн — это пациенты с болезнью Паркинсона. В раннем возрасте распространённость нейродегенеративных заболеваний составляет около 1:1500.
Механизмы развития нейродегенеративных заболеваний
Патофизиологические механизмы нейродегенеративных заболеваний включают в себя синтез и агрегацию белков с патологической конформацией, что, в свою очередь, запускает каскад иммунных и метаболических нарушений, накопление металлов в центральной нервной системе, нарушение энергетического обмена клеток, дефицит ферментов и т.д. При этом, большая роль в развитии нейродегенеративного процесса принадлежит генетическим факторам. Выделяют как моногенные нейродегенеративные заболевания (например, болезнь Гентингтона, спинальные амиотрофии, ряд вариантов спиноцеребеллярных атаксий, болезнь Вильсона, моногенные варианты болезни Паркинсона, спастические параплегии, пароксизмальные дискинезии и пр.), так и болезни, развивающиеся вследствие нарушений функционирования целых ансамблей генов в сочетании с действием неблагоприятных факторов внешней среды (идиопатическая болезнь Паркинсона, атипичный паркинсонизм, болезнь Альцгеймера, боковой амиотрофический склероз, лобно-височная деменция и пр.). При этом, важно помнить, что отсутствие отягощённого семейного анамнеза по нейродегенеративному заболеванию не исключает наличие генетической предрасположенности к его развитию.
Диагностика
Диагностика нейродегенеративных заболеваний складывается из комплексной оценки клинической картины, характера дебюта и прогрессирования заболевания, лабораторно-инструментальных исследований и данных нейровизуализации. Отдельное место занимают в диагностическом процессе методы генетического тестирования. Учитывая сложные взаимоотношения генотипа и фенотипа заболевания, очень часто генетическое тестирование не ограничивается анализом одного-двух генов. В трудных диагностических случаях для удешевления и повышения эффективности диагностического поиска широко применяется метод высокопроизводительного секвенирования, позволяющего анализировать сразу до нескольких сотен генов.
- Деменции (болезнь Альцгеймера, лобно-височная деменция, наследственные прионные заболевания).
- Паркинсонизм (болезнь Паркинсона, мультисистемная атрофия, кортикобазальная дегенерация, болезнь диффузных телец Леви, прогрессирующий надъядерный паралич).
- Наследственные мышечные дистонии.
- Пароксизмальные двигательные расстройства (пароксизмальные дискинезии).
- Нейродегенерация, ассоциированная с накоплением металлов (болезнь Вильсона, нейродегенерации с накоплением железа, синдром Фара, накопление марганца).
- Атаксии (спиноцеребеллярные атаксии, не связанные с экспансией нуклеотидных повторов).
- Наследственные моторно-сенсорные нейропатии (болезнь Шарко-Мари-Тута).
- Нейрометаболические заболеваний (нейрональный цероидный липофусциноз, лизосомные болезни накопления, сфинголипидозы).
- Спастические параплегии (болезнь Штрюмпеля, в т.ч. осложнённые формы).
- Болезни мотонейрона (боковой амиотрофический склероз, спинальные мышечные атрофии).
- Болезни белого вещества (синдром исчезающего белого вещества, прогрессирующие лейкодистрофии).
- Эпилептические энцефалопатии.
- Редкие моногенные синдромы, сопровождающиеся нейродегенерацией (болезнь Айкарди-Гутьерес, синдром Жубер, синдром Кноблоха).
- Митохондриальные заболевания вследствие мутаций в ядерных генах.
Что даёт генетическое исследование?
Завершение диагностического поиска.
Выявление риска передачи заболевания по наследству. Благодаря генетической диагностике можно просчитать риск заболевания в последующих поколениях и избежать повторных случаев в семье.
Подбор правильного лечения. Несмотря на то, что в настоящее время нейродегенеративные заболевания невозможно излечить, ряд из них (например, болезнь Паркинсона, болезнь Вильсона, ряд заболеваний вследствие нарушений метаболизма) имеют специфическое патогенетическое и симптоматическое лечение. Например, при болезни Вильсона применяется пеницилламин, при болезни Ниманна-Пика типа С — миглустат. Правильный диагноз является залогом верно назначенного и эффективного лечения.
Описание и возможности метода
Исследование проводится с помощью метода высокопроизводительного секвенирования (NGS) на секвенаторе нового поколения со средним покрытием не менее 70-100х. В панель «Нейродегенеративные заболевания» входят 723 гена, мутации в которых могут приводить к развитию нейродегенеративных заболеваний. Не стоит забывать, что некоторые заболевания из группы наследственных НДЗ обусловлены экспансией нуклеотидных повторов или хромосомными перестройками, что является ограничением данного метода.
При проведении процедуры строго соблюдается конфиденциальность.
Пациент получает точные результаты, на основании которых специалисты могут составить впечатление о молекулярно-генетическом диагнозе и максимально эффективно скорректировать лечение, а также провести медико-генетическое консультирование.
Как пройти исследование
Подготовка к анализу: нет
Дни забора (приема) материала: по графику работы медицинского центра Примечание: Необходимо иметь с собой направление и выписки/заключения от врачей
Миоклонус-эпилепсия, «рваные» красные волокна (синдром МЕРРР)
Миоклонус-эпилепсия, «рваные» красные волокна (туос1о- П118 ерг1ерз1а, г১ей 1ес! ^Ьгез), или синдром МЕККЕ, впервые описана N РикиЬаы и соавт (1980) при наблюдении 2 сибсов «Рваные» красные волокна были обнаружены при морфоло!ическом исследовании скелетных мышц Обобщив данные литературы и материалы собственных наблюдений, N РикиЬага и соавт обосновали нозологическую самостоятельность синдрома МЕККЕ
Заболевание наследуется внутрисемейным полиморфизмом [ВегкоУ1с 3 Е е!; а1 , 1989, Ко\у- 1апс1 Ь Р е!; а1 , 1991] N ЕикиЬага и соавт (1991) наблюдали мужчину 26 лет, у сестры которого (21 год) клинические признаки синдрома МЕККЕ были типичными, а у него отмечались лишь постуральный тремор рук и изменения ЭЭГ Через 15 лет у больного появились умеренная мозжечковая атаксия, снижение чувствительности в ногах, периодические генерализованные тонико-клонические судороги В другой семье заболевание описано у сына и матери У сына были выявлены миоклонус при движениях, генерализованные тони- ко-клонические приступы и прогрессирующая мозжечковая атаксия с 6-летнего возраста Его мать до 62 лет была здорова, однако в дальнейшем у нее возникли приступы тонико- клонических судорог, а еще через 2 года — миоклонус при движениях, нарушение слуха и постепенно прогрессирующая мозжечковая атаксия Внутрисемейный полиморфизм клинических, биохимических и морфологических проявлений синдрома МЕККЕ может быть обусловлен, по мнению 5 Р Вегкоуш и соавт (1991), вариабельным соотношением между мутантными и нормальными т!;ДНК в различных ооцитах Синдром МЕККЕ обусловлен точечными мутациями в гене лизиновой 1;РНК в позициях 8344 и 8356 [ЗсЬойттег ^ М е
еЬ а1., 1995]. Мутация 8344 не является специфичной для синдрома МЕККР и встречается также при синдроме миоклонус-миопа- тии с туловищными липомами, проксимальной миопатии и у здоровых людей пожилого возраста [01 Маиго §., 1993; Мип- зсЬег С. еЬ а1., 1993].
Клиническая характеристика. Возраст больного при начале заболевания вариабелен — от 3 до 65 лет. Клинические проявления синдрома МЕККР крайне разнообразны. Ранними признаками являются быстрая утомляемость при физических нагрузках, развитие болей в икроножных мышцах, снижение памяти, внимания. Наиболее типичным является симптомокомплекс прогрессирующей миоклонус-эпилепсии, включающий миоклонус, атаксию и деменцию [Ое У1уо О.,
Миоклонус — облигатный признак синдрома МЕККР — наблюдается в 84 —85 % случаев [Вегкхтс 5.Р. е1 а1., 1991]. Миоклонус (греч. «беспорядочное движение мышц») — внезапное, быстрое, кратковременное мышечное сокращение, обусловленное вовлечением в патологический процесс центральной нервной системы. Миоклонические подергивания, как правило, билатеральные, массивные, но иногда выражены незначительно и включают одно или несколько мышечных сокращений. Эпилептический миоклонус при синдроме МЕККР в сравнении с неэпилептическим характеризуется корреляцией с изменениями ЭЭГ, короткими разрядами и синхронной активацией мышц при ЭМ Г-исследовании [Сеп- 1оп Р., Ко§ег .1., 1993].
Помимо миоклоний, наиболее характерными признаками синдрома МЕККР являются атаксия и деменция. Атаксия проявляется шаткостью походки и нарушением выполнения координаторных проб. В ряде случаев атаксию выявить трудно вследствие выраженных миоклоний. Снижение психических функций может предшествовать появлению основных клинических симптомов синдрома МЕККР либо возникает по мере прогрессирования заболевания.
Наряду с симптомами миоклонус-эпилепсии у большинства больных (69 %) наблюдаются генерализованные тонико- клонические судороги [Ое У1уо О., 1993]. Иногда тонико- клонические судорожные приступы возникают за несколько лет до появления основных симптомов синдрома МЕККР — миоклонуса, атаксии, деменции.
У отдельных больных описаны парциальные эпилептические пароксизмы, характеризовавшиеся различными аурами, латерализованными клоничес- кими или тонико-клоническими судорогами, зрительными галлюцинациями [Вегкоую 5 Р е! а1 , 1989]
Нейросенсорная глухота наблюдается при синдроме МЕККР в 52 % случаев и обусловлена поражением периферического отдела слухового анализатора [Ко§ег ^ е! а1 ,
Признаки миопатии (мышечная слабость, гипотрофии мышц, вспомогательные приемы при вставании) выражены незначительно либо умеренно По мнению 5 Р ВегкоУ1с и соавт (1989), данный факт обусловлен разной степенью поражения митохондрий мозговой и мышечной тканей
Сенсорные нарушения у больных проявляются расстройствами вибрационной чувствительности и мышечно-суставного чувства [ВегкоУ1с 5 Р е! а1 , 1991]
Разнообразие клинических проявлений синдрома МЕККР затрудняет его диагностику По мнению 3 Р Вегк- ОУ1С и соавт (1991), «ключевыми» диагностическими признаками синдрома МЕККЕ являются симптомокомплекс прогрессирующей миоклонус-эпилепсии (миоклонус, атаксия, деменция) в сочетании с нейросенсорной глухотой, нарушениями глубокой чувствительности, атрофией зрительных нервов.
Течение заболевания прогрессирующее. Однако тяжесть и темп прогрессирования болезни вариабельны у отдельных больных.
Данные лабораторных и функциональных исследований. При биохимическом анализе крови определяется высокий уровень лактата. В цереброспинальной жидкости возможно умеренное повышение лактата, пирувата, содержания белка [РикиЬага Ы., 1991]. При анализе митохондриальных ферментов дыхательной цепи у больных с синдромом МЕККР обнаружена недостаточность комплексов I и IV в скелетных мышцах [РикиЬага N.. 1991].
При электромиографическом исследовании выявляется первично-мышечный характер изменений, а также снижение скорости проведения импульса по периферическим нервам [РикиЬага N.. 1991].
Электроэнцефалограмма при синдроме МЕККР характеризуется аномалиями основной активности (80 % случаев), спайк-волновыми разрядами, генерализованными комплексами «полиспайк-волна» (73 %), диффузными медленными волнами (33 %). В 40 % случаев регистрируются фокальные аномалии, в 26 % — фотопароксизмальный ответ [Сеп1оп Р., Ко^ег .1., 1993]. При анализе ЭЭГ-характеристик у 13 больных с синдромом в 85 % случаев отмечена дезорганизация основной активности с появлением медленных волн по всем отведениям. У 69 % больных регистрировались единичные и множественные, нерегулярные, билатерально синхронные комплексы «спайк-волна» частотой 2 — 5 Гц. Фокальные комплексы «спайк-волна» в затылочных отведениях обнаружены у 46 % больных.
При компьютерной томографии мозга определяются диффузная атрофия мозга, деструктивные изменения белого вещества, снижение плотности мозговой ткани, иногда кальцификация базальных ганглиев [Вегкоуш 5. Р. ек а1., 1989].
При позитронно-эмиссионной томографии выявляется хроническое снижение метаболизма глюкозы и кислорода в коре головного мозга и подкорковых структурах.
Патоморфологические изменения. В исследованиях в биоптатах скелетных мышц обнаруживаются типичные рваные красные волокна. Ферментно-гистохимический анализ выявляет недостаточность цитохром с-оксидазы.
Отсутствие «рваных» красных волокон при однократной биопсии скелетных мышц не исключает диагноза синдрома МЕККР [Вег- коУ1С З.Р. е! а1., 1989]. Оно может быть обусловлено снижением общего числа митохондрий. При отсутствии «рваных» красных волокон в биоптате скелетной мышцы целесообразны повторная биопсия или морфологический анализ других тканей, в частности электронная микроскопия кожи. При электронной микроскопии наблюдаются увеличение размеров митохондрий, их деформация, липидные включения [РикиЬага 14., 1991].
К 1992 г. опубликованы результаты 9 аутопсий больных с синдромом МЕККР. Наиболее выраженные изменения в виде значительного снижения числа нейронов и глиоза наблюдаются в зубчатых ядрах и верхних ножках мозжечка. Поражаются также бледный шар, красные ядра, кора мозжечка, нижние оливы и черная субстанция. В спинном мозге значительно страдают задние столбы, спиноцеребеллярные пути и пути Кларка, умеренно - кортикоспинальные тракты. При денторубропаллидольюисовой атрофии также обнаруживаются дегенеративные изменения в бледном шаре и зубчатых ядрах, однако поражение спинного мозга отсутствует. Кроме того, при синдроме МЕККЕ зубчатые ядра страдают в большей степени, чем бледный шар, в отличие от денторубропаллидольюисовой атрофии. При электронно-микроскопическом исследовании в коре мозжечка у больных с синдромом МЕККЕ выявляются аномально крупные митохондрии с многочисленными везикулярными кристами и паракристал- лическими включениями. Подобные изменения митохондрий при других митохондриальных энцефаломиопатиях не наблюдаются. Сосудистые аномалии митохондрий церебральных артериол, как и при синдроме МЕЬАЗ, отсутствуют. В периферических нервах выявляется дегенерация аксонов, а также снижение количества миелина, что свидетельствует о возможном поражении леммоцитов (шванновских клеток) [М12и5аша Н. еЬ а1., 1991].
Критерии диагноза. Основными критериями диагноза синдрома МЕККР являются:
— материнский тип наследования;
— дебют заболевания в возрасте 3-65 лет;
— миоклонус, атаксия, деменция в сочетании с нейросенсор- ной глухотой, атрофией зрительных нервов, нарушениями глубокой чувствительности;
— изменения ЭЭГ: генерализованные комплексы «полиспайк-волна»;
— «рваные» красные волокна в биоптатах скелетных мышц;
Эпилептические синдромы, сочетающиеся с миоклони- ческими приступами [5егга1о8а и.М., Ое1дас1о-Е8сие- 1а АЛ/., 1993, в модификации]
Доброкачественные первично-генерализованные эпилептические синдромы
Доброкачественная миоклоническая эпилепсия раннего детского возраста.
Семейная миоклоническая эпилепсия раннего детского возраста. Детская абсансная эпилепсия с легкими миоклониями.
Ювенильная миоклоническая эпилепсия
Тяжелые миоклонические эпилепсии раннего детского возраста
Ранняя миоклоническая энцефалопатия.
Ранняя инфантильная эпилептическая энцефалопатия.
Синдром Леннокса—Гасто Эпилепсия с миоклоническими абсансами.
Тяжелая миоклоническая эпилепсия раннего детского возраста. Миоклонически-астатическая эпилепсия.
Синдром Айкарди. (Згщ-ганглиозидоз. Отг-ганглиозидоз Болезнь Гоше.
Метахроматическая лейкодистрофия Недостаточность бета-галактозидазы. Болезнь Нимана—Пика, тип А Болезнь Менкеса.
Иллюстрацией наиболее типичных клинических проявлений синдрома МЕККР может быть следующее наблюдение
Больной Т , 11 лет, поступил в отделение врожденных и наследственных заболеваний у детей МНИИ педиатрии и детской хирургии М3 РФ с жалобами на частые непроизвольные подергивания различных групп мышц (лица, плечевого пояса, конечностей), приступы потери сознания и тонико-клонические судороги, быструю утомляемость при физических нагрузках Диагноз направившего учреждения эпилепсия
Апашпе513 V 1 I а е Ребенок от 1-й беременности, протекавшей с токсикозом в первой и второй половине Роды в срок, оператив-
ное родораэрешение (кесарево сечение) вследствие острой гипоксии плода и резкого снижения частоты сердечных сокращений Родился с массой тела 3100 г, длиной 54 см Период новорожденное™ протекал без особенностей Задержка темпов раннего психомоторного развития У матери отмечались жалобы на мышечную слабость, быструю утомляемость, мышечные боли (крампи) при физических нагрузках Профессиональных и экологических вредностей нет
Апатпези тогЬ1 Первые признаки заболевания возникли в возрасте 2 лет, манифестными симптомами явились эпилептические по роксизмы в виде кивков, с частотой 2 — 3 раза в сутки В 4 года впервые наблюдался генерализованный тонико-клонический приступ В дальней шем генерализованные тонико-клонические приступы повторялись с частотой 3 — 4 раза в месяц и были резистентны к антиконвульсантной терапии, 3 раза наблюдались эпизоды эпилептического статуса В 6 лет возникли миоклонические подергивания в различных группах мышц Частота миоклоний — до нескольких десятков в день В 10 лет стал жаловать ся на мышечную слабость, быстро уставал при физических нагрузках С 10 лет отмечается снижение памяти, концентрации внимания
Данные объективного исследования При по ступлении обращало на себя внимание наличие диффузных миоклоний в мышцах лица плеч, верхних и нижних конечностей Миоклонии наблюдались сериями (3 — 4 мышечных сокращения) продолжительное тью 5— 10 с Неврологический статус атактическая походка, интенци- онный тремор, диффузная мышечная гипотония, мышечная утомляемость, крампи при физических нагрузках
Данные лабораторных и функциональных исследований Биохимический анализ крови содержание молочной кислоты до нагрузки глюкозой 1,94 ммоль/л (норма 1,0 — 1,7 ммоль/л), через 1 ч — 2,96 ммоль/л, через 3 ч — 2,14 ммоль/л Содержание пировиноградной кислоты до нагрузки — 0,09 ммоль/л (норма 0,05 — 0 09 ммоль/л), через 1ч— 1,22 ммоль/л, через 3 ч — 1,04 ммоль/л рН 7,36 Биохимический анализ мочи без отклонений Глазное дно без патологии На ЭКГ незначительная синусовая тахиаритмия КТ и ЯМР мозга очаговых изменений не выявлено ЭЭГ дезорганизация основной активности, генерализованные комплексы «полиспайк-волна» Патоморфологические изменения Общий план строения скелетной мышечной ткани соответствует возрасту Отмечаются признаки умеренного разрастания соединительной ткани В части мышечных волокон обнаружены признаки региональных некрозов, на личия субсарколеммальных масс, макрофагально лимфоцитарной инвазии, повышения регенераторной активности, обнаруживаются субсарколеммальные скопления кальция, гликогена и липидов (рис 4 4) — косвенный признак митохондриальной недостаточности Выражен феномен ККР (см рис 4 4 6)
На основании данных анамнеза, клинико-лабораторного исследо вания больному был поставлен диагноз синдром МЕККР
Дифференциальный диагноз. При установлении диагноза синдрома МЕККР необходимо учитывать, что миоклонии встречаются также при многих эпилептических синдромах различной этиологии (табл 4 6)
Рис. 4.4. Патоморфологические изменения при синдроме МЕККР.
а — мелкозернистые включения липидов (стрелки) в части мышечных волокон и более крупные — в эпдомизии. Окраска Суданом черным В по Лизопу. х 250; б — умеренно выраженные ККР (стрелки). Выявление сукципатдегидрогеназы
Читайте также: