Морфология и наследственность синдрома Альстрема
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Диабети́ческая нефропати́я (от др.-греч. διαβαίνω , — «перехожу, пересекаю», др.-греч. νεφρός — «почка» и др.-греч. πάθος — «страдание, болезнь») (нефропатия при сахарном диабете, синдром Киммельстил-Уилсона, диабетический гломерулосклероз) — термин, объединяющий весь комплекс поражений артерий, артериол, клубочков и канальцев почек, возникающих в результате нарушения метаболизма углеводов и липидов в тканях почки. Распространённость диабетической нефропатии достигает 75% лиц с сахарным диабетом, чаще других наблюдается следующая патология почек [1] :
- Артериосклероз почечной артерии и её ветвей.
- Артериолосклероз.
- Диабетический «гломерулосклероз»:
- а) узелковый (синдром Киммельстил-Уилсона);
- б) диффузный;
- в) экссудативный.
Содержание
Основой диабетической нефропатии является нефроангиосклероз почечных клубочков, чаще диффузный, реже узелковый (хотя именно узелковый гломерулосклероз был впервые описан Киммельстил и Уилсоном в 1936 году как специфичное проявление диабетической нефропатии). Патогенез диабетической нефропатии сложен, предложено несколько теорий её развития, наиболее изучены три из них:
- метаболическая,
- гемодинамическая,
- генетическая.
Метаболическая и гемодинамическая теории роль пускового механизма отводят гипергликемии, а генетическая — наличию генетической предрасположенности [2] .
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом. Другой почечный клубочек.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом. Другой почечный клубочек.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом.
Гистопатологическая картина диабетического гломерулосклероза с нефротическим синдромом.
Согласно замыслу одного из участников Википедии, на этом месте должен располагаться специальный раздел.
Вы можете помочь проекту, написав этот раздел.Бессимптомное течение заболевания на ранних стадиях приводит к запоздалой диагностике диабетической нефропатии на поздних стадиях. Поэтому у всех пациентов с сахарным диабетом рекомендуется проведение ежегодного скрининга для раннего выявления диабетической нефропатии [3] (анализ крови на креатинин с расчетом скорости клубочковой фильтрации и анализ мочи). В значительном числе случаев при сахарном диабете 2 типа больные изначально обращают внимание на симптомы сахарного диабета (полиурию, кожный зуд, жажду и другие), что вынуждает их обращаться к эндокринологам, а не нефрологам. Выраженность клинических симптомов при диабетической нефропатии (отёки, артериальная гипертензия), ответ на лечение и скорость прогрессирования почечной дисфункции во многом зависит от уровня альбумина в моче и скорости клубочковой фильтрации. При развитии хронической почечной недостаточности и выраженном снижении скорости клубочковой фильтрации замедляется выведение ряда сахароснижающих препаратов и скорость катаболизма инсулина в почках, поэтому следует уделять особое внимание подбору адекватного режима дозирования инсулина и сахароснижающих препаратов [3] , чтобы избежать развития гипогликемических состояний.
Синдром Киммельстил-Уилсона (диабетический гломерулосклероз) — узелковая форма гломерулосклероза, специфическая для сахарного диабета, получила название в честь патологоанатомов Киммельстил и Уилсона, обнаруживших и описавших в 1936 году своеобразное узелковое склеротическое поражение клубочков почек у лиц с сахарным диабетом. При диабете наблюдаются диффузный и узелковый варианты поражения почечных клубочков. Узелковая форма, описанная Киммельстил и Уилсоном, чаще встречается при сахарном диабете 1-го типа уже вскоре после манифестации заболевания, постепенно прогрессирует, приводя в итоге к диабетическому гломерулосклерозу и развитию ХПН. В клинической практике термином «синдром Киммелльстил-Уилсона» характеризуют нефросклеротическую, азотемическую стадию поражения почек при сахарном диабете. Последнее время этим термином пользуются реже, пользуясь диагнозом «Диабетическая нефропатия, хроническая болезнь почек 5 стадии»Ефимов А.С. и соавт.) [2] .
В диагностике диабетической нефропатии и определении прогноза лечения необходимо ориентироваться на два показателя: альбуминурию и скорость клубочковой фильтрации.
Ранним признаком диабетической нефропатии является выявление повышенной экскреции альбумина с мочой (альбуминурии), превышающей нормальные значения (до 30 мг/сутки или менее 20 мкг/минуту в разовой порции мочи). В настоящее время не рекомендуется использовать термины микроальбуминурия (соответствующая экскреции альбумина от 30 до 300 мг/сутки) и макроальбуминурия (экскреция более 300 мг/сутки), а вместо этого использовать градации А2 и А3 современной классификации KDIGO [4] , которые в числовом выражении соответствуют терминам микро- и макроальбуминурия. Альтернативной суточному определению альбуминурии может быть экспресс-диагностика с помощью тест-полосок (которая может давать ложно-положительные или ложно-отрицательные результаты), либо более точное определение соотношения альбумин/креатинин в утренней порции мочи (показатель менее 3 мг/ммоль считается нормальным, значения от 3 до 30 соответствуют градации альбуминурии А2, значения выше 30 мг/ммоль соответствуют градации А3) [4] .
На ранних стадиях диабетической нефропатии возможно выявление повышенной скорости клубочковой фильтрации (140 мл/минуту/1,73м 2 и более), которая при прогрессировании почечной дисфункции снижается. В настоящее время [4] для расчета скорости клубочковой фильтрации рекомендуется использовать специальные формулы (у взрослых: CKD-EPI, MDRD, менее предпочтительна формула Кокрофта-Голта, у детей: формула Шварца) с учетом концентрации креатинина крови. В ряде случаев предпочтительно определение скорости фильтрации на основании клиренса экзогенных препаратов, тогда как использование пробы Реберга-Тареева с определением клиренса эндогенного креатинина за сутки практически не используется.
Главным условием является лечение диабета. Существенную роль играет нормализация артериального давления и липидного метаболизма, одним из основных лечебных мероприятий является диета [4] [5] [6] . При развитии диабетической нефропатии показано назначение нефропротективной терапии, аналогично почечной патологии другой этиологии.
Диета должна ограничивать простые углеводы, и содержать достаточное количество белка (0,8 г/кг массы тела/сут) [5] . Жидкость нельзя ограничивать, но обязательно следует добавлять несладкие соки, содержащие калий. При выраженном снижении скорости клубочковой фильтрации следует рассмотреть вопрос перехода на низкобелковую диету (но не менее 30—40 грамм в сутки) под обязательным регулярным контролем врача и обеспечении достаточной калорийности пищи (последнее необходимо чтобы не допустить развития белково-энергетической недостаточности). При сочетании диабетической нефропатии и артериальной гипертензии важно придерживаться малосолевой диеты (менее 5 г/сутки поваренной соли, что соответствует менее 2 г/сутки элементарного натрия) [4] .
Единственным надёжным фактором профилактики диабетической нефропатии и основой лечения всех её стадий является оптимальная компенсация сахарного диабета (уровень гликозилированного гемоглобина HbA1C < 7,0%) [2] .
Профилактика микроальбуминурии. К факторам риска развития микроальбуминурии относят [2] :
- уровень гликозилированного гемоглобина HbA1C > 7,0%,
- мужской пол,
- длительность течения сахарного диабета 1-го типа более 5 лет (при СД-2 типа появление микроальбуминурии связано с первичным поражением эндотелия клубочков и может появляться задолго до манифестации диабета),
- манифестация сахарного диабета 1-го типа в возрасте до 20 лет (при СД-2 типа возраст значения не имеет),
- отягощённую наследственность по гипертонической болезни,
- полиморфизм гена АПФ (ангиотензин превращающего фермента), кодирующего киназу II типа),
- высокий Na/Li противотранспорт в эритроцитах (более 400 мкмоль/Li/л клеток/ч),
- скорость клубочковой фильтрации более 140 мл/мин для СД-1 типа (для СД-2 типа не установлено),
- отсутствие функционального почечного резерва (СД-1 типа),
- гиперлипидемию,
- наличие ретинопатии,
- курение.
Замедление прогрессирования нефропатии у пациентов с уже повышенным уровнем альбумина в моче и/или сниженной скоростью клубочковой фильтрации. Включает [4] [5] [6] :
- контроль гликемии,
- контроль уровня артериального давления,
- нормализацию липидного профиля крови,
- нормализацию внутрипочечной гемодинамики с помощью блокаторов ренин-ангиотензин-альдостероновой системы (в настоящее время убедительные доказательства эффективности имеются для блокаторов ангиотензин-превращающего фермента и блокаторов рецепторов ангиотензина II,
- низкобелковую диету (требует обязательного врачебного контроля).
Лечение диабетической нефропатии на стадии протеинурии. На этой стадии необходимо минимизировать риск быстрого развития ХПН, к факторам которого относятся [2] :
6.2. Наследственные болезни эндокринной системы
Наследственная патология, в основе которой лежат повреждения отдельных звеньев эндокринной системы, встречается довольно редко. Это объясняется, во-первых, жизненно важной регулирующей ролью данных органов, что при их поражении в пренатальном периоде заканчивается гибелью плода; во-вторых, из-за тесной взаимосвязи между гормонами угнетение синтеза одного из них часто сопровождается гипогонадизмом, отсюда передача болезни по вертикали невозможна.
По клинической картине можно выделить недуги, спровоцировать которые могут:
избыточный эффект гормонов;
резистентность рецепторов к гормону;
опухоли без нарушения его выработки.
При этом генетические повреждения делят на активирующие, наследующиеся по доминантному типу, и подавляющие (инактивирующие) мутации, обычно имеющие рецессивный тип наследования.
Наиболее часто среди клинических признаков, развивающихся вследствие нарушения эндокринного баланса, следует назвать ожирение. Оно может быть вызвано генетическими дефектами в клетках гипоталамуса: синдром Прадэра–Вили (syndrome Prader-Willi) вследствие хромосомных аномалий; синдром Лоуренса-Муна–Барде–Бидла (syndrome Laurence-Moon-Bardet-Biedle) (наследуется по аутосомно-рецессивному типу); синдром Альстрема (syndrome Ahlström), имеющий подобный тип наследования, сопровождающийся также симптомами сахарного диабета, нефропатии; синдром Бабинского-Фрелиха (syndrome Babinski-Fröhlich) из-за наличествующего при этом гипогонадизма иногда называющийся адипозо-генитальной дистрофией. Кроме того, у таких пациентов могут возникнуть симптомы несахарного диабета, низкий рост, инфантилизм; вероятной причиной считают мутации в генах, ответственных за синтез прогормонов аденогипофиза.
К симптомам ожирения обычно присоединяются и другие. Синдром Лоуренса-Муне-Барде-Бидла (syndrome Laurence-Moon-Bardet-Biedle) проявляется гипогонадизмом, полидактилией, пигментной дегенерацией сетчатки, вплоть до потери зрения к 20 годам, сниженным интеллектом. Ожирение с возрастом прогрессирует. Аутосомно-рецессивный тип наследования. Причина, вероятно, генетический дефект образования кортикостероидов. Лечение не разработано.
Для многих наследственных болезней эндокринной системы характерен полиморфизм; причём обязательно регистрируется увеличение размеров различных желёз. Аденоматоз полиэндокринный наследуется по аутосомно-доминантному типу, разделяют на два варианта: 1) синдром Вернера (syndrome Werner), при котором развиваются инсулинома, аденома гипофиза, гиперпаратироз; 2) синдром Сипла (syndrome Sipple), морфология и клиника которого включают рак щитовидной железы, феохромоцитому, гиперплазию паращитовидных желёз.
Логично, что лечение подобной патологии будет включать субтотальные тироидэктомию, паратироидэктомию или гипофизэктомию.
Как для вышеописанных заболеваний, так и для многих других до настоящего времени не описаны локализации генных дефектов, что затрудняет и диагностику, и их терапию. Обычно верификация производится по клиническим признакам.
Но в настоящее время большое внимание привлекают те случаи первичных гипер- и гипотирозов, в основе которых обнаружены мутации гена рецептора ТТГ, расположенного в базальной мембране фолликулярных клеток щитовидной железы, а также генетические дефекты G-белков, сопрягающих рецептор с аденилатциклазой и другими путями внутриклеточной трансдукции гормонального сигнала.
В рецепторах этого семейства выявлены не только активирующие мутации, приводящие к усилению роста и функции железы, наследуемые доминантным способом, но и инактивирующие повреждения, в первую очередь, ферментов, непосредственно участвующих в процессе синтеза тироидных гормонов.
Недавно был клонирован и транскриптон так называемого синдрома Пендрода (syndrome Pendrod). Этот ген, расположенный в длинном плече хромосомы 7, кодирует протеин – пендрин, контролирующий генез тироидной пероксидазы. Поэтому клиника данного недуга представляет из себя сочетание зоба с глухотой; первые симптомы диффузного увеличения щитовидной железы появляются в 5-8 лет, в этом же возрасте может снижаться интеллект. Наследование по аутосомно-рецессивному типу.
Такой же тип наследования имеет синдром Ричардса-Рандля (syndrome Richards-Randle), основными клиническими признаками которого служат гипогонадизм, задержка моторного развития, атаксия, кифосколиоз, глухота, низкий IQ.
Совсем иной картиной отличается адреногенитальный синдром, относящийся к группе наследственных нарушений биосинтеза стероидных гормонов. Так как процесс их образования многоступенчатый, то известно по меньшей мере пять разновидностей генетических дефектов ферментов (21-гидроксилазы, холестерол-десмолазы, 3-β-гидроксистероиддегидрогеназы, 11-β-гидроксилазы, 17-α-гидроксилазы). Дисфункция коры надпочечников сопровождается усиленной секрецией андрогенов, а по принципу «+» - «-» взаимодействия – и АКТГ, что провоцирует опухоль коры надпочечников. Указанная патология наследуется по аутосомно-рецессивному типу. Общая частота 1:5000 – 12 000 новорождённых. Наиболее распространена форма, обусловленная дефицитом 21-гидроксилазы, катализирующей превращение прогестерона в дезоксикортикостерон, а также 17-гидроксипрогестерона в 11-дезоксикортизол. Известны два варианта этого заболевания: соль-теряющая (syndrome Debre-Fibiger) и простая вирильная формы. Первая, [более часто встречающаяся (в 75% случаев)] из-за дефицита минералокортикостероидов проявляется в нарушении минерального обмена с первых дней жизни. У новорождённых отмечаются сонливость, потеря массы тела, полиурия, срыгивание, рвота. Биохимически – ацидоз, гипонатриемия, гиперкалиемия. Без лечения быстрый летальный исход.
Простая вирильная форма характеризуется прогрессирующей вирилизацией, повышенной секрецией тех гормонов коры, синтез которых не нарушен (из-за недостатка кортизола по принципу обратной связи стимулируется выделение АКТГ, что провоцирует гиперплазию коры надпочечников и избыточное образование тех стероидов, активность энзимов генеза которых не изменена). У больных ускорено соматическое развитие. У девочек может быть умеренно гипертрофирован клитор или же полностью сформированы пенис и мошонка. У мальчиков на 5-7-ом годах жизни - преждевременное половое созревание.
Диагноз основывается на увеличенном содержании в крови андрогенов – предшественников кортизола, а в моче – их метаболитов.
Лечение включает введение глюкокортикостероидов.
Ещё одно генетическое поражение надпочечников, также наследующееся по аутосомно-рецессивному типу, в основном, у мальчиков - адренолейкодистрофия; проявляется, в первую очередь, неврологическими и психическими расстройствами (атаксией, судорогами, нарушением поведения и памяти). Прогноз неблагоприятен, смерть в 1-3 года.
Клинический полиморфизм характерен и для наследственной остеодистрофии (псевдогипопаратироза, morbus Martin-Albright), описанной в 1942 году F.Albright и носящей его имя. Наследуется по аутосомно-доминантному типу, характерна генетическая гетерогенность, отсюда выделяют четыре формы.
Тип 1А – в основе - дефект гуаниннуклеотидсвязывающего белка, особенно это сказывается на количестве рецепторов к паратгормону. К 5-10 годам отмечается низкий рост, короткая шея, круглое лицо, укорочение некоторых пальцев (II, IV). Мягкие ткани кальцифицируются, регистрируются подкожные кальцификаты. Кроме паращитовидных желёз, страдают щитовидная, поджелудочная (сахарный диабет), половые (гипогонадизм). Развивающаяся при этом гипокальцемия провоцирует повреждение эмали зубов, остеопороз, нарушение функционирования ЦНС: гиперкинезы, тонические судороги, атаксию, дискоординацию движений. Могут возникать страхи, тревога, беспокойство, плохой сон, у отдельных больных – умственная отсталость. В крови - снижение величин общего и ионизированного кальция, рост концентраций фосфатов, паратгормона.
Тип 1В – сходен с 1А; также регистрируется дефицит рецепторов к паратгормону в клетках-мишенях. Течение более благоприятное, реже встречается остеодистрофия.
Тип 1С и тип 2 отличает поражение нервной системы: моторная неловкость, беспокойство, бессонница.
Терапия мало эффективна. Попытки введения мегадоз витамина Д, ионов кальция, паратгормона слабо эффективны – умственная отсталость, судорожный синдром сохраняются. Отсюда важное место должна занять профилактика: медико- генетическая консультация у будущих родителей из-за высокого риска повторения (50%).
В последние годы описаны случаи полного отсутствия паращитовидных желёз (апаратироз), характер наследования и частота не установлены. Основные симптомы: тетания, поражения кожи, развитие катаракты. Из биохимических изменений – гипокальциемия.
Диагностика и терапия не разработаны. Временное облегчение приносят препараты кальция.
Наследственная гипогликемия (синдром Мак-Куарри, syndrome McQuarrie) имеет в своей основе генетический дефект, обусловливающий нарушение в структуре рецепторов к инсулину, что снижает чувствительность клеток к нему. Наследуется по аутосомно-рецессивному типу. Первые признаки появляются на втором году жизни, особенно симптомы гипогликемии регистрируются по утрам натощак (мышечная слабость, потливость, раздражительность, головокружение, дрожание, нередки судороги). Содержание глюкозы в крови снижается до 0,6-1,1 ммоль/л. В тяжёлых случаях возможны атаксия, задержка умственного развития. Описаны случаи спонтанного выздоровления. Положительный эффект даёт терапия АКТГ, ГКС.
Для отдельных видов тезаурисмозов также характерны признаки поражения эндокринной системы. Синдром Ханд-Шюллер-Крисчена (накопление холестерина) (гл. 4.2.3) сопровождается клиническими проявлениями несахарного диабета, ожирения.
Морфология и наследственность синдрома Альстрема
Морфология и наследственность синдрома Альстрема
При биопсии яичек были обнаружены маленькие гиализировапные канальцы с единичными клетками Лейдига и Сертоли, а также уплотнение lamina propria. Биопсия яичников у 16-летней девушки, умершей от неустановленной причины, патологии не выявила.
Гистологическое исследование почек показало хроническую нефропатию, проявляющуюся утолщением клубочковых и канальцевых мембран. Многие клубочки были гиалинизнрованы (Goldstein, Fialkow).
Наследственность. Несмотря на то что патогенез синдрома сейчас еще не понятен, имеются основания думать, что причиной сахарного диабета, несахарного почечного диабета и первичного гипогонадизма является резистентность к действию некоторых полипептидов — гормонов (инсулин, вазопрессин и гонадотропнны). Goldstein и Fialkow предположили, что генерализованное уплотнение мембран и гиалинизация соединительной ткани может являться важнейшим последствием основной патологии.
Во всех случаях родители больных детей были здоровы. Равная частота синдрома у сибсов обоих полов и увеличение уровня кровнородственных браков между родителями указывают на аутосомно-рецессивный тип наследования.
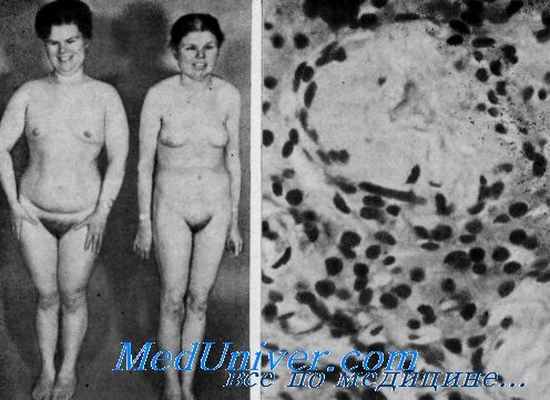
Диагноз. У больных с синдромом Лауренс — Муна отмечаются пигментный ретинит, умственная отсталость, гипогенитализм и спастическая параплегия. У детей с синдромом Барде — Бидля отмечают ожирение и пигментный ретинит, сочетающиеся с полидактилией, гипогонадизмом и умственной отсталостью. У больных с синдромом Альстрёма нет ни умственной отсталости, ни полидактилии. При синдромах Лауренс — Муна или Барде — Бидля глухота и сахарный диабет наблюдаются изредка (Burn, Garstecki et al.). Кроме того, тотальное облысение при синдроме Альстрёма наступает до второго десятилетия жизни, в противоположность выявлению тотального облысения в четвертом десятилетии жизни при синдромах Лауренс — Муна и Барде — Бидля. У сестер, о которых сообщил Weiss, отмечались ожирение, гипогонадизм, умственная отсталость и нейросенсорная глухота, но у них не было пигментного ретинита. До настоящего времени эти случаи не могут быть классифицированы.![синдром альстрема]()
Сестры 36 и 33 лет с синдромом Альстрёма. Слепота была выявлена в возрасте около 1 года, глухота — в шестилетием возрасте, частичное облысение началось в 20-летнем возрасте. У обеих сестер наблюдались нарушения менструального цикла. Левая больная носит парик (из J. L. Goldstein a. P. J. Fialkow, Medicine).
Микрофотография препарата яичка. Видны бледные гиалинизиропанные канальцы. Отмечаются также кристаллоиды Рейнке (из R. L. Weinstein et al., N. Engl. J. Med.).Первичный гипогонадизм, который встречается при синдроме Рейфенштейна, миотонической дистрофии и аплазии герментативных клеток, может быть исключен. Отграничение этих заболеваний от синдрома Альстрёма на основе клинических и микроскопических данных обсуждено Weinstein и соавт..
Мы не можем легко классифицировать описанных Codaccioni с сотр. двух братьев с юношеским диабетом, атрофией зрительных нервов, гопогонадизмом и нейросенсорной глухотой. Эти случаи могут представлять отдельный синдром, так как нет доказательств того, что у больных был пигментный ретинит.
Лечение. При прогрессирующей глухоте можно применять слуховые аппараты. Хотя большая доля дефицита зрения является следствием атипичного пигментного ретинита, путем удаления хрусталика можно добиться улучшения зрения за счет той доли потери зрения, которая была обусловлена полярной катарактой. Ожирение подлежит диетическому контролю, а при сахарном диабете необходимо назначать медикаментозную терапию.
Прогноз. Мало чем можно обнадежить этих больных. В то время как их зрение и слух прогрессивно ухудшаются, интеллект остается нормальным. Они могут погибнуть вследствие нарушения функции почек.
Выводы. Характеристика этого синдрома включает: 1) аутосомно-рецессивное наследование; 2) выявляющийся в младшем возрасте пигментный ретинит с потерей центрального зрения; 3) начинающийся в детстве сахарный диабет; 4) преходящее ожирение; 5) выявляющуюся во втором десятилетии заднюю кортикальную катаракту; 6) выявляющуюся в третьем десятилетии нефропатию; 7) черный акантоз; S) начинающуюся у детей старшего возраста прогрессирующую нейросенсорную глухоту.
Синдромы, включающие пигментный ретинит и неврологические заболевания
Пигментная дегенерация сетчатки, прогрессирующий тетрапарез, умственная отсталость и умеренно глубокая нсйросенсорная глухота у 2 мальчиков сибсов описаны Gordon, Capute и Konigsmark.
Клинические данные. Данные осмотра. Братья отличались низким ростом, атрофичной мускулатурой всех конечностей и тупым, невыразительным лицом. Окружность головы, длина тела и вес были ниже физиологических норм на 30%. Пальцы, особенно средние фаланги, были короткими. Отмечалась клинодактилия V пальцев. Наблюдались умеренные сгибательные контрактуры на ногах.
Орган зрения. Произведенное братьям в возрасте 3 и 4 лет офтальмологическое исследование обнаружило по всей сетчатке диффузное распространение крупных гранул пигмента, маленький и бледный диск зрительного нерва и суженный калибр сосудов (артериол) сетчатки. С возрастом все эти изменения становились более резкими.
Нервная система. Уровень IQ.s, установленный при психометрическом тестировании, был равен 34 и 44. Наиболее постоянным неврологическим симптомом болезни являлся прогрессирующий спастический паралич ног со сгибательными контрактурами бедер и коленей. Отмечались повышение сухожильных рефлексов и сгибательный подошвенный рефлекс. Несмотря на то что масса мышц была резко уменьшена, мышечная сила оставалась вполне хорошей. Больные ходили на широко расставленных ногах. Их походка была неустойчивой. Постепенно они ходили все с большим трудом, и в конце концов передвижение стало совсем невозможным.
Патологических движений и нарушения чувствительности не обнаружено. Выражение лица становилось все более тупым. Нарастали угнетение рвотного и глотательного рефлексов, а также нарушения смыкания век.
Орган слуха. Проведение аудиометрического исследования было затруднено из-за умственной отсталости мальчиков. Отологическое исследование патологии не выявило. ЭЭГ-аудиометрия показала умеренно глубокую пейросенсорную глухоту, охватывающую главным образом высокие частоты. Другие аудиометрические пробы не производились. Речь у обоих братьев не развивалась.
Вестибулярная система. Калорические вестибулярные пробы были нормальными.Пигментный ретинит, прогрессирующий тетрапарез, слабоумие и нейро-сенсорная глухота. Два брата маленького роста с невыразительным лицом и низко посаженными деформированными ушами
Лабораторные данные. Рентгенограммы. На краниограмме обнаружены малые размеры и асимметрия черепа. Отмечались укорочение средних фаланг и гипоплазия V пястной кости.
Другие данные. На ЭЭГ у одного из мальчиков была обнаружена чрезмерно медленная активность, в то время как у его брата ЭЭГ была нормальная. Электроретинографическое исследование зарегистрировало у старшего мальчика почти нормальную реакцию на свет.Рутинные анализы мочи и крови, проба на электролиты и спинномозговая жидкость были нормальными.
Патология. Результаты гистопатологического исследования не представлены.Наследственность. Родители больших детей состояли в отдаленном родстве. У тетки по линии матери был спастический церебральный паралич, но без ретинопатии. Тип наследования более всего похож на аутосомно-рецессивный, но нельзя исключить возможность Х-сцепленного наследования.
Диагноз. При синдроме Ушера моторика и психическая сфера ингактны. Другие заболевания, включающие пигментный ретинит и глухоту, такие как синдром Кокейна, синдром Рефсума и синдром Альстрёма, имеют другие сопутствующие аномалии, отличные от спастической параплегии и умственной отсталости, наблюдающихся при этом заболевании.
Существует несколько синдромов, включающих умственную отсталость и нарушения походки. Синдром Ричарде — Раидля (Richards — Rundle) характеризуется глухотой и медленно прогрессирующей атаксией, но мышечная сила остается вполне хорошей в течение многих лет. Синдром Тройера (Тгоуег) включает спастическую параплегию и дистальиые мышечные атрофии, которые начинаются в детстве и медленно прогрессируют, пока в третьем — четвертом десятилетии жизни ходьба становится совершенно невозможной. Хотя у некоторых больных и выявляют умственную отсталость, у них не доказаны пигментный ретинит или атрофия зрительных нервов (Cross, McKusick).
В одной семье была описана спастическая параплегия с дегенерацией сетчатки в сочетании с нейросеисорной глухотой, но интеллект у больных был нормальным (Louis-Bar, Pirot, Mahloudji, Chuke)Лечение. Так как умственная отсталость у больных выражена резко, применение слуховых аппаратов ограничено.
Прогноз. Заболевание очень медленно прогрессирует, в результате развивается полная идиотия.Выводы. Характеристика этого синдрома включает: 1) вероятное аутосомно-рецессивное наследование, 2) прогрессирующий пигментный ретинит, 3) прогрессирующий тетрапарез, 4) выраженное прогрессирующее слабоумие, 5) умеренную нейросенсорную глухоту.
- Читать далее "Синдром Кокейна: карлик со старческим видом, умственной отсталостью и тугоухостью"
Система восстановления зрения.
Система М. С. Норбекова.
Занятия проводит преподаватель Центра академика М.С. Норбекова Екатерина Зенина.
Бесплатное ознакомительное занятие состоится 17 марта 2014 г. в 18:00.«Пусть во всех медицинских книгах вы прочтете, что ваша болезнь неизлечима, если что-то стоящее удерживает вас в этой жизни – вы обречены на успех».
Так утверждает человек, известный не только в нашей стране, но и далеко за ее пределами, Мирзакарим Санакулович Норбеков. В юности врачи признали его безнадежно больным. Но он сумел победить свой недуг. И теперь этот сильный человек показывает путь к здоровью другим.
Более 20 лет назад академик М.С. Норбеков создал оздоровительную систему, которая позволяет человеку любого возраста за десять занятий научиться самостоятельно наводить порядок в своем организме. В 1998 году по решению Международной ассоциации независимых медицинских экспертов она признана самой эффективной среди других альтернативных оздоровительных систем.
Система ускоренного обучения саморегуляции организма предлагает десять путей выхода из нездоровья. Это и суставная гимнастика, и гимнастика воли, и гимнастика воображения, и тренировка эмоций и многое другое. Но основа – в том, что человек из неудачника превращается в Победителя.
Формула Норбекова: волевое принуждение – мышечный корсет – настроение – вера – результат! Хотите – верьте, хотите – проверьте! Не забудьте, что проверять надо не менее сорока дней и обязательно создавать при этом внутреннее состояние того, что вы Победитель!
Но для получения результата надо потрудиться. Работа над собой требует преодоления лени, поэтому в аудитории только те, кто к этому готов. Это единственное условие выздоровления. Иначе любые теоретические знания бесполезны. И Наставник и Ученик, каждый – должен пройти свою половину пути, суть которого – настрой на добро и жизнь.
За 10 дней Вы сможете: восстановить зрение и слух; избавиться от боли в позвоночнике; нормализовать давление; укрепить середечно-сосудистую и нервную системы; избавиться от бессоницы; укрепить эндокринную и имунную системы; решить гинекологически-урологические проблемы; очистить и омолодить свой организм; устранить дефекты кожи; регулировать дыхание и питание; забыть об апатии и депрессии; наладить семейные отношения; создать личную модель успеха; раскрыть свои внутренние возможности; узнать о секретах древневосточной медицины. Мы учимся исцелять себя и поддерживать свое здоровье естественными методами. Мы освоим навыки: формирования внутренней уверенности; избавления от вредных привычек; создания положительных привычек; развития навыков по Вашему выбору; постановки и достижения целей.
Учебно-оздоровительный курс по методике М.С.Норбекова направлен на обучение наших слушателей базовым возможностям управления своим организмом, вплоть до клеточного уровня.
Результатом практического освоения материалов этого курса является:
- фактическое восстановление физического здоровья;
- повышение сопротивляемости иммунной и прочих жизненно важных систем к неблагоприятным воздействиям окружающей среды;
- ускоренная регенерация тканей и, как следствие, омоложение человека;
- качественное улучшение ежедневного самочувствия, повышение общего тонуса и работоспособности.
Многие вначале сомневаются, что можно быть здоровым при помощи всего лишь каких то физических и мысленных упражнений. Не глотая пачками таблетки и пилюли получать удовольствие от здоровой полноценной жизни. Конечно, Вы можете продолжать дальше жить, как привыкли: не изменяя своим стереотипам, не веря в собственные силы и не используя возможности, которые Природа заложила в нас с детства.
Для тех, кто желает изменять свою жизнь к лучшему, мы предлагаем начать со здоровья, используя проверенную эффективную методику с маленькими восточными хитростями, адаптированными к европейскому восприятию. Она позволит Вам легко и быстро получить ощутимые результаты по улучшению своего здоровья и самочувствия уже в течение первых дней занятий.
СКИДКА 20% на весь курс предоставляется студентам, пенсионерам, инвалидам, а также двум или более представителям одной семьи.
Синдром Альстрема у подростков (первое описание в России)
Синдром Альстрема впервые был описан в 1959 г. Основными симптомами этого прогрессирующего аутосомно-рецессивного расстройства являются врожденная дегенерация сетчатки, ведущая к слепоте, детское ожирение, сахарный диабет 2 типа (СД2). Заболевание встречается среди различных рас и этнических групп. Известны ядерные семьи (2?3 пораженных ребенка). Частота синдрома в популяции остается неизвестной. Полиморфизм клинической картины синдрома Альстрема, сходной с многими другими генетическими синдромами, затрудняет диагностику. Однако молекуляно-генетические исследования способствуют раннему выявлению данного синдрома.
Ключевые слова
Для цитирования:
For citation:
Синдром Альстрема впервые был описан в 1959 г. [1]. Основными симптомами этого прогрессирующего аутосомно-рецессивного расстройства являются врожденная дегенерация сетчатки, ведущая к слепоте, детское ожирение, сахарный диабет 2 типа (СД2). Заболевание встречается среди различных рас и этнических групп. Известны ядерные семьи (2–3 пораженных ребенка). Частота синдрома в популяции остается неизвестной.
Самый обширный материал по фенотипическим особенностям пациентов с синдромом Альстрема представлен в публикации [3]. На основании клинического обследования, стандартизированных опросников, бесед с врачами и родителями получены клинические данные о 182 пациентах в возрасте от 1 года до 48 лет и патологоанатомические описания 5 случаев [3].
При исследовании 2 популяций ген синдрома Альстрема, ALSM1, идентифицирован на коротком плече 2-й хромосомы. В 1999 г. были исследованы 12 неродственных семей с синдромом Альстрема из 6 стран, не являющихся между собой родственниками, и подтверждена локализация гена ALMS1 на хромосоме 2р13 [4]. Всего в гене ALSM1 было обнаружено 15 мутаций, которые при гомозиготном носительстве приводят к развитию синдрома Альстрема.
Наиболее часто встречаются следующие мутации этого гена: 2141delCT, 6571delTCAC, 7132insA, C10483T, 10775delC и G10992A.
Ген ALMS1 кодирует белок, расположенный в основании ворсинок [3]. Его функция пока неизвестна, но предполагается, что он участвует во внутриклеточном транспорте. У сибсов-гомозигот по одной и той же мутации гена ALMS1 клиническая картина может быть различной [6].
Одним из центральных симптомов заболевания является раннее ожирение, которое встречается у 98% больных. Масса тела при рождении ребенка может быть в пределах нормы, однако последующие 2 года отмечается ее интенсивное нарастание [3]. Длина тела при рождении не отличается от нормы, в дальнейшем дети быстро растут, и дифференцировка скелета обычно на 1–3 года опережает хронологический возраст. Однако конечный рост пациентов ниже 3-го процентиля. Исследование уровня IGF-1 и IGF-ВР-3 не выявило нарушений, что позволяет предполагать влияние гиперинсулинемии на рост и костное созревание в раннем детстве [2]. К фенотипическим особенностям следует отнести нарушение осанки (грудной и поясничный сколиоз, кифоз, лордоз), широкие ступни и кисти, короткие пальцы на ногах и руках, глубокопосаженные глаза, аномалии зубов верхней челюсти, аллопецию.
С первых месяцев жизни (5–15 мес.) у ребенка отмечается нистагм и светобоязнь [8]. При офтальмологическом обследовании выявляется пигментная дегенерация сетчатки, менее чем за 10 лет приводящая к слепоте. При электронной микроскопии находят пигментную атрофию эпителия сетчатки, преретинальный фиброз, атрофию диска зрительного нерва, отсутствие палочек и колбочек. У 89% больных развивается тугоухость (средний возраст ее диагностики 5 лет).
Инсулиновая резистентность развивается между 18 мес и 4 годами жизни. Появляющийся аcanthosis nigricans подтверждает этот симптом. Сахарный диабет 2 типа обычно диагностируется во II–III декаде жизни. Однако описаны случаи раннего начала СД2 типа в возрасте 5 лет [9]. Возможно острое развитие гипергликемии с кетоацидозом, требующем неотложной инсулинотерапии. У большинства пациентов отмечается повышенный уровень триглицеридов с нормальным уровнем холестерина в сыворотке. Среди других эндокринных расстройств отмечают гипотиреоз (до 17%), гипер- и гипогонадотропный гипогонадизм, крипторхизм, гирсутизм у девочек с патологическим развитием молочных желез, поликистоз яичников, нерегулярные менструации или аменорею. Пубертат часто задерживается.
Особое место среди симптомов занимает дилатационная кардиомиопатия. Некоторые авторы считают, что в отсутствие патологии сердца диагноз синдрома маловероятен. По мнению других, дилатационная кардиомиопатия может развиться в любом возрасте, иногда до появления основных симптомов заболевания, и часто спонтанно исчезает.
У больных с синдромом Альстрема описаны легочные и мочеполовые расстройства, медленно прогрессирующая хроническая нефропатия. У ряда больных наряду с проявлениями синдрома Альстрема имелись симптомы, характерные для метаболического синдрома, включая артериальную гипертонию.
Патогномоничными для синдрома Альстрема являются нейросенсорная тугоухость, дегенерация сетчатки, ведущая к слепоте, нефропатия, а также СД2. Полидактилия, синдактилия и брахидактилия являются патогномоничными для синдромов Лоренса-Муна-Барде-Бидла, Барде-Бидла, а для синдрома Прадера-Вилли – акромикрия. Поражения глаз также характерны для синдромов Лоренса-Муна-Барде-Бидла и Барде-Бидла. Для синдрома Прадера-Вилли характерна мышечная гипотония новорожденных. При синдромах Прадера-Вили, Лоренса-Муна-Барде-Бидла, Барде-Бидла рост в норме, выше или ниже ее границ. СД2 типа отмечен при синдроме Прадера-Вили. При синдромах Прадера-Вили, Лоренса-Муна-Барде-Бидла, Барде-Бидла выявлено сильное отставание в умственном развитии, тогда как у больных с синдромом Альстрема достаточно часто отмечается нормальный интеллект. При синдромах Прадера-Вилли, Лоренса-Муна-Барде-Бидла не отмечалось патологии почек, а при синдроме Барде-Бидла выявлены аномалии развития почек.
Дифференциальный диагноз рассматриваемых синдромов представлен в табл. 1.
В доступной литературе нам не встретилось описаний синдрома Альстрема в Российской Федерации. Приводим два собственных наблюдения (см. рисунок).
Пациент К.К., 16 лет, поступил в ЭНЦ с жалобами на слепоту, головные боли, избыточный вес, сонливость, нестабильную гликемию, подъем АД до 140/90 мм рт. ст., баланопостит, фимоз.
Ребенок от 2-й беременности, вторых родов. Первая беременность закончилась родами. Родился мальчик с массой тела 3600 г, длиной 54 см., умер в 3,5-месячном возрасте от острой сердечной недостаточности. Вторая беременность протекала с токсикозом в I половине и нефропатией во II половине. Роды на 42-й нед. Родился мальчик с массой тела 3500 г, длиной 53 см. К груди приложен в первые сутки. На грудном вскармливании до 5 мес., докорм с 3 мес. В 6 мес при плановом осмотре выявлен врожденный нистагм, гипертензионно-гидроцефальный синдром. В 7 мес при обследовании в связи с судорожным синдромом диагностирована перинатальная энцефалопатия, задержка психомоторного развития. В 11 мес родители отметили снижение остроты зрения (рассматривал игрушки на близком расстоянии). К году установили наличие избыточной массы тела. В 3-летнем возрасте окулистом установлена врожденная патология обоих глаз: сходящееся косоглазие, колбочковая дисфункция, дистрофия сетчатки.
В 4 года проконсультирован генетиком. На основании таких симптомом, как ожирение, задержка психомоторного развития, дистрофия сетчатки, диагностирован синдром Лоуренса-Муна-Барде-Бидла.
В 6 лет стали беспокоить боли в области печени; при обследовании выявлено увеличение печени при отрицательных результатах определения маркеров вирусных гепатитов в крови. В возрасте 8 лет диагностирована тугоухость III ст. В 12-летнем возрасте выявлена гипергликемия натощак (9,0 ммоль/л) и через 2 ч после пищевой нагрузки (14 ммоль/л). Диагностировано нарушение толерантности к глюкозе. В 14 лет диагностирован СД2. В течение 2 лет получал лечение метформином, затем глюренормом.
При обследовании в ЭНЦ длина тела 153 см, масса тела – 67 кг, ИМТ=28,6. Кожные покровы бледные сухие с выраженной потливостью ладоней и подмышечных областей; угревая сыпь на спине, лице; acanthosis nigricans на шее. Подкожный жировой слой развит избыточно с перераспределением жира в области туловища. Костных деформаций нет.
Тоны сердца приглушены, ЧСС – 92 в мин. АД 160/110 мм рт. ст. Печень увеличена (+12 см по L. Medioclavricularis dextra), плотная. Пальпируется край селезенки. Щитовидная железа не пальпируется.
Половое развитие: Таннер 2 (Ах 3, Р 3, яички 6-8 мл). Половой член – состояние после circumcizio, размеры соответствуют возрасту. Дифференцировка скелета соответствует половозрелому субъекту. Зоны роста закрыты.
При УЗИ сердца данных за дилатационную кардиомиопатию нет. Отмечен диффузный гипокинез стенок левого желудочка (ЛЖ), снижение систолической функции ЛЖ, нарушение диастолической функции ЛЖ по 2 типу.
Осмотр окулиста: Vis OD = 0,005 Vis OS = 0,005. OU – спокоен, роговицы прозрачные, в хрусталике начальное помутнения в задней капсуле; на глазном дне: диск зрительного нерва бледный однотонный; границы нечеткие. Резкое сужение сосудов, артерии облитерированы, дегенерация сетчатки с отложением пигмента от центра до периферии сетчатки.
При УЗИ брюшной полости отмечено увеличение печени, селезенки, почек. Повышение эхогенности печени. Протеинурия 1,525 г/л.
Результаты гормональных и биохимических исследований представлены в табл. 2.
Анамнез, клиническая картина, лабораторные данные позволили диагностировать синдром Альстрема. Из неописанных ранее симптомов мы отметили множественный папилломатоз, себорейный дерматит.
В семье пациента были обследованы оба родителя и пробанд. У матери обнаружена мутация 2141delCT. Других мутаций из изученных локусов гена ALMS1 в этой семье обнаружено не было.
Пациент М.А., 17 лет. Ребенок от 11-й беременности, протекавшей на фоне нефропатии, пятых срочных родов в головном предлежании. Первая беременность закончилась рождением живого мальчика, который умер в 2 мес от сердечной недостаточности; 2-я беременность – роды девочкой, 3–4-я беременности – медицинские аборты, 5-я беременность – роды двойней, 6-я беременность – роды двойней, 8–10-я беременности - медицинские аборты. Мать страдала ожирением, гипертонической болезнью, хроническим пиелонефритом; умерла в возрасте 55 лет от сердечно-легочной недостаточности; 2 сестры и 3 брата здоровы.
Масса тела больного при рождении 4350 г, длина 53 см. С рождения – на искусственном вскармливании. С первых месяцев жизни отмечено быстрое увеличение массы тела. В возрасте 1 г 8 мес имел массу тела 17,8 кг, рост 85 см, ИМТ – 25,4 кг/см2.
В 6 мес выявлены нистагм, гипоплазия диска зрительного нерва, в 1 год – светобоязнь. В 13 лет обнаружена глюкозурия и протеинурия (ортостатическая, по предположению уролога). В 15 лет выявлены аcanthosis nigricans, гипергликемия натощак 8–10 ммоль/л ; через 2 ч после нагрузки глюкозой – 12,9–14,9 ммоль/л. Диагностирован СД2. Уровень С-пептида 6,8 нг/мл; ИРИ- 66,2 мЕд/л. Лечился Сиофором по 500 мл 2 раз в день, нерегулярно. Гликемия в течение суток снизилась до 4-8 ммоль/л.
При обследовании в ФГУ ЭНЦ: рост – 157 см, масса тела 91 кг, ИМТ – 35 кг/м2. Гиперстеническое телосложение. Кожа сухая, аcanthosis nigricans на шее, множественные папилломы в подмышечной области на фоне гиперпигментации. Равномерное ожирение. Череп башенной формы, сглаженный затылок, короткая шея, низкий рост волос на лбу. Пальцы на руках и ногах короткие. Широкая грудная клетка.
Тоны сердца ритмичные, приглушены, ЧСС – 90 в мин, АД 150/100 мм рт. ст. Отеков нет. Живот вздут. Печень не пальпируется из-за метеоризма. Запоров нет. Щитовидная железа не увеличена. Клинически эутиреоз. Половое развитие: Таннер 2-3.
На ЭКГ: синусовый ритм с ЧСС 86-104 в мин. Отклонение электрической оси сердца влево. Неполная блокада правой ножки пучка Гиса и передней ветви левой ножки пучка Гиса. Изменения предсердного компонента.
УЗИ сердца: камеры сердца не расширены, миокард желудочков не утолщен. Уплотнение стенки кольца аортального клапана. Уплотнение стенок митрального клапана. Заключение: крепление хорд митрального клапана к базальному сегменту межжелудочковой перегородки. Турбулентный трансаортальный ток. Максимальная скорость 1,0 м/с. Перикард без особенностей. Признаки дилатационной кардиомиопатии отсутствуют.
УЗИ брюшной полости: гепатомегалия, жировой гепатоз, диффузные изменения почек, липоматоз поджелудочной железы.
Заключение окулиста: Vis OD, Vis OS на уровне светоощущения. Роговицы прозрачны. Радужки структурны. Хрусталик с помутнениями в ядре. Глазное дно: диск зрительного бледно-розовый, монотонный, границы нечеткие. Артерии сужены. Сетчатка с фиброзными включениями, «пестрая». Протеинурия 5,3 г/л.
Результаты гормональных и биохимических исследований представлены в табл. 2.
Молекулярно-генетическое обследование: оба родителя гетерозиготные носители мутации 2141delCT гена ALMS1, а ребенок унаследовал мутантные аллели от обоих родителей и является гомозиготным носителем этой мутации.
Таким образом, в обоих случаях первые беременности у матерей закончились рождением живых мальчиков, которые погибли на 2–3-м мес жизни от острой сердечно-сосудистой недостаточности. Можно предположить, что новорожденные погибли от дилатационной кардиомиопатии, которая развилась раньше классических симптомов синдрома Альстрема. Наблюдаемые подростки не имели указанной патологии, возможно, вследствие ее транзиторности.
Фенотипически больные мало отличаются друг от друга, но при молекулярно-генетическом исследовании были выявлены различные мутации. Как отмечалось ранее, даже у сибсов при фенотипическом сходстве выявляются разные мутации гена ALMS1. Наличие у родителей одних мутаций не исключает появления у детей других. Поэтому отсутствие у отца и ребенка (К.К.) выявленной у матери мутации 2141delCT требует дальнейших молекулярно-генетических исследований.
Полиморфизм клинической картины синдрома Альстрема, сходной с многими другими генетическими синдромами, затрудняет диагностику. Однако молекуляно-генетические исследования способствуют раннему выявлению данного синдрома.
Выражаем благодарность московскому представительству фирмы «Ново Нордиск» за спонсирование молекулярно-генетических исследований.
Читайте также: