Множественная эндокринная неоплазия
Добавил пользователь Владимир З. Обновлено: 07.01.2026
Множественная эндокринная неоплазия (МЭН) проявляется опухолями органов эндокринной системы — паращитовидной железы, поджелудочной железы, надпочечников, гипофиза — и делится на 4 подтипа: МЭН 1 (синдром Вермера), МЭН 2А (синдром Сиппла), МЭН 2B (синдром Горлина) и семейный медуллярный рак щитовидной железы. При подозрении на любой из типов МЭН рекомендовано определение характерных мутаций.
Приём и исследование биоматериала
Когда нужно сдавать анализ Диагностика семейного медуллярного рака щитовидной железы и синдромов МЭН 1 и 2?
Диагностика и дифференциация семейного медуллярного рака щитовидной железы и синдромов МЭН 1 и 2.
Подробное описание исследования
Синдромы множественных эндокринных неоплазий (МЭН, MEN) — представляют собой группу генетических патологий, ассоциированных с развитием новообразований эндокринных внутренней секреции (наиболее часто щитовидной железы (ЩЖ). Заболевание наследуется аутосомно-доминантно. Этот термин означает, что патологические гены имеют приоритет в их воспроизведении организмом и находятся в неполовых хромосомах: у человека в норме 46 хромосом, из которых 2 половые – X и Y, а остальные неполовые — аутосомы. МЭН делится на две большие группы: MEN1 и MEN2.
MEN1 (синдром Вермера) — это заболевание, вызванное мутациями (изменениями) в гене-супрессоре опухоли MEN1, который кодирует белок менин. Гены-супессоры подавляют возникновение того или иного признака, а в данном случае развитие эндокринных новообразований. Таким образом, обнаружение MEN 1 у пациента связано с возникновением патологии, а именно: опухолей паращитовидной железы, островков поджелудочной железы, а также онкопатологии передней доли гипофиза.
Также у некоторых больных могут развиваться:
- Карциноидные (нейроэндокринные) новообразования;
- Опухоли надпочечников;
- Менингиомы;
- Ангиофибромы лица;
- Коллагеномы и липомы.
У лиц с МЭН 1 снижена ожидаемая продолжительность жизни. Прогноз для этих больных может быть улучшен путем выявления неоплазии и проведения лечения до появления клинических симптомов. Поэтому пациентам с МЭН 1 и членам их семей рекомендуется находиться под наблюдением врачей различных специализаций. Также в лечении таких пациентов рекомендуется применять мультидисциплинарный подход.
МЭН 2 состоит из трех различных клинических подтипов: MEN-2a, MEN-2b и семейный медуллярный рак (карцинома) ЩЖ (МРЩЖ). МЭН 2 — это достаточно редкая патология, встречаемая в 1 случае на 200 000 новорожденных. Лица с МЭН 2 имеют 100 % риск развития МРЩЖ к возрасту 70 лет, также данное новообразование является наиболее частой причиной смерти у этих больных.
Общими признаками MEN-2a и MEN-2b являются двусторонняя МРЩЖ, встречающаяся у всех пациентов, и двусторонние феохромоцитомы (доброкачественные опухоли надпочечников), встречающиеся у половины.
MEN-2a (синдром Сиппла) — наиболее частое проявление 2-го типа, которое встречается в 55 % случаев. Рак ЩЖ часто является первым проявлением MEN-2a и обычно возникает в возрасте 20–30 лет.
MEN-2b (синдром Горлина) — это более редкая и тяжелая форма данного синдрома. При ней отмечается раннее начало заболевания и более агрессивные формы карциномы ЩЖ, которые появляются и прогрессируют в течение первого года жизни ребенка. Пациенты имеют проблемы с опорно-двигательным аппаратом (марфаноидная внешность), множественные невриномы на губах, щеках, языке и в ЖКТ и обычно умирают в возрасте до 30 лет.
Семейная медуллярная карцинома ЩЖ — это второй по распространенности вариант MEN-2 (35% случаев). Этот подтип заболевания имеет относительно легкое течение, больные обычно не умирают от него.
Как отмечалось ранее, МЭН наследуется аутосомно-доминантно, поэтому болезнь появляется в каждом поколении семьи с вероятностью 50 %. Своевременная диагностика и лечение могут сыграть важную роль в улучшении качества и увеличении продолжительности жизни пациентов. Данный анализ позволяет с высокой вероятностью определить наличие семейного МРЩЖ, а также синдромов MEN 1 и 2.
Генодиагностика изменений при данной патологии имеет решающее значение для выявления лиц, находящихся в группе риска, и направления их для своевременного наблюдения и хирургического лечения. Тест проводится в Москве, а также в других городах и населенных пунктах.
Синдром множественной эндокринной неоплазии 1-го типа: клинический случай
Синдром множественной эндокринной неоплазии 1-го типа (МЭН1) – это наследственная патология, характеризующаяся наличием двух и более опухолей (или гиперплазии) эндокринных желез, наиболее часто поражаются паращитовидные железы. Данная патология встречается довольно редко и изучена недостаточно, однако, учитывая высокую распространенность первичного гиперпаратиреоза в популяции, требует знания подходов к ведению таких больных. Важное прогностическое значение имеет выявление больных из группы риска, поскольку заболевание часто диагностируется спустя годы после возникновения первых клинических симптомов, что может отрицательно повлиять на прогноз. С целью своевременного скрининга и лечения, а также всестороннего динамического контроля за больными с синдромом МЭН1 необходима повышенная настороженность в отношении этой наследственной патологии. Комплексный междисциплинарный подход к ведению больного позволяет улучшить выявляемость заболевания и увеличить продолжительность жизни. В статье представлено собственное клиническое наблюдение больной с синдромом МЭН1 с гетерозиготной заменой с.1261Т>С:р.С421R гена MEN1, не описанный ранее в литературе.
Ключевые слова: синдром множественной эндокринной неоплазии 1-го типа, первичный гиперпаратиреоз, гиперкальциемия, паратгормон, паращитовидные железы, нейроэндокринные опухоли, аденома гипофиза.
Для цитирования: Тевосян Л.Х., Древаль А.В., Крюкова И.В., Барсуков И.А. Синдром множественной эндокринной неоплазии 1-го типа: клинический случай. РМЖ. 2017;1:61-63.
The syndrome of multiple endocrine neoplasia type 1: clinical case
Tevosyan L.H., Dreval’ A.V., Kryukova I.V., Barsukov I.A.
M.F. Vladimirskiy Moscow Regional Research and Clinical Institute, Moscow
MEN syndrome type 1 is a hereditary pathology characterized by the presence of two or more tumors (or hyperplasia) of the endocrine glands most commonly located in the parathyroid glands. This pathology is quite rare and poorly studied, however, given the high prevalence of primary hyperparathyroidism in the population, it requires certain knowledge for the treatment of such patients. Identification of patients at risk has an important prognostic significance, because the disease is often diagnosed years after the occurrence of the first clinical symptoms, which may adversely affect the prognosis for the patient. It is necessary to increase vigilance against this hereditary disease for a prompt screening and treatment, as well as comprehensive dynamic monitoring of patients with the syndrome of multiple endocrine neoplasia type 1. An integrated, multidisciplinary approach to patient management can improve the detection of the disease and increase life expectancy. The article presents the clinical observation of a patient with syndrome of MEN 1 type and heterozygous substitution s.1261T> C: r.S421R MEN1 gene, which hasn’t been described in the literature yet.
Key words: multiple endocrine neoplasia type 1, primary hyperparathyreodism, hypercalcemia, parathyroid hormone, parathyroid glands, neuroendocrine tumors, pituitary adenoma.
For citation: Tevosyan L.H., Dreval’ A.V., Kryukova I.V., Barsukov I.A. The syndrome of multiple endocrine neoplasia type 1: clinical case // RMJ. 2017. № 1. P. 61–63.
В статье представлен клинический случай синдрома множественной эндокринной неоплазии 1-го типа
Синдром множественной эндокринной неоплазии (МЭН) – генетически детерминированная патология с аутосомно-доминантным типом наследования, проявляющаяся доброкачественными или злокачественными опухолями (гиперплазией) двух и более эндокринных желез [1–3]. Синдром МЭН характеризуется высокой степенью пенетрантности, которая к 20-летнему возрасту составляет 50%, к 40 годам – 95% [4, 5]. Риск развития синдрома МЭН в семьях с данной патологией составляет около 75%. В 50% случаев синдром развивается спорадически, т. е. обусловлен вновь возникшей (de novo) мутацией в половых или соматических клетках. Риск развития синдрома МЭН у ребенка со спорадической формой составляет 50%.
Выделяют несколько типов синдрома МЭН, которые отличаются генетической основой, локализацией и сочетанием поражения внутренних органов. Синдром МЭН 1–го типа (МЭН1) – синдром Вермера – наиболее часто характеризуется наличием патологии околощитовидных желез (гиперплазии или аденомы), нейроэндокринной опухолью (НЭО) поджелудочной железы и опухолью аденогипофиза. Для синдрома МЭН 2-го типа характерно наличие медуллярного рака щитовидной железы как постоянного признака, сочетание которого с феохромоцитомой и паратиромой относят к синдрому МЭН 2А типа (синдром Сиппла), а при сочетании феохромоцитомы с множественными ганглионейромами слизистой оболочки желудочно-кишечного тракта – МЭН 2Б типа (синдром Горлина). Различают также синдром МЭН 4-го типа, который характеризуется наличием паратиромы, опухолей аденогипофиза, репродуктивных органов, почек и надпочечников [6, 7].
Синдром МЭН1 наследуется аутосомно-доминантно, но может возникать и спорадически. Распространенность синдрома составляет до 15–30 случаев на 100 000 населения [8]. Причиной развития синдрома является мутация в гене – супрессоре опухолей, расположенном на 11-й хромосоме (11q13). Ген кодирует белок менин, регулирующий пролиферацию клеток. Синдром МЭН1 чаще манифестирует в молодом возрасте (20–25 лет) первичным гиперпаратиреозом, который характеризуется более мягким течением, при этом высокой частотой рецидивов после паратиреоидэктомии (50% от 8 до 12 лет после операции) и морфологически проявляется гиперплазией трех или четырех паращитовидных желез [1]. Первичный гиперпаратиреоз выявляется в 90% случаев синдрома МЭН1 к 40 годам.
В 30–80% случаев синдрома МЭН1 встречаются энтеропанкреатические опухоли, вырабатывающие различные гормоны: соматостатин, грелин, вазоактивный интестинальный пептид, серотонин, кальцитонин, нейротензин, гастрин, хромогранин А или Б, инсулин, проинсулин, глюкагон. Они характеризуются мультицентрическим ростом, чаще манифестируют после 40 лет.
Опухоли гипофиза при синдроме МЭН1 в 2 раза чаще встречаются у мужчин, чем у женщин. Среди опухолей гипофиза при синдроме МЭН1 наиболее часто встречаются пролактиномы (60%), а также опухоли, секретирующие гормон роста (25%). Реже встречаются макроаденомы, которые характеризуются агрессивным ростом и низкой чувствительностью к терапии. Менее чем в 5% случаев встречаются нефункциональные опухоли, кортикотропиномы [2].
При синдроме МЭН1 также могут диагностироваться опухоли, расположенные в бронхах и тимусе. Опухоли тимуса чаще являются нефункциональными, характеризуются агрессивным ростом и преимущественно встречаются у курильщиков [3]. У женщин чаще наблюдаются карциноиды бронхов, которые могут секретировать соматостатин, серотонин, кортикотропин и гормон роста. Липомы при синдроме МЭН1 могут располагаться подкожно, ретроперитонеально, висцерально и плеврально [4]. У 20–40% больных встречаются опухоли надпочечников, чаще всего доброкачественные (аденомы, диффузная или узловая гиперплазия) и гормонально неактивные [4].
Ожидаемая продолжительность жизни больных с синдромом МЭН1 снижена. Благоприятный прогноз имеется при раннем выявлении синдрома и своевременном лечении, что предотвращает инвалидизацию. У больных с множественными злокачественными опухолями (глюкагономой, инсулиномой, випомой, гастриномой) прогноз хуже. Смертность составляет 50% у больных старше 50 лет, примерно в половине случаев – вследствие злокачественных НЭО и карциноидных опухолей тимуса [5, 9]. В связи с этим очень важно наблюдение пациентов специалистами различного профиля (гастроэнтерологов, онкологов, хирургов-эндокринологов, радиологов, генетиков), имеющих опыт лечения данного заболевания.
Клинический случай
Больная М., 35 лет, в 2016 г. обратилась к кардиологу консультативно-диагностического отделения МОНИКИ им. М.Ф. Владимирского в связи повышением артериального давления максимально до 200/100 мм рт. ст., которое стало отмечаться с 20-летнего возраста. При лабораторном обследовании впервые была выявлена гиперкальциемия, больную направили на консультацию к эндокринологу. При проведении лабораторных исследований для уточнения наиболее вероятных причин гиперкальциемии отмечалось повышение паратгормона (ПТГ) до 69,6 пг/мл (референсные значения 15,0–65,0) и 10,1 пмоль/л (реф. зн. 1,26–7,58). При двухкратном повторном исследовании общего кальция крови было подтверждено его повышение до 2,63 и 2,69 ммоль/л (реф. зн. 2,10–2,55), а также выявлено снижение неорганического фосфора до 0,64 ммоль/л (реф. зн. 0,87–1,45). В биохимическом анализе крови исследовались общий белок, альбумин, щелочная фосфатаза, уровень которых был в пределах референсных значений, а также креатинин – 72 мкмоль/л (реф. зн. 44–80) с расчетом скорости клубочковой фильтрации по формуле Коккрофта – Голта, которая составила 93 мл/мин. Также при определении кальция в суточной моче была отмечена гиперкальциурия – 8,8 ммоль/сут (реф. зн. 0–6,2). У больной диагностирован первичный гиперпаратиреоз.
У пациентки исследован витамин D сыворотки крови 25(ОН)D, уровень которого составил 18,9 нг/мл, что говорит о его дефиците. Несмотря на наличие легкой гиперкальциемии, под контролем общего кальция крови и ПТГ был назначен препарат колекальциферола в стандартной насыщающей дозе 7000 МЕ/сут курсом 8 нед. После восполнения дефицита витамина D (до 31,3 нг/мл) показатели кальция крови и ПТГ оставались в исходных пределах.
Дальнейшее обследование с применением инструментальных методов диагностики было направлено на поиск возможных костных и висцеральных проявлений первичного гиперпаратиреоза и визуализацию паращитовидных желез.
Была проведена двухэнергетическая рентгеновская абсорбциометрия, при которой выявлено незначительное снижение минеральной плотности кости (по Z-критерию) по сравнению с возрастной нормой только в проксимальном отделе бедра – 1,4SD (neck), при отсутствии в анамнезе переломов и деформаций скелета. При эзофагогастродуоденоскопии диагностированы дистальный катаральный эзофагит и гастродуоденит. В проекции хвоста поджелудочной железы выявлено гипоэхогенное округлое образование неоднородной структуры размером 10×8 мм по данным ультразвукового исследования брюшной полости и почек. Компьютерная томография органов брюшной полости и забрюшинного пространства с контрастированием также выявила объемное образование в хвосте поджелудочной железы, размеры которого составили 14,5×14,5×15 мм, кроме этого были отмечены признаки гиперплазии левого надпочечника.
Ультразвуковое исследовании щитовидной и паращитовидных желез показало наличие гипоэхогенного округлого образования с четкими контурами размером 6×4×4 мм позади нижнего полюса левой доли щитовидной железы, что трактовалось как вероятное образование левой нижней паращитовидной железы, требующее дифференцирования с лимфатическим узлом. На мультиспиральной компьютерной томографии шеи и средостения с контрастированием вдоль задне-нижнего контура левой доли щитовидной железы также выявлено образование неправильной округлой формы размером 6,0×6,1 мм, что было расценено как вероятная аденома паращитовидной железы. В переднем средостении, кпереди от восходящей части аорты и легочного ствола, определялось мягкотканное образование неправильной округлой формы с четкими контурами, не реагирующее на контрастное усиление, размером 17×40×21 мм, прилежащее к восходящему отделу аорты и легочному стволу. При сцинтиграфии с однофотонной эмиссионной томографией шеи и средостения определялся очаг гиперфиксации радиофармпрепарата по заднему контуру левой доли щитовидной железы на уровне ее нижней трети, что расценено как аденома левой нижней околощитовидной железы.
Учитывая молодой возраст больной, подтвержденный первичный гиперпаратиреоз, а также выявленное образование поджелудочной железы, было высказано предположение о наличии синдрома МЭН1 и рекомендовано скрининговое обследование родственников первой линии родства. Проведены диагностические тесты для исключения первичного гиперпаратиреоза как наиболее частого проявления предполагаемой наследственной патологии. У матери выявлено повышение общего кальция до 3,09 ммоль/л (реф. зн. 2,1–2,55) и ПТГ 78,4 пг/мл (реф. зн. 12–65); у родного брата – кальций ионизированный составил 1,22 ммоль/л (реф. зн. 1,03–1,23), ПТГ – 6,91 пмоль/л (реф. зн. 1,6–6,9). В настоящее время проводится дальнейшее обследование родственников.
Продолжено обследование больной для исключения гормональной активности выявленного образования поджелудочной железы, гиперфункции коры надпочечников, а также других возможных проявлений синдрома МЭН1 при отсутствии каких-либо специфических жалоб и симптомов. При магнитно-резонансной томографии гипофиза с контрастированием патологии не выявлено. Несмотря на это, гормональное обследование включило определение гормонов аденогипофиза: адренокортикотропного гормона, тиреотропного гормона, пролактина, а также инсулиноподобного фактора 1 (ИРФ-1), кортизола крови – отклонений от референсных значений не выявлено. Указание на подъем артериального давления и вероятная гиперплазия надпочечника послужили показанием к исследованию уровня альдостерона крови и активности ренина плазмы с расчетом альдостерон-ренинового соотношения, а также метилированных катехоламинов в суточной моче (метанефрина и норметанефрина). Повышения данных параметров не выявлено. Уровень исследованного хромогранина А также был в пределах нормальных значений, другие маркеры НЭО по техническим причинам не определялись. Отсутствие отклонений исследуемых лабораторных параметров позволило в настоящее время исключить гормональную активность образования поджелудочной железы и гиперфункцию коры надпочечников.
В дальнейшем больной было проведено генетическое исследование методом полимеразной цепной реакции (ПЦР) в Эндокринологическом научном центре. По результатам ДНК-диагностики (ПЦР, прямое секвенирование экзонов 2–10 гена MEN1) выявлена гетерозиготная замена с.1261Т>С:р.С421R. Мутация в том же кодоне описана при синдроме МЭН1, однако конкретная мутация ранее не была описана.
Таким образом больной был поставлен диагноз: «Множественная эндокринная неоплазия 1-го типа. Первичный гиперпаратиреоз, мягкая форма. Аденома левой нижней околощитовидной железы. Гормонально неактивное образование поджелудочной железы. Образование переднего средостения».
Следует отметить наличие у больной осложненного акушерско-гинекологического анамнеза. Первая неразвивающаяся беременность была прервана на сроке 5 нед. гестации в возрасте 29 лет. Вторая беременность в 32 года завершилась экстренным оперативным вмешательством путем кесарева сечения на сроке 24–25 нед. в связи с тяжелой преэклампсией. Было рекомендовано проведение генотипирования, по результатам которого выявлен полиморфизм, ассоциированный со снижением фибринолитической активности и риском тромбозов в гетерозиготной форме SERPINE1: 4G/5G (PAI1:4G/5G4; Ins/Del G) и полиморфизм, предрасполагающий к тромбозам в гетерозиготной форме MTHFR: C677T (Ala222Val).
Молодой возраст является одним из абсолютных показаний к оперативному лечению первичного гиперпаратиреоза. Однако неоднозначность современных подходов относительно объема хирургического вмешательства на паращитовидных железах у больных с синдромом МЭН1 и мягкое течение гиперпаратиреоза у данной больной позволили придерживаться в настоящее время консервативной тактики ведения. Возможное планирование беременности должно послужить поводом для повторного решения вопроса о необходимости оперативного лечения в связи с риском утяжеления гиперкальциемии, а сочетание с выявленной генетической предрасположенностью к тромбообразованию требует всестороннего дальнейшего наблюдения с участием специалистов различного профиля.
Заключение
Данное клиническое наблюдение свидетельствует о важности скринингового исследования общего кальция крови при наличии неспецифических проявлений первичного гиперпаратиреоза. Молодой возраст больных с подтвержденным диагнозом первичного гиперпаратиреоза, в особенности сочетающегося с плохо контролируемым течением артериальной гипертензии, является безусловным поводом для проведения диагностического поиска синдрома множественной эндокринной неоплазии. Выявленная мутация гена MEN1 у представленной больной, возможно, способствует менее агрессивному течению заболевания. Выбранная консервативная тактика ведения должна сопровождаться активным динамическим контролем за развитием клинической картины для своевременной оценки показаний к оперативному лечению.
1. Eller-Vainicher C., Chiodini I., Battista C. et al. Sporadic and MEN1-related primary hyperparathyroidism: differences in clinical expression and severity // J Bone Miner Res. 2009. Vol. 24(8). Р. 1404–1410.
2. Trouillas J., Labat-Moleur F., Sturm N. et al. Pituitary tumors and hyperplasia in multiple endocrine neoplasia type 1 syndrome (MEN1): a case-control study in a series of 77 patients versus 2509 non-MEN1 patients // Am J Surg Pathol. 2008. Vol. 32(4). Р. 534–543.
3. Ferolla P., Falchetti A., Filosso P. et al. Thymic neuroendocrine carcinoma (carcinoid) in multiple endocrine neoplasia type 1 syndrome: the Italian series // J Clin Endocrinol Metab. 2005. Vol. 90(5). Р. 2603–2609.
4. Thakker R.V., Newey P.J., Walls G.V. et al. Clinical Practice Guidelines for Multiple Endocrine Neoplasia Type 1 (MEN1) // J Clin Endocrinol Metab. 2012. Vol. 97(9). Р. 2990–3011.
5. Brandi M.L., Gagel R.F., Angeli A. et al. Guidelines for diagnosis and therapy of MEN type 1 and type 2 // J Clin Endocrinol Metab. 2001. Vol. 86(12). Р. 5658–5671.
6. Эндокринология. Национальное руководство под редакцией акад. РАН И.И. Дедова. М.: ГЭОТАР-Медиа, 2016. С. 1112 [Еhndokrinologiya. Nacionalnoe rukovodstvo pod redakciej akad. RAN I.I. Dedova. M: Gehotar-Media. 2016. S. 1112 (in Russian)].
7. Древаль А.В. Эндокринология: руководство для врачей. М.: ГЭОТАР-Медиа. 2016. С. 544 [Dreval A.V. Еndokrinologiya: rukovodstvo dlja vrachej. M.: Geotar-Media. 2016. S. 544 (in Russian)].
8. Vasilev V., Daly A.F., Petrossians P., Zacharieva S., Beckers A. Familial pituitary tumor syndromes // Endocrine Practice. 2011. Vol. 17(S. 3). 41–46.
9. Goudet P., Murat A., Binquet C. et al. Risk factors and causes of death in MEN1 disease. A GTE (Groupe d'Etude des Tumeurs Endocrines) cohort study among 758 patients // World J Surg. 2010. Vol. 34(2). Р. 249–255.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Множественная эндокринная неоплазия
ФГУ Эндокринологический научный центр, Москва
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Множественная эндокринная неоплазия 2-го типа
Журнал: Проблемы эндокринологии. 2011;57(6): 21‑26
Юкина М.Ю., Гончаров Н.П., Бельцевич Д.Г., Трошина Е.А. Множественная эндокринная неоплазия 2-го типа. Проблемы эндокринологии. 2011;57(6):21‑26.
Iukina MIu, Goncharov NP, Bel'tsevich DG, Troshina EA. Multiple type 2 endocrine neoplasia (case report). Problemy Endokrinologii. 2011;57(6):21‑26. (In Russ.).
ФГУ Эндокринологический научный центр, Москва
Наследственная форма феохромоцитомы отличается склонностью к рецидиву, двустороннему, мультицентричному и первично-множественному поражению. Для пациентов с синдромом множественных эндокринных неоплазий 2-го типа не характерны вненадпочечниковая локализация феохромоцитомы и наличие метастазов. Обследование и лечение данной категории пациентов необходимо проводить с учетом этих особенностей. На примере семьи с синдромом множественных эндокринных неоплазий 2-го типа освещены вопросы ведения пациентов с генетически-детерминированной феохромоцитомой.
ФГУ Эндокринологический научный центр, Москва
ФГУ Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Эндокринологический научный центр, Москва
Множественная эндокринная неоплазия 2-го типа (МЭН 2) — синдром с аутосомно-доминантным характером наследования и с предполагаемой распространенностью 1: 30 000 населения. МЭН 2 обусловлена герминативными мутациями гена RET, который расположен на хромосоме 10q11.2 и включает 21 экзон. RET (REarranged during Transfection) был открыт в 1985 г. как протоонкоген, способный активироваться после генетической перестановки [1]. Его продуктом является тирозинкиназа рецепторного типа отвечающая за рост, дифференцировку и выживание клетки. Мутация гена RET в эмбриональных клетках приводит к экспрессии патологически измененного сверхактивного RET-протеина в нейроэндокринных тканях, что влечет за собой бесконтрольную клеточную пролиферацию [2—8]. Более чем в половине случаев причиной синдрома является герминативная мутация RET, которая возникает только в отцовском аллеле [9—11].

Синдром МЭН 2 включает типы 2А (синдром Сиппла) и 2В (синдром Горлина); выделяют также семейную форму МРЩЖ (медуллярный рак щитовидной железы — МРЩЖ) [12, 13]. Эти формы отличаются друг от друга распространенностью, возрастом манифестации заболевания, типом мутации, ассоциацией с другими заболеваниями, агрессивностью МРЩЖ и прогнозом [2, 3] (см. таблицу).
Синдром МЭН 2A более распространен (90% пациентов с МЭН 2) [14—18], тогда как синдром МЭН 2B (5—10% всех случаев) — более агрессивный вариант.
МЭН 2 имеет высокую пенетрантность (90—100%) относительно MРЩЖ (кальцитонинпродуцирующей опухоли из парафолликулярных клеток щитовидной железы) [19, 20]. На долю МРЩЖ приходится 2—8% всех раков щитовидной железы [21—23]. Лучший маркер опухоли — кальцитонин [24, 25]. Уровень кальцитонина как базальный, так и стимулируемый пентагастрином или глюконатом кальция при МРЩЖ почти всегда повышен [26—28]. МРЩЖ — самая частая причина смерти больных с МЭН 2 [29]. Агрессивность МРЩЖ уменьшается в следующем порядке: МЭН 2В > МЭН 2А > семейный вариант МРЩЖ [30]. При МЭН 2B клинические проявления МРЩЖ манифестируют уже на первом году жизни, при МЭН 2А — в возрасте от 2 до 25 лет в зависимости от мутации [31, 32]. Наследственный МРЩЖ обычно бывает двусторонним и мультифокальным [33]. МРЩЖ является быстрометастазирующей опухолью с тенденцией к медленному росту [34]. Метастазы определяются в лимфоузлах средостения, центральных и боковых лимфоузлах шеи или имеют более отдаленную локализацию: в легкие, печень, кости [35]. Выживаемость зависит от распространения опухоли: 10-летняя выживаемость 95% для пациентов с опухолью, не выходящей за пределы щитовидной железы, и 40% для пациентов с отдаленными метастазами [12]. МРЩЖ относительно устойчив к лучевой терапии и стандартной химиотерапии [36, 37]. Хирургическое лечение остается единственным лечением для пациентов с МРЩЖ [38]. Недавние молекулярно-биологические исследования выявили определенные генетические изменения, связанные с МРЩЖ и привели к модификациям диагностических и терапевтических алгоритмов [39]. Так, профилактическая тиреоидэктомия оказалась эффективной лечебно-профилактической мерой при наследственном МРЩЖ у молодых пациентов [35].
Феохромоцитома манифестирует приблизительно у 50% пациентов с МЭН 2A или 2B. Феохромоцитома у пациентов с МЭН 2 всегда локализована в надпочечниках [40] и в 30% случаев уже при первичном диагнозе присутствует с обеих сторон [41]. У 50% пациентов с односторонней феохромоцитомой в течение 10 лет выявляется феохромоцитома контралатерального надпочечника [42]. Для диагностики используют измерение метанефрина и норметанефрина в плазме или суточной моче, КТ или МРТ надпочечников [43]. При выявлении феохромоцитомы хирургическое лечение проводится после соответствующей предоперационной подготовки.
ПГПТ отмечается у 20—30% больных с МЭН 2А [44]. Большинство случаев протекают бессимптомно, хотя могут выявляться гиперкальциурия и мочекаменная болезнь. При МЭН 2А отмечается более мягкое течение ПГПТ, чем при МЭН 1. Средний возраст выявления заболевания — 38 лет [45, 46]. Обследование на ПГПТ включает определение уровня кальция и паратиреоидного гормона в сыворотке. При определении показаний и тактики хирургического лечения (резекция увеличенных желез, субтотальная паратиреоидэктомия, тотальная паратиреоидэктомия с аутотрансплантацией) необходимо учитывать множественное поражение околощитовидных желез и высокую вероятность рецидива ПГПТ [43].
Герминальная мутация RET в 10—40% обусловливает болезнь Гиршпрунга, причем чаще — семейные ее формы [47, 48]. Болезнь Гиршпрунга (врожденное отсутствие иннервации тонкого кишечника) приводит к кишечной непроходимости в раннем детстве [49].
Кожный амилоидоз по типу красного плоского лишая или кожный зуд в верхней части спины может указывать на наличие мутации в 634 кодоне RET. При гистологической верификации у данных пациентов рекомендуется генетическое исследование. При наличии лихеноидного амилоидоза кожи показано симптоматическое лечение, направленное на уменьшение кожного зуда [43].
Фенотипические признаки синдрома МЭН 2В включают костно-мышечные нарушения: марфаноподобную внешность, ганглионейромы губ, переднелатеральной поверхности языка и конъюнктивы, полую стопу, воронкообразную грудную клетку, слабость проксимальных мышц, миелинизацию роговичного нерва, ганглионейроматоз и мальформацию мочевого пузыря, интестинальный ганглионейроматоз [31,50—55].
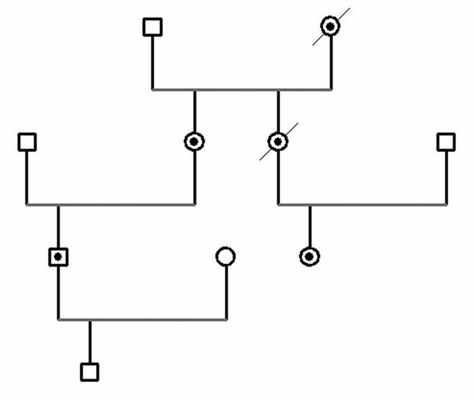
В рамках обсуждаемой темы, представляем клиническое наблюдение семьи с синдромом МЭН 2А (см. рисунок). Рисунок 1. Родословная семьи Н/Г.
Пациентка Н., 1958 г. рождения; из семейного анамнеза известно, что ее сестра умерла в 30-летнем возрасте от острого нарушения мозгового кровообращения во время родов (анамнез ее дочери описан ниже). Впервые обратилась за медицинской помощью в 1978 г. (после родов, анамнез сына описан ниже) с жалобами на повышение АД (максимально до 260/130 мм рт.ст.), сопровождающееся повышенным потоотделением, чувством жара, дрожью в руках, учащенным сердцебиением, болями в области сердца, (симптомы купировались самопроизвольно). В 1979 г. проведена двусторонняя резекция надпочечников с феохромоцитомой; в 1980, 1988 и 1995 гг. — операции по поводу рецидива феохромоцитомы правого надпочечника, в 1987 г. — адреналэктомия с феохромоцитомой слева. Назначена заместительная терапия надпочечниковой недостаточности. В 1996 г. проведено молекулярно-генетическое исследование; выявлена гетерозиготная мутация в 11-м экзоне RET-протоонкогена TGC→CGC Cys634Arg. В 1997 г. произведена экстрафасциальная тиреоидэктомия по поводу МРЩЖ (T1N1вM0). Назначен левотироксин, препараты кальция. При УЗИ и КТ надпочечников данных за рецидив феохромоцитомы не выявлено. В 2001 г. на основании положительной пробы с глюконатом кальция (кальцитонин >100 пг/мл) произведено удаление клетчатки и лимфоузлов левого и правого боковых треугольников шеи. С 2006 г. начала вновь отмечать подъемы АД (максимально до 160/90 мм рт.ст.), повышение гликемии до 8 ммоль/л. При УЗИ в проекции правого надпочечника определяется гипоэхогенное образование диаметром 1,9 см. Уровень норметанефрина в моче — 537 мкг/сут (норма 30—440 мкг/сут), метанефрина — 371 мкг/сут (норма 20—345 мкг/сут). Учитывая спаечный процесс после неоднократных оперативных вмешательств в области проекции объемного образования в надпочечнике, небольшой размер опухоли и отсутствие риска ее метастазирования при данном синдроме, решено придерживаться выжидательной тактики. Назначен доксазозин (4 мг/сут). С августа 2008 г. повышение АД максимально до 260/130 мм рт.ст. При МРТ забрюшинного пространства надпочечники не визуализируются (удалены); в проекции правого надпочечника объемное образование округлой формы с четкими несколько неровными контурами, размерами до 2,3×2,5 см. Назначен доксазозин (6 мг утром и 6 мг вечером). Уровень метанефрина в моче — 490 мкг/сут, норметанефрина — 670 мкг/сут. В ноябре 2008 г. выполнена правосторонняя поперечная боковая лапаротомия. При ревизии за нижним краем правой доли печени, располагаясь по задней поверхности нижней полой вены в сращениях, связанных с предыдущими операциями, обнаружена опухоль около 2,5 см в диаметре. Опухоль удалена. Гистологическое заключение: феохромоцитома без прорастания капсулы и сосудов. В послеоперационном периоде возникло кровотечение диффузного характера из рубцовой ткани, что привело к развитию ДВС-синдрома. Явления полиорганной недостаточности постепенно нарастали и пациентка скончалась.
Пациент Н., 1978 г. рождения, сын пациентки Н. В 1999 г. выявлена мутация в кодоне Cys634Arg (TGC>CGC) 11-го экзона протоонкогена RET (у сына пациента Н. мутация не выявлена). Проведена тиреоидэктомия. Гистологическое заключение: микрофокусы медуллярной аденокарциномы. Назначен левотироксин, препараты кальция. В 2003 г. выполнена правосторонняя адреналэктомия по поводу феохромоцитомы правого надпочечника. В декабре 2008 г. на фоне тяжелой физической нагрузки возник приступ длительностью около 15 мин, сопровождавшийся слабостью, сердцебиением, дрожью в руках; АД 90/60 мм рт.ст., приступ купировался самостоятельно. В марте 2009 г. уровень метанефрина — 789 мкг/сут, норметанефрина — 352 мкг/сут. МСКТ брюшной полости и забрюшинного пространства: из типично расположенного левого надпочечника исходит овоидной формы образование неоднородной структуры с участками кистозной трансформации и некроза в центре, размерами 3,54×3,57×4,82 см. В апреле 2009 г. была произведена лапароскопическая левосторонняя адреналэктомия с опухолью. Послеоперационный период протекал без осложнений. Гистологическое исследование послеоперационного материала: феохромоцитома надпочечника альвеолярного строения. Назначена заместительная терапия надпочечниковой недостаточности.
Пациентка Г., 1988 г. рождения, дочь сестры пациентки Н. В 1996 г., учитывая семейный анамнез, проведено молекулярно-генетическое исследование, результаты которого подтвердили наличие гетерозиготной мутации в 11-м экзоне RET-протоонкогена TGC→CGC Cys634Arg. В 11-летнем возрасте выполнена превентивная тиреоидэктомия (без удаления околощитовидных желез). При гистологическом исследовании удаленной ткани признаков С-клеточной гиперплазии не обнаружено. Назначен левотироксин. В августе 2008 г. обратилась с жалобами на повышение АД в ночное время (максимально до 180/100 мм рт.ст.), сопровождающееся чувством жара, головной болью, перебоями в сердце, чувством страха, потливостью, головокружением, дрожью в руках. Симптомы самостоятельно купировались через 15—30 мин. Уровень метанефрина в моче — 2366 мкг/сут, норметанефрина — 1616 мкг/сут. При МРТ в области правого надпочечника определяется неоднородно изогипоинтенсивное по Т1 и гиперинтенсивное по Т2 объемное образование неправильной овоидной формы с четкими контурами без перифокального отека размером 2,7×1,9×2,0 см; в проекции левого надпочечника визуализируется кистозное образование с гипоинтенсивной капсулой, четкими контурами, гиперинтенсивным по Т2 содержимым, округлой формы без перифокальной реакции до 4,7 см в диаметре. В предоперационном периоде получала доксазозин 4 мг/сут. Произведена лапароскопическая двусторонняя адреналэктомия с опухолями. Послеоперационный период протекал без осложнений. Гистологическое заключение: феохромоцитомы надпочечников. Назначена заместительная терапия хронической надпочечниковой недостаточности.
Данное наблюдение является хорошим примером неэффективности органосохраняющих операций при генетически-детерминированной феохромоцитоме. Генетическая составляющая синдрома, предполагающего прогрессирование первично-множественного процесса, не позволяет сомневаться в необходимости очень осторожного решения относительно органосохраняющих операций. Семейный анамнез и время манифестации заболевания, агрессивность течения, сопряженная с определенными вариантами мутации RET-протоонкогена, возраст и комплаентность пациента могут определять тактику лечения. Хирургическое вмешательство с сохранением коркового вещества надпочечника может считаться показанным лишь при наличии только одного надпочечника, а при двусторонней феохромоцитоме — в случаях «неагрессивных» мутаций с клинической манифестацией в позднем возрасте. Важно максимально информировать пациента о необходимости пожизненной заместительной терапии и ее недостатках (в случае тотальной адреналэктомии), а также о вероятности рецидива феохромоцитомы при органосохраняющих операциях и высокого интра- и послеоперационного риска повторных вмешательств.
Приведенные описания позволяют внести коррективы в интерпретацию результатов определения уровня метилированных катехоламинов у пациентов с генетически-детерминированной феохромоцитомой. Обычно 2- или 4-кратное превышение верхней границы нормы или уровень норметанефрина в моче более 1500 мкг/сут и метанефрина более 600 мкг/сут с большой долей вероятности предполагает наличие феохромоцитомы. При таких значениях биохимических показателей вероятность феохромоцитомы у пациента настолько высока, что проведение дополнительных лабораторных исследований не требуется. Следующая задача — определение местонахождения опухоли [56]. При меньшем увеличении экскреции катехоламинов, не достигающей диагностического уровня (в пределах «серой зоны»), проводят дополнительные исследования: повторное определение метанефрина и норметанефрина с исключением влияния лекарственных препаратов, тест с клонидином или определение уровня хромогранина А. Тактика обследования пациентов из группы риска (с известным наследственным синдромом) несколько иная. Известно, что при наличии опухоли могут иметь место отрицательные биохимические результаты (26—29%) или повышение показателей в пределах «серой зоны»[57, 58]. Наиболее вероятными причинами является небольшой размер опухоли и ее невысокая метаболическая активность на ранней стадии [58]. Таким образом, у пациентки Н. повышение уровней метанефрина и норметанефрина, остающихся в пределах «серой зоны», было правильно интерпретировано как манифестация феохромоцитомы. Учет ее генетического анамнеза позволил исключить необходимость дополнительных уточняющих исследований.
Выводы
1. Хирургическое вмешательство с сохранением коркового вещества надпочечника может рассматриваться при наличии только одного надпочечника, а при двусторонней феохромоцитоме — в случаях получения данных о «неагрессивных» мутациях с манифестацией в позднем возрасте.
2. Повышенный уровень метилированных катехоламинов в пределах «серой зоны» при известном генетическом синдроме можно рассматривать как показатель манифестации феохромоцитомы. В отличие от спорадических феохромоцитом, в данном случае возможна инициация топографического поиска без проведения дополнительных исследований.
Особенности множественной эндокринной неоплазии 1 типа у пациента 48 лет (МЭН-1 у пациента 48 лет)
Приведено уникальное наблюдение нетипичной множественной эндокринной неоплазии 1го типа (МЭН1) (смешанный вариант) у мужчины 48 лет, обусловленной наличием у него 9 опухолей и множественных гиперплазий в 8 различных органах (околощитовидных железах, поджелудочной (ПЖ) и щитовидной (ЩЖ) железах, в двенадцатиперстной кишке, легком, желудке, коже, надпочечниках). Особенностью данного случая является медуллярная карцинома ЩЖ, которая обычно ассоциируется с синдромом МЭН2. У пациента имела место типичная клиника гипогликемического синдрома, однако среди всех исследованных нейроэндокринных опухолей инсулинпродуцирующих клеток не оказалось, что не исключает наличия невыявленной (возможно, очень маленькой) опухоли ПЖ. В то же время известно, что клинические проявления МЭН1 могут быть обусловлены суммарным эффектом всего комплекса гормонов, продуцируемых одновременно несколькими множественными опухолями.
Ключевые слова
Для цитирования:
For citation:
Синдром множественной эндокринной неоплазии 1-го типа (МЭН-1), впервые описанный P. Wermer в 1954 г., возникает в результате мутации 11-й хромосомы в перицентрической области (11q13), которая затем наследуется по аутосомно-доминантному типу [1]. МЭН-1 чаще всего манифестирует в 3–4-й декаде жизни, редко после 60 лет. Она обычно характеризуется гиперплазией или неопластической трансформацией двух или более эндокринных желез: одной или всех четырех околощитовидных желез (ОЩЖ), наличием аденомы передней доли гипофиза, одной или множеством нейроэндокринных опухолей (НЭО) поджелудочной железы (ПЖ) и/или двенадцатиперстной кишки (ДПК) [2, 3]. Реже у пациентов возникают НЭО желудочно-кишечного тракта, тимуса, легких, щитовидной железы (ЩЖ), надпочечников [4]. Клинические проявления МЭН-1, количество и локализация опухолей у разных пациентов различаются. В литературе описано более 20 различных вариантов этого синдрома. Из гиперфункциональных синдромов при МЭН-1 чаще всего встречаются синдромы Золлингера–Эллисона и гиперинсулинизма [5]. Представляем уникальное клиническое наблюдение синдрома МЭН-1.
КЛИНИЧЕСКИЙ СЛУЧАЙ
Клинический диагноз. Синдром МЭН-1. Первичный гиперпаратиреоз, субклиническая форма, аденома нижней левой ОЩЖ; органический гиперинсулинизм, инсулинома хвоста ПЖ; карциноидный синдром (стертая форма), карциноиды ДПК; множественные нейрофибромы кожи; метастаз злокачественного карциноида в правое легкое .
12.03.2012. Операция: удаление аденомы ОЩЖ слева; энуклеация узла из правой доли ЩЖ; дуоденотомия, удаление опухолей ДПК и парадуоденальной зоны; дистальная резекция ПЖ, спленэктомия.
При срединной лапаротомии в брюшной полости выраженное абдоминальное ожирение. Желудок громадных размеров, стенки его утолщены, через серозную оболочку пальпируются грубые гипертрофированные складки слизистой желудка. В луковице ДПК интрамурально определяются два опухолевидных, эластичных, четко отграниченных образования по внутренне-передней стенке. В области дуоденального окна в клетчатке имеется инкапсулированная опухоль, не связанная с ПЖ, расположенная рядом с нисходящей частью ДПК, размерами 7 × 6 см, мягкой консистенции. Головка ПЖ интактна. Выполнена передняя продольная дуоденотомия в области луковицы ДПК над пальпируемым в ее стенке образованием. Под слизистой и субсерозно (в интрамуральном слое) обнаружено два образования плотно-эластической консистенции, размерами 2 × 2,5 и 1,1 × 1,5 см, которые энуклеированы со стороны слизистой. ПЖ диффузно изменена, плотно-дольчатой структуры. При тщательной пальпации в области хвоста в толще железы обнаружено образование до 1,5 см. Предпринята резекция хвоста ПЖ. После резекции ПЖ на операционном столе уровень сахара крови поднялся с 2,0–2,5 до 8 ммоль/л. Показатели Са и Р крови после операции нормализовались (Са – 1,96 ммоль/л; Р – 1,7 ммоль/л).
Послеоперационный период протекал тяжело: уровень сахара крови колебался от 12,7 до 6,8 ммоль/л, периодически снижаясь до 2,3–3,5 ммоль/л. 02.04.2012 диагностирована перфорация задней стенки желудка. Выполнены ушивание дефекта стенки желудка и гастростомия с целью его разгрузки. Интенсивное лечение в условиях реанимационного отделения оказалось неэффективным, и 04.04.2012 больной скончался.
Заключительный клинический диагноз: синдром МЭН-1 ( аденома ОЩЖ, первичный субклинический гиперпаратиреоз; инсулинома ПЖ, гиперинсулинизм; карциноиды ДПК, карциноидный синдром; медуллярный рак ЩЖ с метастазом в правое легкое; множественные нейрофибромы кожи, гипертрофический гастрит по типу болезни Менетрие).
Результаты патоморфологического и иммуногистохимического (ИГХ) исследований
Все НЭО были положительны к нейроэндокринным маркерам – синаптофизину (Син) и хромогранину А (ХрА).
1. Поджелудочная железа
А) Операционный материал. Множественные узелки 0,5–0,8 см, желтовато-коричневого цвета. При гистологическом исследовании выявлены три нейроэндокринные опухоли: 1 – опухоль сложного строения, где участки из светлых клеток чередуются с участками из темных клеток, окружена капсулой, в которой располагаются скопления нейроэндокринных островков, при ИГХ исследовании большинство клеток продуцируют панкреатический полипептид (ПП), единичные – соматостатин (Сом); 2 – микроопухоли трабекулярного строения, без капсулы, с локальной инвазией окружающей ткани. При ИГХ исследовании в большинстве клеток микроопухолей выявлена продукция глюкагона (Гл), в единичных клетках – Сом и ПП; индекс Ki67 всех опухолей – от 1 до 1,5%, G1.
Б) На аутопсии в ткани ПЖ дополнительно выявлены множественные узелки размером до 0,8 см, плохо отграниченные, серо-желтые. Гистологически опухоль трабекулярного строения, в капсуле, до 30% клеток продуцируют Гл, 5–8% – ПП, индекс Ki67 = 6,5%. Во всех четырех выявленных опухолях ПЖ продукция инсулина и гастрина не обнаружена. В окружающей ткани ПЖ картина незидиобластоза (НЗБ) с большим количеством микроаденом различного калибра и ПП-клеточной гиперплазией островков.
Заключение: множественные (4) высокодифференцированные нефункционирующие НЭО ПЖ, на фоне НЗБ эндокринной ткани ПЖ: 2 глюкагономы, 1 ПП-ома, G1; 1 мультигормональная, G2.
2. Опухоли двенадцатиперстной кишки
На операции в стенке ДПК выявлены две опухоли в капсуле, мягко-эластической консистенции, серовато-желтого цвета, размерами 2 × 2,5 и 1,0 × 1,5 см, обе преимущественно трабекулярного строения с преобладанием гиалинизированной стромы, в их клетках выявляется интенсивная продукция гастрина, а также виллина и CDX-2 (которые подтверждают их кишечное происхождение); индекс Ki67 = 2 и 2,2%.
Заключение: множественные (2) гастриномы ДПК, G1.
3. Околощитовидные железы
На операции удален узел в капсуле, 2 см, серо-желтый. Аденома ОЩЖ солидного строения из мелких клеток, в которых при ИГХ исследовании выявлена продукция паратгормона, индекс Ki67 = 2%. На аутопсии дополнительно выявлены два узла (в переднем и заднем средостении), 0,6–1,5 см, серо-коричневой окраски, мягкой консистенции, в капсуле, аденома и гиперплазия ОЩЖ из мелких клеток, которые экспрессируют паратгормон, индекс Ki67 менее 1%.
Заключение: аденомы и гиперплазия нижних ОЩЖ.
4. Щитовидная железа
На операции выявлен узел ЩЖ размером 6 × 4 мм, окруженный жировой тканью. Узел представлен двумя разными компонентами: 1) компонент солидно-трабекулярного строения с наличием розеткоподобных структур, по строению мало отличимый от описанных выше опухолей ДПК; его клетки интенсивно экспрессируют гастрин, CDX-2 и не экспрессируют паратгормон, кальцитонин, тиреоглобулин; индекс Ki67 = 2,5%; 2) компонент солидного строения из мелких клеток с амилоидозом стромы, его клетки экспрессируют кальцитонин и TTF-1 (положителен в ткани ЩЖ и легкого) и не экспрессируют паратгормон и тиреоглобулин. На аутопсии в ЩЖ дополнительно выявлен плотноватый, белесо-коричневый, коллоидный узел в капсуле размером 0,6 см, по иммунофенотипу аналогичный второму компоненту.
Заключение: медуллярный рак ЩЖ и метастаз гастриномы ДПК в ЩЖ. Аутоиммунный тиреоидит.
5. Опухоль легкого
На аутопсии в средней доле правого легкого выявлен серо-розовый узел размером 2 см, хорошо отграниченный от окружающей ткани; опухоль трабекулярного строения, ее клетки экспрессируют нейроэндокринные маркеры (Син и ХрА) и TTF-1, единичные клетки экспрессируют кальцитонин, индекс Ki67 = 1,5%.
Заключение: типичный карциноид легкого низкой степени злокачественности.
6. Желудок
На аутопсии: слизистая оболочка серая, рельефная, “мозговидная”, толщина стенки до 1,5 см. Гипертрофический гастрит.
7. Надпочечники
Обнаружена диффузно-узелковая гиперплазия коры. В гипофизе патологии не выявлено.
Клинико-морфологический диагноз. Атипичный синдром МЭН-1 с множественными НЭО: аденомы и гиперплазия нижних левой и правой ОЩЖ, первичный субклинический гиперпаратиреоз; 4 нефункционирующие НЭО ПЖ, G1 и G2 (3 и 1) (Гл- и ПП-продуцирующие), гиперинсулинизм; 2 гастриномы ДПК, G1; медуллярный рак правой доли ЩЖ, аутоиммунный тиреоидит, метастаз гастриномы ДПК в ЩЖ; типичный карциноид правого легкого; диффузно-узелковая гиперплазия коры надпочечников; множественные нейрофибромы кожи, гипертрофический гастрит.
ЗАКЛЮЧЕНИЕ
Приведено уникальное наблюдение нетипичной МЭН-1, смешанного варианта, обусловленной наличием у пациента 9 опухолей и/или гиперплазий в 8 различных органах (ОЩЖ, ПЖ, ДПК, ЩЖ, легкое, желудок, кожа, надпочечники). Особенностью данного случая является медуллярная карцинома ЩЖ, которая обычно ассоциируется с синдромом МЭН-2. У пациента имела место характерная клиника гипогликемического синдрома, однако среди исследованных НЭО инсулином не оказалось, что не исключает наличия невыявленной (возможно, очень маленькой) опухоли ПЖ. В то же время известно, что клинические проявления МЭН-1 могут быть обусловлены суммарным эффектом всего комплекса гормонов, которые одновременно продуцируют множественные опухоли.
Множественная эндокринная неоплазия 1 типа. Клиническое наблюдение Текст научной статьи по специальности «Клиническая медицина»
Аннотация научной статьи по клинической медицине, автор научной работы — Фархутдинова Л. М., Гусева П. С., Калимуллин Н. Н., Шарипов Т. Н.
Похожие темы научных работ по клинической медицине , автор научной работы — Фархутдинова Л. М., Гусева П. С., Калимуллин Н. Н., Шарипов Т. Н.
PLURAL ENDOCRINE NEOPLASIA OF THE FIRST TYPE. CLINICAL OBSERVATION
Plural endocrine neoplasia is the combination of tumours or hyperplasias of two and few endocrine glands. Plural endocrine neoplasia of the first type, or Vermers syndrome , is the most frequent form of this pathology. The diagnosis of this syndrome is complicated enough because of atypical symptomatics, gradual involving of endocrine organs. The presented information reproduces the stages and complications of diagnosis using the own clinical observation.
Текст научной работы на тему «Множественная эндокринная неоплазия 1 типа. Клиническое наблюдение»
лентности выше в сравнении со здоровым ребенком.
Полученные данные могут быть использованы при разработке методических материалов по оказанию консультативной помощи матерям, имеющим ребенка с нарушениями здоровья и планированию психотерапевтических мероприятий с целью гармонизации материнско - детских взаимоотноше-
ний, оказывающих существенную роль как в развитии индивидуальности ребенка, так и в оптимизации соматического и психологического состояний его матери.
Перспектива исследования видится нам в проведении сравнительного анализа и определении специфики представлений матери о ребенке в связи с различными его заболеваниями, а также различий в представлениях матери и других членов семьи.
1. Бовина И.Б. Социальные представления молодежи о больных СПИДом // Вопросы психологии. - 2008. - №5. - С. 96-104.
2. Журавлев А.Л. Психологические факторы физического и психического здоровья человека // Психологический журнал. - 2004. - №3. - С.107 - 117.
3. Захаров А.И. Происхождение детских неврозов и психотерапия. - СПб., 2000. - С. 238, 253296.
4. Коротовских Л.А. Представления матери о ребенке как форма познания его индивидуальности: материалы международной научно-практической конференции «Психологическое познание: актуальные проблемы» (27 ноября, 2008). - Пермь, 2008. - С. 123 - 128.
5. Психология здоровья /под ред. Г.С. Никифорова. - СПб., 2003. - С. 65.
6. Ромицына Е.Е. Здоровье глазами детей: опыт психологического анализа детских рисунков //Вопросы психологии. - 2006. - № 1. - С.39 - 47.
7. Собкин В.С., Марич Е.М. Опыт структурного анализа ценностных ориентаций родителей дошкольников //Вопросы психологии. - 2003. - № 1. - С.3-12.
© Л.М. Фархутдинова, П.С. Гусева, Н.Н. Калимуллин, Т.Н. Шарипов, 2009
Л.М. Фархутдинова, П.С. Гусева, Н.Н. Калимуллин, Т.Н. Шарипов МНОЖЕСТВЕННАЯ ЭНДОКРИННАЯ НЕОПЛАЗИЯ 1 ТИПА.
ГОУ ВПО «Башкирский государственный медицинский университет Росздрава», г. Уфа Республиканская клиническая больница им. Г.Г. Куватова МЗ РБ
Ключевые слова: гиперпаратиреоз, множественная эндокринная неоплазия, синдром Вермера.
L.M. Farkhutdinova, P.S. Guseva, N.N. Kalimullin, T.N. Sharipov PLURAL ENDOCRINE NEOPLASIA OF THE FIRST TYPE.
Plural endocrine neoplasia is the combination of tumours or hyperplasias of two and few endocrine glands. Plural endocrine neoplasia of the first type, or Vermers syndrome, is the most frequent form of this pathology. The diagnosis of this syndrome is complicated enough because of atypical symptomatics, gradual involving of endocrine organs. The presented information reproduces the stages and complications of diagnosis using the own clinical observation.
Key words: hyperparathyroidism, plural endocrine neoplasia, Vermers syndrome.
Множественная эндокринная неоплазия (МЭН) представляет собой сочетание опухолей и (или) гиперплазий двух и более эндокринных желез. Наиболее частая форма МЭН -
множественная эндокринная неоплазия 1 типа (МЭН 1), или синдром Вермера, названный в честь автора, впервые описавшего заболевание в 1954 г. Распространенность синдрома
колеблется от 1 до 10 на 100 000 населения. Это генетическая патология, обусловленная мутацией гена супрессии опухолей, с чем связывают дебют заболевания в раннем возрасте, до 20 лет, и полиорганный характер поражения. Частота наследственной передачи 50 %.
Клиническая картина синдрома разворачивается в течение ряда лет, а ранняя диагностика позволяет оптимизировать прогноз. Вместе с тем своевременная диагностика синдрома МЭН 1 затруднена в связи с постепенным вовлечением в процесс эндокринных органов и стертостью, атипичностью симптомов за счет взаимного влияния основных компонентов синдрома.
Наиболее частая локализация аденома-тоза при МЭН 1 - паращитовидные железы (90-100 %), поджелудочная железа (80 %) и гипофиз (65 %). Гиперпаратиреоз в рамках МЭН, как правило, обусловливает его первые клинические проявления и составляет до 4 % среди пациентов с первичным гиперпаратире-озом. Гораздо реже, до 10 % случаев, заболевание манифестирует с аденомы гипофиза, чаще всего пролактинсекретирующей. Возможна сочетанная гиперпродукция пролакти-на и соматотропного гормона. Крайне редкий вариант - кортикотропинома.
В 20-40 % случаев может наблюдаться аденома или гиперплазия надпочечников, чаще двусторонняя, гормонально неактивная, в 20 % - поражение щитовидной железы. К редким нарушениям относятся карциноид ви-лочковой железы, злокачественные опухоли в легком, желудке и 12-перстной кишке 3.
Приводим собственное наблюдение.
Больной 27 лет впервые в 1997г обратился за медицинской помощью в связи с головными болями, когда на компьютерной томографии (КТ) головного мозга была обнаружена макроаденома гипофиза, не подлежащая хирургическому удалению, поскольку в толще образования визуализировалась правая внутренняя сонная артерия. Лечение бром-криптином по 2 таблетки 2 раза в сутки дало положительный эффект в виде купирования цефалгии. С 2002г. больной стал отмечать избыточный вес, появление стрий, повышение артериального давления до 140 и 100 мм рт. ст. и сахара в крови до 8,0 ммоль/л. Диагностирован сахарный диабет, рекомендован прием диабетона МВ, а с гипотензивной целью - ингибитор АПФ.
рапию. Кроме того, у него обнаружены ги-перпролактинемия 4500 мМЕ/л (норма 105540), снижение тестостерона до 7,5 нмоль/л (12,1-38,3), несколько повышенный уровень кортизола крови - 960 нмоль/л (150-860). На КТ выявлены узелковая гиперплазия надпочечников, больше слева, и макроаденома гипофиза по результатам магнитно-резонансной томографии (МРТ) головного мозга. Установлен диагноз: аденома гипофиза, гиперпролак-тинемия, гиперплазия надпочечников, синдром Кушинга (?), ожирение, сахарный диабет. Рекомендовано продолжить прием агонистов дофамина, инсулинотерапию и повторить КТ надпочечников в динамике.
На фоне рекомендованной терапии отмечалось улучшение самочувствия - нормализовались гликемия и и артериальное давление, что позволило обследуемому вести достаточно активный образ жизни: работать, не ограничивать физические нагрузки. Однако в 2007г. состояние значительно ухудшилось: появились интенсивные боли в правом коленном и правом голеностопном суставах, не купируемые нестероидными противовоспалительными средствами и вынудившие пользоваться костылями. Кроме того, появились периодические боли в гастродуоденальной области преимущественно натощак и чувство тяжести после приема пищи. Лечение по месту жительства не дало эффекта, и больной был направлен в эндокринологическое отделение Республиканской клинической больницы им. Г. Г. Куватова для уточнения диагноза и коррекции лечения.
Из анамнеза жизни: дед оперирован по поводу мочекаменной болезни, опухоли поджелудочной железы, умер в 70-летнем возрасте в результате язвенного кровотечения. При осмотре больного обращала на себя внимание избыточная масса тела (110 кг) при росте 165 см (индекс массы тела - 40,7 кг/м2), матео-ризм, гиперемия лица, бледно-розовые стрии в области живота. Артериальное давление -130 и 80 мм рт. ст. Определялась умеренная болезненность при пальпации в эпигастральной области, а также выраженные боли при активных и пассивных движениях в правом коленном и правом голеностопном суставах.
В процессе обследования выявлено увеличение суточного диуреза до 5 л., удельный вес мочи в разовой порции оказался на нижней границе нормы - 1010. Обнаруженная глюкозурия 2,96 %, следы ацетона наряду с гипергликемией 17,9-15,5-12,2 ммоль/л и высоким уровнем гликированного гемоглобина 10,4 % свидетельствовали о декомпенсации
сахарного диабета, при этом уровень антител к декарбоксилазе глутаминовой кислоты не превышал диапазона нормы 0,56 МЕ/мл (при норме до 1,0).
Биохимическое исследование крови определило гиперхолестеринемию 6,5 ммоль/л (норма 3,6-5,2), гипертриглицеридемию 3,1 ммоль/л (0,15-1,71) и повышенный уровень общего кальция 2,7-3,0 ммоль/л (2,15-2,56), вместе с тем содержание фосфора было в пределах референтных значений и составило 1,24 ммоль/л (0,87-1,45). Гормональные исследования выявили повышение базальной корти-золемии при сохранном ритме секреции, высокий уровень пролактина и С-пептида, а также низкий - тестостерона (см. таблицу).
Показатели гормонального статуса обследуемого
Дата Тест Пока- затель Единицы измерения Норма
19.01.08 Кортизол (8.00 утра) 1171,0 150-860
Кортизол (20.00 вечера) 475,4 84-660
17.01.08 АКТГ 28,0 пг/мл 9,0-46,0
ВМК 1,0 мкг/мл 0,6-3,0
- Пролактин 4500,0 мМЕ/л 105-540
- Тестостерон 7,6 нмоль/л 12,1-38,3
- ФСГ 4,2 мМЕ/мл 1,0-11,8
- сТ4 17,7 пмоль/л 10,0-23,2
- ТТГ 3,0 мкМЕ/мл 0,23-3,4
С-пептид 1401,0 пмоль/л 216,0-1025,0
Примечание. ВМК - ванилилминдальная кислота, ЛГ - лютеи-низирующий гормон, ФСГ - фолликулостимулирующий гормон, сТ4 - свободный тироксин, ТТГ - тиреотропный гормон.
Рентгенография черепа в боковой проекции обнаружила увеличение турецкого седла до 35*25 мм, размытость задней стенки. По данным МРТ головного мозга размеры макроаденомы составили: вертикальный - 15 мм, поперечный - 32 мм и переднезадний - 24. Отмечались эндо-, пара- и супраселлярный рост, смещение ножки гипофиза влево. При сравнении с результатом МРТ от 2006 г. наблюдалось уменьшение размеров аденомы. Учитывая положительную динамику нейрохирургом было рекомендовано продолжить лечение агонистами дофамина под наблюдением эндокринолога.
По данным фиброгастродуоденоскопии выявлена хроническая язва луковицы 12-перстной кишки 5*6 мм, хронический эрозивный гастрит, рубцово-язвенная деформация луковицы, в связи с чем агонисты дофамина были временно отменены, а лечение дополнено эрадикационной и антисекреторной терапией, однако болевой синдром сохранялся.
УЗИ органов брюшной полости обнаружило в области хвоста поджелудочной железы объемное гипоэхогенное образование с
неровным четким контуром до 30 мм в диаметре с единичными сосудами. В почках выявлены щелевидное расширение чашечнолоханочной системы слева и участки кальцинации.
КТ брюшной полости подтвердило наличие патологического образования в поджелудочной железе 45*27 мм неоднородной структуры, с кальцинатами, а также выявило множественные изоденсивные округлые образования в надпочечниках до 20 мм в диаметре.
На рентгенограмме кистей рук определялся остеопороз с ячеистой перестройкой, стоп и нижней трети берцовых костей - множественные очаги просветления, местами сливающиеся, грудного и поясничного отделов позвоночника - признаки остеопороза и остеохондроза. Тяжесть поражения костной системы с выраженной деминерализацией, деструкцией и интенсивным болевым синдромом, требовавшим применения трамала, диктовала необходимость диагностического поиска. Учитывая многоочаговость поражения эндокринной системы (аденома гипофиза, гиперплазия надпочечников, вновь выявленное объемное образование в поджелудочной железе), было высказано предположение о множественной эндокринной неоплазии, в связи с чем проведено ультразвуковое исследование паращитовидных желез для исключения аденомы.
Действительно, по данным УЗИ щитовидной железы у нижнего полюса правой доли были обнаружены образования пониженной эхогенности 17*14*7 мм и 5 мм диаметром с повышенной васкуляризацией. Исследование паратгормона подтвердило гиперфункцию паращитовидной железы: уровень паратиреоидного гормона составил 168,1 пг/мл (норма 16-62). Проведение КТ органов грудной клетки и средостения для исключения опухоли тимуса выявило наличие диффузного остеопороза, сросшихся переломов 2,
3, 4 и 6 ребер справа.
По результатам обследования установлен диагноз: синдром множественной эндокринной неоплазии 1 типа: аденома паращито-видных желез, диффузный остеопороз, язва луковицы 12-перстной кишки; макроаденома гипофиза, пролактинсекретирующая, вторичный гипогонадизм, ожирение III степени; опухоль хвоста поджелудочной железы; гиперплазия надпочечников. Сопутствующий диагноз: сахарный диабет 2 типа, вторично инсулинопотребный, средней тяжести, осложненный ретинопатией I степени, дисталь-
ной полинейропатией II степени, гепатозом.
Хронический пиелонефрит, латентное течение, без нарушения азотвыделительной функции.
Больной был переведен в отделение сосудистой хирургии, где произведена парати-реоидэктомия пораженных желез. Гистологическое исследование определило смешанноклеточную аденому паращитовидной железы с железистыми, трабекулярными и солидными структурами.
1. Валдина Е.А. Заболевания щитовидной железы: Руководство. - СПб.: Питер, 2006. - 368 с.
2. Гуревич Л.Е. Диагностика нейроэндокринных опухолей желудочно-кишечного тракта // Практическая онкология. - 2005. - №4. С. 193-201.
3. Дедов И.И., Мельниченко Г.А., Фадеев В.В. Эндокринология.- М.: ГЭОТАР-Медиа, 2007. -432 с.
4. Дядя Г.И., Лазарева Г.Ю., Краснова М.А. и др. Полный справочник эндокринолога. - М.: Изд-во Эксмо, 2005. - С 412-431.
5. Иловайская И. А. Синдром множественных эндокринных неоплазий 1-го типа // Эндокринология: национальное руководство / под. ред. И.И. Дедова, Г.А. Мельниченко. - М.: ГЭОТАР-Медиа, 2008. - С 944-949.
6. Кеттайл В.М., Арки Р.А. Патофизиология эндокринной системы: пер. с англ. - М.: Издательство БИНОМ, 2007. - 336 с.
7. Котова И.В., Калинин А.П. Первичный гиперпаратиреоз и синдром множественных эндокринных неоплазий: лекция // Проблемы эндокринологии. - 2003. - № 3. - С. 37-39.
8. Латкина Н.В., Лысенко М.А., Иловайская И.А., Егорова О.Ю., Кузнецов Н.С. Синдром множественный эндокринных неоплазий 1 типа // Эндокринная хирургия. - 2007. - № 1. - С. 4346.
9. Сайзмо Г.В. Множественная эндокринная неоплазия // Доказательная эндокринология: пер. с англ. - 2-е изд. - М.: ГЭОТАР-Медиа, 2008. - С. 525-559.
Диагностика множественной эндокринной неоплазии - достаточно сложная задача и для ее осуществления в ранние сроки необходима настороженность при выявлении у лиц молодого возраста гиперкальциемии как возможного гиперпаратиреоидного компонента множественной эндокринной неоплазии 1 типа.
Читайте также: