Митохондриальные заболевания
Добавил пользователь Skiper Обновлено: 27.01.2026
Митохондриальные заболевания клинически представляют собой гетерогенную группу состояний, развивающихся в связи с нарушением работы ферментов дыхательной цепи митохондрий. Нарушения могут быть связаны как с мутациями, появляющимися в геноме человека, так и с мутациями в митохондриальной ДНК (мтДНК). Наиболее часто наблюдаются повреждения мтДНК, которые характеризуются протяженными делециями и дупликациями, а также точечными мутациями (чаще всего 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C).
Митохондриальные заболевания могут манифестировать в любом возрасте. При некоторых митохондриальных заболеваниях поражается только один орган (наследственная оптическая нейропатия Лебера), однако в большинстве данная группа состояний имеет системные проявления, чаще всего связанные с поражением нервной и мышечной системы. Довольно часто клинические проявления митохондриальных заболеваний могут подпадать под специфические синдромы: MELAS-синдром (митохондриальная миопатия, энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды), MERRF-синдром (миоклоническая эпилепсия, ассоциированная с порванными красными волокнами), прогрессирующая наружная офтальмоплегия, синдром Кернса-Сейра, синдром Лея, нейрогенная слабость с атаксией и пигментным ретинитом. Однако довольно много клинических случаев митохондриальных заболеваний не подпадают ни под одну из перечисленных категорий, имея перемежающийся спектр проявлений и симптомов, что возможно объясняется гетероплазмией мутаций в мтДНК (вариация количества «мутантных» митохондрий в одной клетке).
Частыми клиническими проявлениями митохондриальных заболеваний являются блефароптоз, офтальмоплегия, проксимальная миопатия, кардиомиопатия, нейросенсорная тугоухость, атрофия глазного нерва, пигментная ретинопатия, энцефалопатия, судороги, деменция, мигрень, инсультоподобные эпизоды, атаксия, спастичность.
Мутации в мтДНК могут передаваться по материнской линии. При наличии мутаций в мтДНК мужчины риск передачи аберрации детям отсутствует. Делеции в мтДНК чаще всего возникают de novo (единственный случай в семье), тогда как точечные аберрации и дупликации часто передаются по материнской линии.
Специальной подготовки не требуется. Взятие крови желательно проводить не ранее 4 часов после приема пищи.
- подозрение на митохондриальную патологию;
- дифференциальный диагноз синдрома Девика (оптическая нейропатия);
- дифференциальный диагноз миопатии;
- дифференциальный диагноз энцефалопатии, судорожного синдрома.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Делеций и дупликаций митохондриальной ДНК, а также мутаций 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C обнаружено не было.
В связи с тем, что одна и та же мутация может вызывать различные клинические проявления у разных пациентов, иногда сложно определить точно корреляцию генотипа и фенотипа. Однако считается, что делеции и дупликации мтДНК чаще всего встречаются при синдроме Кернса-Сейра, прогрессирующей офтальмоплегии и синдроме Пирсона. Мутация 3243A>G характерна для синдрома MELAS, 11778G>A, 14484T>C, 3460G>A - для наследственной оптической нейропатии Лебера Отрицательный результат исследования не исключает диагноз митохондриального заболевания в связи с возможностью наличия менее распространенных точечных мутаций, а также появления генетических аберрации в участках геномной ДНК, кодирующих митохондриальные белки.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации. ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Митохондриальные заболевания

Митохондриальные заболевания (МЗ) — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках.
Историческая справка:
Понятие «митохондриальные болезни» сформировалось в медицине в конце ХХ века. В первую очередь были изучены болезни, связанные с мутациями митохондриальной ДНК, открытой в начале 60-ых годов. Полная первичная структура митохондриальной ДНК человека была опубликована в 1981 го¬ду и уже в конце 80-ых годов была доказана ведущая роль ее мутаций в развитии ряда наследственных заболеваний. К последним относятся: наследственная атрофия зрительных нервов Лебера, синдром NARP (нейропатия, атаксия, пигментный ретинит), синдром MERRF (миоклонусэпилепсия с "рваными" красными волокнами в скелетных мышцах), синдром MELAS (митохондриальная энцефаломиопатия, лактат-ацидоз, инсультоподобные эпизоды), синдром Кернса-Сейра (пигментный ретинит, наружная офтальмоплегия, блокада сердца, птоз, мозжечковый синдром), синдром Пирсона (поражение костного мозга, панкреатическая и печеночная дисфункции) и многие другие.
В меньшей степени изучены наследственные митохондриальные дефекты, связанные с повреждением ядерного генома.
Патогенез.
Митохондрии отвечают за выработку большей части энергии, необходимой для функционирования клеток. Фактически они являются настолько важным источником энергии, что в каждой клетке их сотни. При МЗ могут «выключиться» как часть митохондрий, так и все они, что приводит к прекращению выработки необходимой энергии
Поскольку наиболее энергоемкими являются нервные и мышечные клетки, при МЗ наиболее распространены мышечные и неврологические проблемы, такие, как мышечная слабость, непереносимость физических нагрузок, потеря слуха, нарушения баланса и координации, эпиприступы.
Митохондриальные зааболевания, вызывающие выраженные мышечные проблемы, именуют митохондриальными миопатиями (myo - означает «мышца», а pathos – «болезнь»), а те, которые вызывают как мышечные, так и неврологические проблемы – митохондриальными энцефаломиопатиями (encephalo – «мозг»)
Когда клетка заполнена дефектными митохондриями, она не только лишена АТФ, но в ней могут накапливаться неиспользуемые молекулы топлива и кислород, что приводит к катастрофическим последствиям. В этом случае избыточные молекулы топлива используются для синтеза АТФ неэффективно, в результате чего могут образовываться потенциально опасные продукты, такие, как молочная кислота (Это также происходит, когда клетки испытывают недостаток кислорода, например – мышечные клетки при усиленных физических нагрузках). Накопление молочной кислоты в крови – лактатацидоз – ассоциировано с мышечной усталостью, и может вызывать повреждение нервной и мышечной тканей.
При этом неиспользуемый в клетке кислород может трансформироваться в разрушительные соединения, именуемые реактивными формами кислорода, включая т. н. свободные радикалы (Они являются мишенью для т. н. антиоксидантных препаратов и витаминов).
Синтезированная в митохондриях АТФ – основной источник энергии для сокращения мышечных и возбуждения нервных клеток (т. к. клетки этих тканей наиболее метаболически активны, энергетически зависимы). Таким образом, нервные и мышечные клетки особенно чувствительны к дефектам митохондрий. Комбинированный эффект от потери энергии и накопления токсинов в этих клетках, надо полагать, и вызывает развитие симптомов митохондриальных миопатий и энцефаломиопатий
Клиника
В случаях, когда человек с мутацией в митохондриальном гене несет смесь нормальной и мутантной ДНК - мутации поначалу могут вообще не иметь внешних проявлений. Нормальные митохондрии до поры до времени обеспечивают клетки энергией, компенсируя недостаточность функции митохондрий с дефектами. На практике это проявляется более или менее длительным бессимптомным периодом при многих митохондриальных заболеваниях. Однако рано или поздно наступает момент, когда дефектные формы накапливаются в количестве, достаточном для проявления патологических признаков. Возраст манифестации заболевания варьирует у разных больных. Раннее начало заболевания приводит к более тяжелому течению и неутешительному прогнозу.
Характерные признаки митохондриальных цитопатий:
•скелетные мышцы: низкая толерантность к физической нагрузке, гипотония, проксимальная миопатия, включающая фациальные и фарингеальные мышцы, офтальмопарез, птоз
•сердце: нарушения сердечного ритма, гипертрофическая миокардиопатия
•центральная нервная система: атрофия зрительного нерва, пигментная ретинопатия, миоклонус, деменция, инсультоподобные эпизоды, расстройства психики
•периферическая нервная система: аксональная нейропатия, нарушения двигательной функции гастроинтестинального тракта
•эндокринная система: диабет, гипопаратиреоидизм, нарушение экзокринной функции панкреас, низкий рост
Таким образом, типичны для митохондриальных заболеваний вовлеченность разных органов и одновременное проявление внешне не связанных между собой аномалий. Примерами служат:
1. Мигрени с мышечной слабостью
2. Наружная офтальмоплегия с нарушением проводимости сердечной мышцы и мозжечковой атаксией
3. Тошнота, рвота с оптической атрофией и кардиомиопатией
4. Низкорослость с миопатией и инсультоподобным и эпизодами
5. Экзокринная дисфункция поджелудочной железы с сидеробластной анемией
6. Энцефало- миопатия с диабетом
7. Диабет с глухотой
8. Глухота с наружной офтальмоплегией, птозом и ретинопатией
9. Задержка развития или потеря навыков и офтальмоплегия, офтальмопарез
Характер и тяжесть клинических проявлений митохондриальных болезней определяется:
• тяжестью мутации мтДНК;
• процентным содержанием мутантной мтДНК в конкретных органах и тканях;
• энергетической потребностью и функциональным резервом органов и тканей, содержащих мтДНК (их “порогом чувствительности” к дефектам окислительного фосфори лирования).
Миопатия
Основные симптомы митохондриальной миопатии – истощение мышц и их слабость, и непереносимость физических нагрузок.
У некоторых индивидов слабость наиболее выражена в мышцах, контролирующих движения глаз и век. Два наиболее частых следствия такой слабости – это постепенный паралич движения глаз (прогрессирующая наружная офтальмоплегия, ПНО), и опущение верхних век (птоз). Зачастую люди автоматически компенсируют ПНО движениями головы для того, чтобы смотреть в различных направлениях, и могут даже не подозревать о каких либо проблемах. Птоз потенциально более неприятен, поскольку может ухудшить зрение, а также придает лицу апатичное выражение, но он может быть скорректирован хирургическим путем, либо использованием специальных очков с устройством для подъема века
Митохондриальные миопатии могут также вызывать слабость других мышц лица и шеи, что приводит к заплетающейся речи и трудностям с глотанием. В этих случаях могут помочь речевая терапия (занятия с логопедом) или включение в рацион питания таких продуктов, которые легче проглатываются.
Непереносимость физических нагрузок, также именуемая усталостью напряжения - это необычное чувство утомления в ответ на физическую активность. Степень этой непереносимости существенно варьируется у разных людей. Некоторые могут испытывать проблемы только при занятиях физкультурой, таких например, как оздоровительный бег, в то время как у других возникают сложности с выполнением повседневных дел, например с выходом к почтовому ящику или поднятием пакета молока.
Энцефаломиопатия
Митохондриальная энцефаломиопатия, как правило, включает некоторые из вышеупомянутых симптомов миопатии, дополненными одним или несколькими неврологическими симптомами. Также как и при миопатии, наблюдается значительная вариабельность симптомов обоего типа и тяжести течения у разных индивидов.
Среди наиболее частых симптомов митохондриальной энцефаломиопатии – нарушения слуха, мигренеподобные головные боли и эпиприступы. По крайней мере, в одном синдроме головные боли и эпиприступы часто сопровождается инсультоподобными эпизодами
Дополнительно к поражению глазных мышц, митохондриальная энцефаломиопатия может поражать как сами глаза, так и участки головного мозга, ответственные за зрение. Например, потеря зрения вследствие оптической атрофии (дегенерации зрительного нерва) или ретинопатии (дегенерации некоторых клеток, выстилающих глазное дно) – обычные симптомы митохондриальной энцефаломиопатии. По сравнению с мышечными проблемами, эти эффекты с большей вероятностью приводят к серьезным нарушениям зрения
Довольно часто митохондриальная энцефаломиопатия вызывает атаксию, или сложности с балансом и координацией.
Диагностика.
Ни один из отличительных симптомов митохондриального заболевания – мышечная слабость, непереносимость нагрузок, ухудшение слуха, атаксия, эпиприступы, неспособность к обучению, катаракта, диабет и низкорослость – не является уникальным именно для такого заболевания. Однако комбинация трех или более из этих симптомов у одного индивида свидетельствует в пользу митохондриального заболевания, особенно если симптомы затрагивают более одной системы организма
Физикальное обследование обычно включает в себя тесты на силу и выносливость, такие например, как повторяющиеся сжатия-разжатия кулака, или подъем и спуск по небольшой лестнице. Неврологическое обследование может включать в себя проверку рефлексов, зрения, речи и базовых когнитивных способностей.
Существует ряд рутинных клинических методов исследования, которые можно использовать при подозрении на митохондриальную цитопатию:
•лактатный ацидоз является практически постоянным спутником митохондриальных болезней (только этот признак является недостаточным для постановки диагноза, так как он может выявляться и при других патологических состояниях; в этом отношении может быть полезным измерение уровня лактата в венозной крови после умеренной физической нагрузки, например на велоэргометре)
•ЭМГ-исследование - само по себе данное исследование также не могут быть маркером митохондриальной цитопатии; вместе с тем нормальная или близкая к нормальной ЭМГ у пациентов с выраженной мышечной слабостью может быть подозрительной в отношении митохондриальной патологии.
•ЭЭГ – данные ЭЭГ не является достаточно специфическими
•биопсия скелетных мышц - является наиболее информативным методом при постановке диагноза митохондриальной цитопатии - помимо обнаружения RRF при трехцветной окраске по Гомори, полезными являются другие гистохимические и иммунологические исследования: окраска на цитохромс-оксидазу и сукцинатдегидрогеназу, иммунногистохимические исследования с применением антител к отдельным субъединицам дыхательного комплекса; мышечная ткань удобна для биохимического исследования респираторной цепочки, а также как материал для генетического исследования.
Образцы мышечных биоптатов целесообразно делить на три части – одна для микроскопического исследования (гистология, гистохимия и электронная микроскопия), вторая для энзимологического и иммунологического анализа (изучение характеристик компонентов дыхательной цепи) и третья – непосредственно для молекулярно-генетического анализа. Поиск известных мутаций на мышечном материале позволяет в большинстве случаев успешно осуществлять ДНК-диагностику болезни. При отсутствии из вестных мутаций мтДНК в мышечной ткани следующим этапом является развернутый молекулярно-генетический анализ – секвенирование всей цепи мтДНК (или кандидатных генов ядерной ДНК) с целью выявления нового варианта мутации.
•электронно-микроскопическое исследование скелетных мышц - дает прекрасные результаты, поэтому данный метод надо использовать, если имеется такая возможность
Лечение.
Что касается терапии митохондриальных цитопатий, то речь может идти пока только о симптоматической.
Лечение митохондриальных болезней проводится обычно по двум основным направлениям:
•повышение эффективности энергетического обмена в тканях (тиамин, рибофлавин, никотинамид, коэнзим Q10 (кудесан), L-карнитин (элькар), препараты кальция и магния. , витамин С, цитохром С)
•предупреждение повреждения митохондриальных мембран свободными радикалами с помощью антиоксидантов (витамин Е, a-липоевая кислота) и мембранопротекторов.
В практику входят всё новые препараты комбинированного действия, такие, например, как идебенон (Нобен) – улучшенный структурный аналог коэнзима Q10, благоприятно влияющий на активность дыхательного пути и обладающий выраженным антиоксидантным, антиапоптотическим и нейротрофическим действием.
Очевидно, что расширение терапевтического арсенала при митохондриальных болезнях диктует настоятельную необходимость того, чтобы практические врачи различных специальностей (неврологи, психиатры, педиатры, генетики, гематологи и др. ) были хорошо знакомы с алгоритмом диагностики этих заболеваний.
IX Международная студенческая научная конференция Студенческий научный форум - 2017

Митохондриальные заболевания — неоднородная группа наследственных заболеваний, которые вызваны структурными, генетическими или биохимическими дефектами митохондрий, приводящих к нарушениям энергетических функций в клетках эукариотических организмов. У человека при митохондриальных заболеваниях в первую очередь поражается мышечная и нервная система.
Митохондриальные заболевания как отдельный тип патологий выделены в конце ХХ века после выявления мутации генов, которые ответственны за синтез митохондриальных белков.
Открытые в 1960-х годах мутации митохондриальной ДНК и вызванные этими мутациями болезни более изучены, чем заболевания, вызванные нарушениями ядерно-митохондриальных взаимодействий (мутации ядерной ДНК).
По имеющимся на сегодняшний день данным не менее 50 известных медицине заболеваний связано с митохондриальными нарушениями. Распространенность этих заболеваний составляет 1:5000.
Виды митохондриальных заболеваний.
Митохондрии являются уникальными клеточными структурами, которые обладают собственным ДНК.
Согласно мнению многих исследователей, митохондрии – потомки архебактерий, превратившиеся в эндосимбионтов (микроорганизмы, которые живут в организме «хозяина» и приносят ему пользу). В результате внедрения в эукариотические клетки они постепенно утратили или передали ядру эукариотического хозяина большую часть генома, и это учитывается при классификации. Также принимается во внимание и участие дефектного белка в биохимических реакциях окислительного фосфорилирования, которое позволяет запасать энергию в виде АТФ в митохондриях.
Единой общепринятой классификации не существует.
Обобщенная современная классификация митохондриальных заболеваний выделяет:
Заболевания, которые возникают при мутациях митохондриальной ДНК. Дефекты могут быть вызваны точечными мутациями белков, тРНК или рРНК (обычно наследуются по материнской линии), или структурными перестановками – спорадическими (нерегулярными) дупликациями и делециями. Это первичные митохондриальные заболевания, к которым относятся наследственные ярко выраженные синдромы — синдром Кернса — Сейра, синдром Лебера, синдром Пирсона, синдром NAPR, синдром MERRF и др.
Заболевания, которые вызваны дефектами ядерной ДНК. Ядерные мутации могут нарушать функции митохондрий – окислительное фосфолирование, работу электронтранспортной цепи, утилизацию или транспорт субстратов. Также мутации ядерной ДНК вызывают дефекты ферментов, которые необходимы для обеспечения циклического биохимического процесса — цикла Кребса, являющегося ключевым этапом дыхания всех использующих кислород клеток и центром пересечения в организме метаболических путей. К данной группе относят гастроинтестинальное митохондриальное заболевание, синдром Люфта, атаксию Фридриха, синдром Альперса, болезни соединительной ткани, диабет и др.
Заболевания, которые возникают в результате нарушений в ядерной ДНК и вызванных этими нарушениями вторичных изменений в митохондриальной ДНК. Вторичными дефектами являются тканеспецифические делеции или дупликации митохондриальной ДНК и уменьшение количества копий митохондриальной ДНК или их отсутствие в тканях. В данную группу входят печеночная недостаточность, синдром Де Тони-Дебре-Фанкони и др.
Причины данных заболеваний.
Митохондриальные заболевания вызываются дефектами находящихся в клеточной цитоплазме органелл — митохондрий. Основной функцией этих органелл является выработка энергии из поступающих в цитоплазму продуктов клеточного обмена веществ, которая происходит благодаря участию около 80 ферментов. Выделяющаяся энергия запасается в виде молекул АТФ, а затем преобразуется в механическую или биоэлектрическую энергию и т.д.
Причины митохондриальных заболеваний – нарушение выработки и аккумуляции энергии из-за дефекта одного из ферментов. В первую очередь при хроническом дефиците энергии страдают самые энергозависимые органы и ткани – ЦНС, сердечная мышца и скелетные мышцы, печень, почки и эндокринные железы. Хронический дефицит энергии вызывает патологические изменения в данных органах и провоцирует развитие митохондриальных заболеваний.
Этиология митохондриальных заболеваний имеет свою специфику – большинство мутаций происходит в генах митохондрий, поскольку в этих органеллах интенсивно протекают окислительно-восстановительные процессы и образуются повреждающие ДНК свободные радикалы. У митохондриальной ДНК механизмы восстановления повреждений несовершенны, так как ее не защищают белки-гистоны. В результате дефектные гены накапливаются быстрее в 10-20 раз, чем в ядерной ДНК.
Мутировавшие гены передаются при делении митохондрий, поэтому даже в одной клетке находятся органеллы с разным вариантом генома (гетероплазмия). При мутации митохондриального гена у человека наблюдается смесь мутантной и нормальной ДНК в любом соотношении, поэтому даже при наличии одинаковой мутации митохондриальные заболевания у людей выражены в разной степени. Наличие 10% дефектных митохондрий не оказывает патологического влияния.
Мутация может длительное время не проявляться, так как нормальные митохондрии компенсируют на начальном этапе недостаточность функции дефектных митохондрий. Со временем дефектные органеллы накапливаются, и проявляются патологические признаки заболевания. При раннем манифесте течение болезни более тяжелое, прогноз может быть негативным.
Митохондриальные гены передаются только от матери, так как содержащая эти органеллы цитоплазма присутствует в яйцеклетке и практически отсутствует в сперматозоидах.
Митохондриальные заболевания, которые вызваны дефектами ядерной ДНК, передаются благодаря аутосомно-рецессивному, аутосомно-доминантному или Х-сцепленному типу наследования.
Некоторые митохондриальные болезни.
Митохондриальная миопатия
Митохондриальная миопатия – это одно из генетических заболеваний, которое в мире встречается относительно редко. Его особенность состоит в том, что причина болезни – это нарушение работы митохондрий, при этом степень тяжести заболевания будет зависеть от того, какое количество их не выполняет свою положенную работу. Существует несколько разновидностей этой болезни, и каждая из них имеет свои симптомы.
Как и любое другое заболевание, митохондриальная миопатия имеет свои причины. И здесь виноваты не вирусы или бактерии, и даже не действия беременной женщины или её окружения, а мутации в генах. А это значит, что такие гены с «поломками» ребёнок получает от своих родителей, а следовательно — эту болезнь можно назвать наследственной.
Хотя иногда бывает, что мутация в генах и рождение больного митохондриальной миопатией ребёнка происходит у совершенно здоровых родителей.
Когда клетка заполнена дефектными митохондриями, то она просто не в состоянии нормально работать. Кроме того, в ней могут постепенно накапливаться и не удаляться опасные вещества, которые в итоге приводят к смерти человека с этим диагнозом. А самыми чувствительными клетками к таким дефектам в митохондриях являются как раз мышечные и нервные. Именно они острее всего реагируют на полное отсутствие или нехватку такого вещества, как АТФ, а ведь именно его и должны вырабатывать митохондрии, для того, чтобы организм работал без перебоев.
Сложность диагностики этого недуга состоит в том, что для постановки правильного диагноза нужно провести сразу несколько анализов, результаты которых будут получены далеко не в течение одного дня.
Так, для диагностики любого из перечисленных синдромов обязательно проводится генеалогический анализ, то есть определяются родственники, у которых была или есть эта патология. Также делается клинический и биохимический анализ крови, анализ состояния мышц, для чего требуется биопсия, и, конечно — исследование генов, которое и даёт представление о том, где именно случилась «поломка», которая и привела к развитию такой миопатии.
На сегодняшний день лечение митохондриальных миопатий не разработано. Но доказано, что в большинстве случаев пациентам продляет жизнь лечение при помощи витаминов.
Атрофия зрительного нерва Лебера.
Атрофия зрительного нерва Лебера, является наследственной митохондриальной дегенерацией ганглионарных клеток сетчатки и их аксонов, что приводит к острой или почти острой потере центрального зрения; это влияет преимущественно на молодых мужчин.
Распространенность этого заболевания точно неизвестна, но оценивается в 2-4 случая на 100 000 населения.
Заболевание проявляется внезапной, безболезненной, острой/подострой потерей центрального зрения, обычно в возрасте от 18 до 30 лет.
При атрофии зрительного нерва Лебера поражаются или оба глаза одновременно или последовательно с интервалом в несколько недель или месяцев после первого. Чаще потеря зрения происходит подостро в течение нескольких недель, затем состояние стабилизируется. Однако у многих пациентов в течение нескольких лет продолжаются расширяться размеры центральной скотомы, приводя к глубокой слепоте.
На ранних этапах поражения зрения могут наблюдаться нарушения цветового восприятия красного и зелёного и контрастности.
Могут присутствовать также и другие неврологические симптомы. Эти нарушения известны как Лебера плюс, и включают двигательные расстройства, дистонию, постуральный тремор и мозжечковую атаксию.
На данный момент лечения не существует. Основной поддерживающей терапии являются препараты для слабовидящих. Несколько веществ показали положительные результаты в восстановлении зрения. Синтетический аналог коэнзима Q10 - идебенон улучшил зрение после года применения.
Синдром MELAS
Синдром MELAS - заболевание, обусловленное точковыми мутациями в митохондриальной ДНК.
Симптомы синдрома MELAS. Возраст, в котором манифестирует заболевание, широко варьирует от младенческого до взрослого, однако чаще всего первые симптомы появляются в периоде от 5 до 15 лет. Начало болезни часто характеризуется инсультоподобными эпизодами, злокачественными мигренями или задержкой психомоторного развития. Инсульты локализуются чаще в височной, теменной или затылочной областях головного мозга, сопровождаются гемипарезом и имеют тенденцию к быстрому восстановлению. Они обусловлены митохондриальной ангиопатией, характеризующейся избыточной пролиферацией митохондрий в стенках артериол и капилляров сосудов мозга. По мере прогрессирования болезни, на фоне повторных инсультов нарастает неврологическая симптоматика. Присоединяются мышечная слабость, судороги, миоклонии, атаксия и нейросенсорная тугоухость. Иногда развиваются эндокринные расстройства (сахарный диабет, гипофизарный нанизм).
«Мальчик, который не переставал расти… и другие истории про гены и людей»
Порой серьезные заболевания достаются нам по наследству: муковисцидоз, шизофрения, болезнь Альцгеймера и многие другие. К сожалению, устранять их причины мы пока не умеем — зато можем узнать, носителями каких мутаций, вызывающих болезни, мы являемся. И в некоторых случаях даже вылечиться. В книге «Мальчик, который не переставал расти… и другие истории про гены и людей» (издательство «Альпина нон-фикшн»), переведенной на русский язык Марией Елиферовой, специалист по медицинской генетике и по генетическим патологиям Эдвин Керк рассказывает о работе врача-генетика на примере историй реальных пациентов. Предлагаем вам ознакомиться с фрагментом, посвященным недугам, связанным с отсутствием в организме участка митохондриальной ДНК.

У Фелисити было что-то не так с митохондриями, причем всю жизнь. Однако первые признаки неблагополучия стали проявляться только теперь, когда ей было под 40. Болезнь прогрессировала настолько медленно, настолько коварно, что Фелисити даже не замечала ее, пока однажды муж не обратил внимание, что у нее стали плохо подниматься веки. Она пошла к окулисту, который понял, что дело не только в веках. Ее глазные яблоки не могли нормально двигаться, потому что глазные мышцы постепенно слабели. Это означало, что в будущем она вообще не сможет двигать глазами и ей придется поворачивать голову, чтобы посмотреть в сторону. Однако во всех остальных отношениях она была совершенно здорова.
Ахмед всегда был неуклюжим ребенком и не мог угнаться за товарищами на детской площадке. Когда ему исполнилось лет десять, его мать забеспокоилась — он все хуже держался на ногах и начал ходить на цыпочках. Он плохо учился в школе — все предметы как будто стали для него труднее, чем раньше. Мать отвела Ахмеда к педиатру, который обнаружил у него мышечную слабость и выяснил, что ребенок ходит на цыпочках из-за напряжения ахиллова сухожилия, не позволяющего поставить на землю всю стопу. Ахмед тоже страдал параличом век и не мог нормально двигать глазами.
Джейкоб не дожил до своего первого дня рождения. Еще в младенчестве у него обнаружили анемию — его костный мозг производил недостаточно красных кровяных клеток для организма. Анемия была настолько тяжелой, что ему требовались регулярные переливания крови. Его поджелудочная железа плохо работала, и он не мог как следует усваивать пищу. В крови у него был высокий уровень молочной кислоты, а печень была больной с самого рождения, пока наконец не отказала совсем — ему тогда не исполнилось и года.
Джейкоб, Ахмед и Фелисити не имели между собой ничего общего, кроме одной и той же глубинной проблемы: в некоторых митохондриях у них не хватало кусочка кольцевой ДНК. По причинам, которые еще не изучены, если у кого-то не хватает большого участка митохондриальной ДНК, то у этого человека, скорее всего, разовьется одно из трех родственных заболеваний: хроническая прогрессирующая внешняя офтальмоплегия (ХПВО) * , как у Фелисити, синдром Кернса — Сейра, как у Ахмеда, либо синдром Пирсона, как у Джейкоба. Если бы Джейкоб прожил дольше, у него тоже почти наверняка появились бы те же проблемы с глазами, что у Ахмеда и Фелисити. Хотя эти три заболевания называются по-разному, на самом деле это одно заболевание разной степени тяжести, из которых ХПВО — самая легкая, а синдром Пирсона — самая тяжелая форма.
* Звучит заумно, но это всего лишь профессиональный жаргон. «Хроническая» значит «длительная». «Прогрессирующая» — такая, которая со временем становится тяжелее. «Офтальмо-» по-гречески означает «глаз», а «плегия» — «слабость». «Внешняя» в пояснениях не нуждается. Сложите все вместе, и получится «длительное заболевание, которое со временем становится тяжелее и поражает мышцы с внешней стороны глазного яблока (отвечающие за движения глаз)».
Генетика изобилует случаями, когда мутации ДНК, которые, казалось бы, должны вести к широкому спектру последствий, в реальности проявляются крайне специфически, и это как раз типичный тому пример. Замена единственного из 16 569 оснований ДНК, составляющих митохондриальный геном, может привести к катастрофическому поражению многих органов, а порой к смерти в первые дни жизни. Так почему же выпадение ¾ митохондриальной ДНК у кого-то дает всего лишь проблему с глазами, причем это проявляется лишь через десятилетия? Почему Джейкоб умер в младенчестве, Ахмед испытывал серьезные и разнообразные проблемы со здоровьем в детстве, а Фелисити была совершенно здорова, если не считать нарушения подвижности глаз?
Нам лишь частично известны ответы на эти вопросы. Наиболее понятная часть связана с тем фактом, что внутри клетки митохондриальные геномы независимы друг от друга. Следовательно, одна клетка вполне может иметь более одной версии митохондриального генома. Есть вероятность, что такая изменчивость окажется безвредной, но может быть и так, что одни копии нормальны, тогда как с другими что-то не в порядке, например не хватает кусочка. У такой смеси есть название: гетероплазмия. Нетрудно догадаться, что, если большинство копий митохондриального генома нормальны, это приведет к менее тяжелому заболеванию, чем когда почти вся митохондриальная ДНК поражена.
По крайней мере отчасти это и объясняет разницу в судьбах Фелисити и Джейкоба: если бы мы могли заглянуть в каждую их клетку и сосчитать нормальные и аномальные копии, то, скорее всего, оказалось бы, что у Фелисити преобладают нормальные копии, а у Джейкоба — аномальные. Чем меньше нормальных копий в наличии, тем хуже работают митохондрии и, соответственно, тем тяжелее проблемы со здоровьем и тем раньше они начинаются.
Отсюда возникает следующий вопрос. Как могло случиться, что Фелисити отлично себя чувствовала на протяжении десятилетий, хотя как минимум часть митохондрий в ее клетках была повреждена? Ответ состоит в том, что по мере нашего старения в митохондриях накапливаются поломки, в том числе делеции, наблюдаемые при ХПВО. Фелисити родилась с определенным процентом поврежденных копий митохондриального генома. У нее было достаточно здоровых, функциональных копий, чтобы клетки глазных мышц успешно работали. С годами поломки накапливались, и клетки перешли некий критический уровень повреждений, после чего уже не могли полноценно функционировать. Постепенно началось опущение век. У бедного Джейкоба клетки с самого рождения находились за красной чертой — большая часть его митохондриальной ДНК была патологической.
Разные типы клеток по-разному справляются с этой проблемой, что объясняет различия в характере симптомов у наших трех пациентов. У Джейкоба с самого начала отказывало множество разных тканей, потому что в их клетках содержалась непосильно высокая доля поломанной митохондриальной ДНК, и это было необратимо. Ахмед попал в середину между двумя крайностями. Неловкость движений, замеченная матерью в его раннем детстве, была признаком того, что некоторые нервные клетки испытывают затруднения. Уже на момент рождения у него, по-видимому, было много клеток, едва справлявшихся с выработкой нужного количества энергии. Всего несколько лет спустя добавилось чуть больше поломок — и проявились симптомы.
На этом месте у вас может возникнуть вопрос по поводу собственных митохондрий. Они что, тоже накапливают поломки? Будьте уверены, накапливают. В ходе исследования 2006 г. сотрудники Университета штата Висконсин взяли образцы мышечной ткани у людей в возрасте от 49 до 93 лет, которым не были диагностированы митохондриальные заболевания. Мышечные волокна с поломанными митохондриями обнаружились у всех людей в выборке, и чем старше был человек, тем заметнее проблема. У пациентов в возрасте от 40 до 50 лет около 6 процентов мышечных волокон содержали дефекты; у тех, кому было за 90, эта доля достигала 30 процентов. Обнаружился также постоянный рост доли делеций того типа, который наблюдается у таких пациентов, как Фелисити, Ахмед и Джейкоб (пусть и в гораздо меньших масштабах). Потеря мышечной силы, которую мы все ощущаем по мере старения, отчасти связана с этим постепенным нарушением работы митохондрий. Не стоит ожидать от поломанной электростанции, что она обеспечит мышцам достаточно энергии.
Если у отдельных особей накапливаются митохондриальные поломки, как с этим обстоят дела у вида в целом? Как это возможно, что люди в течение многих поколений рождались и проживали всю жизнь, как правило, без всяких признаков митохондриальных заболеваний, если их митохондриальная ДНК постоянно разрушается? Почему мы все не вымерли еще миллионы лет назад?
Как ни странно, ответ заключается в «бутылочном горлышке». Бутылка в данном случае метафорическая: митохондриальное бутылочное горлышко — это событие, а не физический предмет. Оно происходит в жизни каждой женщины еще до ее рождения и связано с количеством митохондрий в яйцеклетке. Типичная клетка содержит несколько тысяч копий митохондриального генома, однако в человеческой яйцеклетке около 100 000 копий. После оплодотворения яйцеклетка делится быстрее, чем успевают размножаться митохондрии, поэтому количество митохондрий на каждую новую клетку быстро падает до более «нормального» уровня. Среди разнообразных типов клеток растущего эмбриона (девочки) есть и такой, который в будущем даст яйцеклетки; следующее поколение планируется чуть ли не с самого начала. В этом процессе участвует много клеточных делений. В определенный момент количество митохондрий резко сокращается, а затем вновь возрастает — это и есть бутылочное горлышко. Точно неизвестно, насколько оно сокращается, то есть насколько узкое это горлышко, и неизвестно даже, одно ли оно или, условно говоря, сужение и расширение наблюдаются несколько раз. Так или иначе, суть в том, что, если у вас мало митохондрий, скажем две сотни, а затем они размножаются, достигая численности в 100 000, любая опасная мутация, затесавшаяся в ДНК одной из 200 митохондрий, тоже размножится. На первый взгляд, это неразумная идея, но именно она спасает от постепенного накопления поломок в череде поколений.
Дело в том, что, если какая-нибудь поломанная ДНК проскочит сквозь бутылочное горлышко, она размножится и, скорее всего, составит значительную долю митохондриальной ДНК образовавшейся яйцеклетки. Такая яйцеклетка с большим грузом вредных мутаций, как правило, лишается возможности передать эту поврежденную ДНК потомкам женщины. В идеале для этого понадобилось бы сделать яйцеклетку нежизнеспособной — неспособной к оплодотворению или делению с последующим превращением в эмбрион. Вероятно, чаще всего так и происходит. Но с точки зрения выживания вида не имеет особого значения, если время от времени рождается детеныш, который не доживет до обзаведения собственным потомством. Главное, чтобы дефектная ДНК не передалась дальше по наследству, тогда вид убережется от ее негативного воздействия и продолжит существование. С этой точки зрения митохондриальные заболевания — наша всеобщая плата за то, что мы не вымерли как вид.
Конечно, это дорогая цена для тех, кому приходится расплачиваться лично. Мы не знаем наверняка, почему так происходит, но тот тип митохондриальной делеции, который вызывает проблемы как у Фелисити, Ахмеда и Джейкоба, практически никогда не передается от матери к детям. По-видимому, бутылочное горлышко очень эффективно устраняет такие поломки. Однако это не относится к другим типам митохондриальных мутаций. Замена единственного основания в митохондриальной ДНК может вызвать проблемы, во всех отношениях столь же тяжелые, как у Джейкоба, с такой же изменчивостью по соотношению здоровой и мутантной митохондриальной ДНК. Чаще всего это, по-видимому, разовое событие: определенной клеточной линии не повезло, и в ней появилась одна яйцеклетка с высокой долей патологической митоходриальной ДНК; в семье это больше не повторится. Но иногда заболевание поражает нескольких братьев и сестер, а порой даже передается следующим поколениям.
Подробнее читайте:
Керк, Э. Мальчик, который не переставал расти… и другие истории про гены и людей / Эдвин Керк ; Пер. с англ. [Марии Елиферовой] — М.: Альпина нон-фикшн, 2022. — 312 с.
Митохондриальные заболевания
Митохондриа́льные заболева́ния — группа наследственных заболеваний, связанных с дефектами в функционировании митохондрий, приводящими к нарушениям энергетических функций в клетках эукариотов, в частности — человека.
Содержание
Общие сведения
Митохондриальные заболевания обусловлены генетическими, структурными, биохимическими дефектами митохондрий, приводящими к нарушениям тканевого дыхания. Они передаются только по женской линии к детям обоих полов, так как сперматозоиды передают зиготе половину ядерного генома, а яйцеклетка поставляет и вторую половину генома, и митохондрии. Патологические нарушения клеточного энергетического обмена могут проявляться в виде дефектов различных звеньев в цикле Кребса, в дыхательной цепи, процессах бета-окисления и т. д.
Не все ферменты и другие регуляторы, необходимые для эффективного функционирования митохондрий, кодируются митохондриальной ДНК. Большая часть митохондриальных функций контролируется ядерной ДНК [1] .
Можно выделить 2 группы митохондриальных заболеваний:
- Ярко выраженные наследственные синдромы, обусловленные мутациямигенов, ответственных за митохондриальные белки (синдром Барта, синдром Кернса-Сейра, синдром Пирсона, синдром MELAS, синдром MERRF и другие).
- «Вторичные митохондриальные заболевания», включающие нарушение клеточного энергообмена как важное звено формирования патогенеза (болезни соединительной ткани, синдром хронической усталости, гликогеноз, кардиомиопатия, мигрень, печеночная недостаточность, панцитопения, а также гипопаратиреоз, диабет, рахит и другие).
Наследование митохондриальных болезней
Митохондрии наследуются иначе, чем ядерные гены. Ядерные гены в каждой соматической клетке обычно представлены двумя аллелями (за исключением большинства сцепленных с полом генов у гетерогаметного пола ). Один аллель унаследован от отца, другой от матери. Однако митохондрии содержат собственную ДНК, причем в каждой митохондрии человека обычно содержится от 5 до 10 копий кольцевой молекулы ДНК (см. Гетероплазмия), и все они наследуются от матери. Когда митохондрия делится, копии ДНК случайным образом распределяются между ее потомками, а затем происходит удвоение ДНК. Если только одна из исходных молекул ДНК содержит мутацию, в результате случайного распределения такие мутантные молекулы могут накопиться в некоторых митохондриях. Митохондриальная болезнь начинает проявляться в тот момент, когда заметное число митохондрий во многих клетках данной ткани приобретают мутантные копии ДНК (пороговая экспрессия).
Мутации в митохондриальной ДНК происходят, по разным причинам, намного чаще, чем в ядерной. Это означает, что митохондриальные болезни достаточно часто проявляются из-за спонтанных вновь возникающих мутаций. Иногда темп мутирования увеличивается из-за мутаций в ядерных генах, кодирующих ферменты, которые контролируют репликацию ДНК митохондрий.
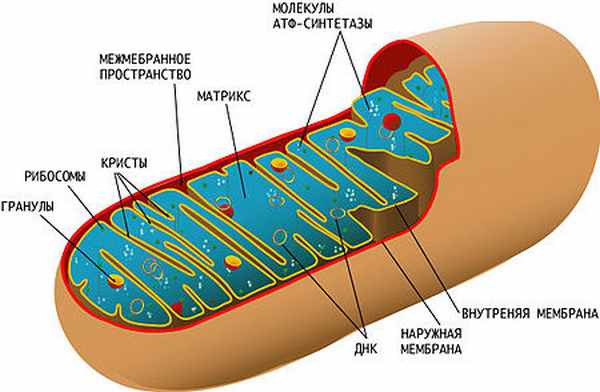
Схема строения митохондрии. Сложная структура митохондрии и наличие собственной кольцевой хромосомы, кодирующей некоторые компоненты митохондрии, усложняет выяснение причин митохондриальных заболеваний
Дефекты и симптомы
Эффекты митоходриальных заболеваний очень разнообразны. Из-за различного распределения дефектных митохондрий в разных органах, мутация у одного человека может привести к заболеванию печени, а у другого — к заболеванию мозга. Величина проявления дефекта может быть большой или малой, и она может существенно изменяться, медленно нарастая во времени. Некоторые небольшие дефекты приводят лишь к неспособности пациента выдерживать физическую нагрузку, соответствующую его возрасту, и не сопровождаются серьёзными болезненными проявлениями. Другие дефекты могут быть более опасны, приводя к серьёзной патологии.
В общем случае, митоходриальные заболевания проявляются сильнее при локализации дефектных митохондрий в мышцах, мозге, нервной ткани, [2] поскольку эти органы требуют больше всего энергии для выполнения соответствующих функций.
Несмотря на то, что протекание митохондриальных заболеваний сильно отличаются у разных пациентов, на основании общих симптомов и конкретных мутаций, вызывающих болезнь, выделено несколько основных классов этих заболеваний.
Типы заболеваний
Помимо относительно распространённой митохондриальной миопатии, встречаются:
- , сопровождающийся глухотой (DAD, MIDD, синдром MELAS) — это сочетание, проявляющееся в раннем возрасте, может быть вызвано мутацией митохондриального гена MT-TL1, но сахарный диабет и глухота могут быть вызваны как митохондриальными заболеваниями, так и иными причинами;
- наследственная оптическая нейропатия Лебера (en:Leber's hereditary optic neuropathy (LHON)), характеризующийся потерей зрения в раннем пубертатном периоде; (en:Wolff-Parkinson-White syndrome)
- рассеянный склероз и подобные ему заболевания; [источник не указан 371 день] (Leigh) или подострая некротизирующая энцефаломиопатия : После начала нормального постнатального развития организма болезнь обычно развивается в конце первого года жизни, но иногда проявляется у взрослых. Болезнь сопровождается быстрой потерей функций организма и характеризуется судорогами, нарушенным состоянием сознания, деменцией, остановкой дыхания
- «Нейропатия, атаксия, retinitis pigmentos и птоз» en:Neuropathy, ataxia, retinitis pigmentosa, and ptosis (NARP): прогрессирующие симптомы нейропатии, атаксии, тунельное зрение и потеря зрения, птоз, деменция;
- Митохондриальная нейрогастроинтенстинальная энцефалопатия en:Mitochondrial neurogastrointestinal encephalomyopathy (MNGIE): гастроинтестинальная псевдообструкция и кахексией, нейропатия, энцефалопатия с изменениями белого вещества головного мозга
Диагностика
Для постановки диагноза митохондриального заболевания важен комплексный генеалогический, клинический, биохимический, морфологический и генетический анализ.
Эпидемиология
Изначально мутации мтДНК считались крайне редкими, однако исследование 3000 здоровых новорожденных на 10 наиболее известных патогенных мутаций, проведённое в 2008 году, выявило таковые у одного человека из 200. [3] «Горячей точкой» в мтДНК оказалась позиция 3243, здесь часто происходит замена A-G, изменяющая функционирование гена MT-TL1.
Лечение
В настоящее время лечение митохондриальных заболеваний находится в стадии разработки, но распространённым терапевтическим методом служит симптоматическая профилактика с помощью витаминов [4] . Также в качестве одного из методов применяются пируваты [5] .
Поскольку в сперматозоиде, который вносит половину хромосом будущего организма, содержится мало митохондрий, митохондриальная наследственность определяется, в основном, митохондриями яйцеклетки. Сейчас проводятся экспериментальные работы по экстракорпоральному (in vitro) оплодотворению с использованием переноса ядра оплодотворённой яйцеклетки в безъядерную цитоплазму другой яйцеклетки с нормально функционирующими митохондриями (замена ядра).
Читайте также:
- Шизофрения у детей и подростков
- Систематика бактерий. Окраска по Граму. Грамположительные бактерии. Грамотрицательные бактерии. Кислотоустойчивые бактерии.
- ЭКГ при фибрилляции желудочков и его лечение
- Топография мочевого пузыря. Мочевой пузырь. Строение мочевого пузыря.
- Хроническая лимфатическая лейкемия (хронический лимфолейкоз) - факторы риска