МРТ при хронической воспалительной демиелинизирующей полиневропатии (ХВДП)
Добавил пользователь Владимир З. Обновлено: 27.01.2026
МРТ при хронической воспалительной демиелинизирующей полиневропатии (ХВДП)
а) Терминология:
• Клинически гетерогенная группа нейропатий, характеризующихся в основном симметричными сенсорными и моторными нарушениями и развивающихся как монофазное, рецидивирующее или прогрессирующее заболевание:
о Продолжительность заболевания > 8 недель
б) Визуализация хронической воспалительной демиелинизирующей полиневропатии (ХВДП):
• Утолщение и патологическое изменение Т2-сигнала корешков спинного мозга, нервных сплетений и периферических нервов
• Изменения корешков спинного мозга и периферических нервов (экстрафораминальные > интрадуральные участки корешков)
• Поясничный > шейный отдел позвоночника, плечевое сплетение, грудной отдел/межреберные нервы > черепные нервы
(Слева) Аксиальный срез, Т1-ВИ FS с КУ: симметричное утолщение и некоторое контрастное усиление сигнала поясничных корешков спинного мозга. Классическая картина хронической воспалительной демиелинизирующей полиневропатии (ХВДП) включает чувствительные и двигательные нарушения проксимальных и дистальных сегментов всех конечностей с арефлексией, развивающиеся более восьми недель. Типично также увеличение концентрации белка в ликворе и неравномерное замедление нервной проводимости.
(Справа) Аксиальный срез, Т2-ВИ: умеренное утолщение и изо- или гиперинтенсивность сигнала поясничных корешков и нервных стволов поясничного сплетения с обеих сторон. (Слева) Сагиттальный срез, Т2-ВИ: утолщение и патологическое усиление Т2-сигнала экстрадуральных участков спинномозговых нервов поясничного и крестцового отделов позвоночника. Диффузная гипертрофия и усиление сигнала корешков при хронической воспалительной демиелинизирующей полиневропатии (ХВДП) могут быть достаточно распространенными.
(Справа) Аксиальный срез, Т2-ВИ: утолщение и патологическое усиление Т2-сигнала экстрадуральных участков спинномозговых нервов пояснично-крестцового отдела позвоночника. В типичных случаях наблюдается прогрессирующее или возвратно-ремиттирующее течение заболевания, сопровождающееся увеличением концентрации белка в ликворе и электрофизиологическими изменениями, характерными для демиелинизирующего процесса.
в) Дифференциальная диагностика:
• Синдром Гийена-Барре (ОВДП)
• Наследственная демиелинизирующая нейропатия
• Нейрофиброматоз 1 типа
г) Патология:
• Аутоиммунное заболевание, опосредованное клеточным и гуморальным звеньями иммунной системы
• Характерные особенности хронической воспалительной демиелинизирующей полиневропатии (ХВДП): утолщение нервов с формированием «луковиц», демиелинизация
д) Клинические особенности:
• Диагноз хронической воспалительной демиелинизирующей полиневропатии (ХВДП) обычно ставится клинически на основании картины прогрессирующих моторных/сенсорных нарушений и положительного эффекта терапии глюкокортикоидами:
о Типичная форма: симметричная слабость мышц проксимальных и дистальных отделов конечностей, чувствительные нарушения
• Патологические изменения при ЭМГ/ЭНМГ: ключевые изменения электрофизиологических показателей → блокада проведения по нервам, замедление проводимости, позволяющие заподозрить демиелинизирующий процесс
• Диагноз основывается в первую очередь на клинических данных, результатах электрофизиологического исследования и подтверждается биопсией нерва
Магнитно-резонансная томография конского хвоста при хронической воспалительной демиелинизирующей полинейропатии
Введение. Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП) является курабельным аутоиммунным заболеванием. Своевременная диагностика и лечение позволяют улучшить долгосрочный прогноз, отсрочить инвалидизацию. В атипичных случаях заболевания и при дифференциальной диагностике необходимо расширение объема исследований, включающих нейровизуализационные методы.
Цель работы – оценка диагностической роли магнитно-резонансной томографии (МРТ) конского хвоста при ХВДП.
Материалы и методы. В исследование включены 8 пациентов с первоначальным диагнозом ХВДП в соответствии с критериями Европейской федерации неврологических сообществ и Сообщества периферической нервной системы: 6 больных соответствовали достоверному диагнозу ХВДП, 1 пациентка – вероятной ХВДП и у 1 пациента в последующем была выявлена смешанная криоглобулинемия, связанная с гепатитом С. МРТ конского хвоста с контрастным усилением гадолинием выполнена всем первично включенным больным основной группы и 8 пациентам с метаболическими полинейропатиями группы контроля. Через 12 мес МРТ проведена повторно пациентам основной группы.
Результаты. Утолщения корешков конского хвоста (протяженные или фокальные) выявлены в основной группе у 6 пациентов с достоверной ХВДП и у 1 пациентки с вероятной ХВДП и ни в одном случае в группе контроля. Выраженность качественных изменений соотносилась с длительностью болезни. Накопление контрастного вещества и признаки гипертрофии корешков обнаружены у всех пациентов основной группы, при этом накопление гадолиния не отражало активность процесса. Контрастное усиление в области конского хвоста, выявленное у пациентов группы контроля, было обусловлено контрастированием медуллярной артерии.
Заключение. МРТ конского хвоста с контрастым усилением позволяет улучшить диагностику ХВДП, при этом магнитно-резонансная картина не отражает активность болезни. МРТ при ХВДП является методом, заслуживающим дальнейшего изучения и стандартизации.
Ключевые слова
Об авторах
ФГБОУ ВО «Южно-Уральский государственный медицинский университет» Минздрава России
Россия
Россия, 454048 Челябинск, ул. Воровского, 64
ФГБОУ ВО «Южно-Уральский государственный медицинский университет» Минздрава России
Россия
Россия, 454048 Челябинск, ул. Воровского, 64
ФГБОУ ВО «Южно-Уральский государственный медицинский университет» Минздрава России
Россия
Россия, 454048 Челябинск, ул. Воровского, 64
ФГБОУ ВО «Южно-Уральский государственный медицинский университет» Минздрава России
Россия
Россия, 454048 Челябинск, ул. Воровского, 64
Список литературы
1. Bergh P.Y., Hadden R.D., Bouche P. et al. European Federation of Neurological Societies/Peripheral Nerve Society Guideline on management of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force of the European Federation of Neurological Societies and the Peripheral Nerve Society-First Revision. J Peripher Nerv Syst 2010;15(1):1–9. DOI: 10.1111/j.1529-8027.2010.00245.x. PMID: 20433600.
3. Gasparotti R., Lucchetta M., Cacciavillani M. et al. Neuroimaging in diagnosis of atypical polyradiculoneuropathies: report of three cases and review of the literature. J Neurol 2015;262(7):1714–23. DOI: 10.1007/s00415-015-7770-z. PMID: 25957643.
4. Lee S.E., Park S.W., Ha S.Y. et al. A case of cauda equina syndrome in early-onset chronic inflammatory demyelinating polyneuropathy clinically similar to Charcot–Marie–Tooth disease type 1. J Korean Neurosurg Soc 2014;55(6): 370–4. DOI: 10.3340/jkns.2014.55.6.370.
5. PMID: 25237436.
6. Viala K. Diagnosis of atypical forms of chronic inflammatory demyelinating polyradiculoneuropathy: a practical overview based on some case studies. Int J Neurosci 2016;126(9):777–85. DOI: 10.3109/00207454.2015.1096786. PMID: 26392214.
7. Grimm A., Schubert V., Axer H. et al. Giant nerves in chronic inflammatory polyradiculoneuropathy. Muscle Nerve 2017;55(2):285–9. DOI: 10.1002/mus.25272. PMID: 27463360.
8. De Silva R., Willison H., Doyle D. et al. Nerve root hypertrophy in chronic inflammatory demyelinating polyneuropathy. Muscle Nerve 1994;17(2):168–70. DOI: 10.1002/mus.880170206. PMID: 8114785.
9. Midroni G., de Tilly L.N., Gray B., Vajsar J. MRI of the cauda equina in CIDP: clinical correlations. J Neurol Sci 1999;170(1):36–44. PMID: 10540034.
12. Hiwatashi A., Togao O., Yamashita K. et al. Lumbar plexus in patients with chronic inflammatory demyelinating polyneuropathy: evaluation with 3D nerve-sheath signal increased with inked rest-tissue rapid acquisition of relaxation enhancement imaging (3D SHINKEI). Eur J Radiol 2017;93:95–9. DOI: 10.1016/j.ejrad.2017.05.031. PMID: 28668438.
13. Oguz B., Oguz K., Cila A., Tan E. Diffuse spinal and intercostal nerve involvement in chronic inflammatory demyelinating polyradiculoneuropathy: MRI findings. Eur Radiol 2003;13(Suppl 4):L230–4. DOI: 10.1007/s00330-003-1996-3. PMID: 15018192.
14. Tazawa K., Matsuda M., Yoshida T. et al. Spinal nerve root hypertrophy on MRI: clinical significance in the diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy. Intern Med 2008;47(23):2019–24. DOI: 10.2169/internalmedicine.47.1272. PMID: 19043253.
15. Bradley L.J., Wilhelm T., King R.H. et al. Brachial plexushypertrophy in chronic inflammatory demyelinating polyradiculoneuropathy. Neuromuscul Disord 2006;16(2):126– 31. DOI: 10.1016/j.nmd.2005.11.006. PMID: 16427285.
16. Echaniz-Laguna A., Dietemann J.L. Brachial plexus atrophy in chronic inflammatory demyelinating polyradiculoneuropathy. J Clin Neuromuscul Dis 2012;13(4):243–5. DOI: 10.1097/CND.0b013e31822b1993. PMID: 22622171.
18. Zaidman C.M., Al-Lozi M., Pestronk A. Peripheral nerve size in normals and patients with polyneuropathy: an ultrasound study. Muscle Nerve 2009;40(6):960–6. DOI: 10.1002/mus.21431. PMID: 19697380.
19. Kerasnoudis A. Ultrasonographic findings in chronic inflammatory demyelinating polyneuropathy. Am J Phys Med Rehabil 2014;93(1):94. DOI: 10.1097/PHM.0b013e318282cfe9. PMID: 23370587.
20. Дружинин Д.С., Наумова Е.С., Никитин С.С. Ультразвуковая визуализация периферических нервов при мультифокальной моторной нейропатии и хронической воспалительной демиелинизирующей полинейропатии. Нервно-мышечные болезни 2016;(1): 63–73. [Druzhinin D.S., Naumova E.S., Nikitin S.S. Nerve sonography in multifocal motor neuropathy and chronic inflammatory demyelinating polyneuropathy. Nervno- myshechnye bolezni = Neuromuscular Diseases 2016;(1):63–73. (In Russ.)]. DOI: 10.17650/2222‑8721‑2016‑6‑1-63-73.
21. Ginsberg L., Platts A.D., Thomas P.K. Chronic inflammatory demyelinating polyneuropathy mimicking a lumbar spinal stenosis syndrome. J Neurol Neurosurg Psychiatry 1995;59(2):189–91. PMID: 7629539.
22. Schady W., Goulding P., Lecky B.R. et al. Massive nerve root enlargement in chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry 1996;61(6):636–40. PMID: 8971116.
23. Duggins A.J., McLeod J.G., Pollard J.D. et al. Spinal root and plexus hypertrophy in chronic inflammatory demyelinating polyneuropathy. Brain 1999;122(Pt 7): 1383–90. PMID: 10388803.
24. Rossi D.P., Doria Lamba L., Pistorio A. et al. Chronic inflammatory demyelinating polyneuropathy of childhood: clinical and neuroradiological findings. Neuroradiology 2013;55(10):1233–9. DOI: 10.1007/s00234-013-1240-z. PMID: 23893072.
Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП) (информация для пациента)
"Хроническая" - означает, что заболевание имеет длительный курс течения, а симптоматика может неуклонно прогрессировать и рецидивировать. Для установления диагноза хронической полинейропатии должно пройти более 8 недель от момента появления первых симптомов.
"Воспалительная" - подразумевает "воспаление" как основной механизм повреждения периферических нервов вследствие нарушения комплексной работы иммунной системы, поэтому данное заболевание также можно назвать “аутоиммунным”.
"Демиелинизирующая" - характеризует тип повреждения периферических нервов, при котором преимущественно страдает миелиновая оболочка нерва.
"Полирадикулонейропатия" - означает, что патологический процесс вовлечено более одного нерва, а также корешки спинного мозга и стволы сплетений.
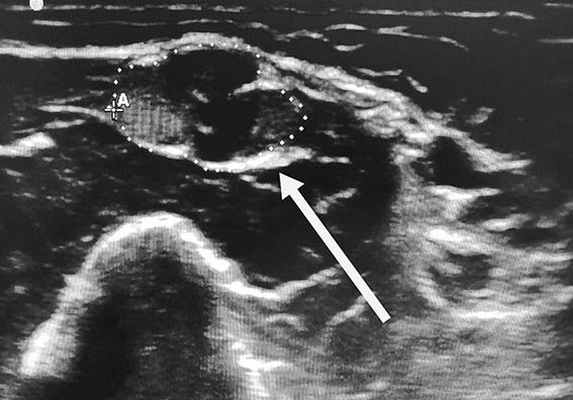
Как часто встречается это заболевание?
ХВДП является довольно редким заболеванием. Средняя распространенность пациентов с ХВДП в мире в среднем составляет до 0,81-1,90 случаев на 100 000 человек. Мужчины заболевают несколько чаще женщин. Дебютировать ХВДП может в любом возрасте, даже в детском, однако пик заболеваемости приходится на средний возраст - 40-50 лет.
Каковы причины развития заболевания?
Причины развития ХВДП до сих пор полностью не изучены. Однако результаты многочисленных исследований и эффективность иммуномодулирующей терапии указывают на нарушение работы иммунной системы, как ключевой причины развития заболевания. Иммунная система представляет собой очень сложный и гармоничный механизм. Ключевыми звеньями иммунной системы являются антитела, ряд белков сыворотки крови и белые клетки крови, лейкоциты. В норме иммунная система борется с чужеродными агентами (белки, вирусы, бактерии). Однако при ряде заболеваний (аутоиммунные заболевания) компоненты иммунной системы по ошибке начинают работать против собственного организма. К одним из таких заболеваний и относится ХВДП. В данном случае развивается реакция против компонентов оболочки периферических нервов, что проявляется в виде специфических симптомов, характерных для ХВДП.
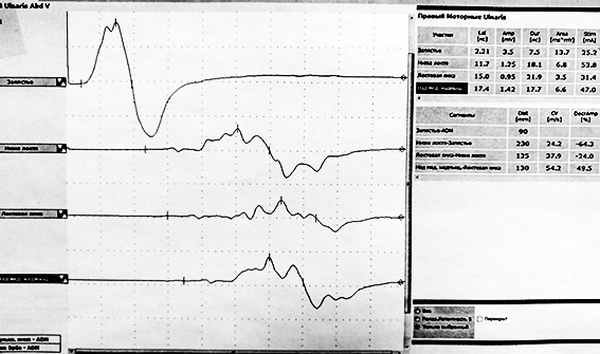
Есть ли факторы риска заболевания?
В качестве триггерных факторов описаны респираторно-вирусные заболевания, оперативные вмешательства, беременность, вакцинация и другие причины, тем не менее прямая связь вышеуказанных факторов с развитием заболевания пока не доказана.
Возможна ли передача заболевания по наследству?
Нет. Существует ряд исследований, в которых выявлены гены, предполагаемые в развитии заболевания. Тем не менее, пока не доказано их участие в риске развития заболевания у потомков.
Как проявляется ХВДП и в чем особенности этого заболевания?
ХВДП - это заболевание периферических нервов. В подавляющем большинстве в патологический процесс вовлекаются так называемые “толстые” нервные волокна, имеющие толстый слой миелиновой оболочки, которая и является мишенью при данном заболевании. Эти волокна несут от головного и спинного мозга к нашей скелетно-мышечной системе информацию о двигательной команде и в обратном направлении о положении тела в пространстве для обеспечения равновесия. Поэтому самым распространенными симптомами являются:
- слабость в руках и ногах
- неустойчивость при ходьбе
- ощущение онемения в кистях и стопах
- похудание мышц и снижение их тонуса
Точный механизм ХВДП до конца не раскрыт ввиду комплексности иммунных реакций, а потому симптоматика и характер течения заболевания могут варьировать. Отсюда выделены так называемые атипичные формы ХВДП, которые несколько отличаются от классического течения заболевания и могут иметь особенности прогноза и лечения. Диагностика таких форм может быть затруднена. К атипичным формам ХВДП относят:
- мультифокальную форму ХВДП (синдром Льюиса-Самнера)
- дистальную форму ХВДП
- чисто сенсорную или чисто моторную формы ХВДП
- ХВДП с острым началом
- хроническую иммунную сенсорную полирадикулонейропатию
Течение заболевания также может быть вариабельным - у части пациентов может развиваться грубая симптоматика, приводящая к инвалидизации, у других - минимальные неврологические нарушения; ряд пациентов может испытывать частые обострения, в то время как встречаются случаи с единственным обострением в жизни.
На основании чего мне установили диагноз ХВДП?
Ключевым в постановке диагноза является клинический осмотр неврологом. Существуют критерии заболевания, предложенные Европейской федерацией неврологических сообществ в 2010 г. Для подтверждения диагноза и исключения альтернативных причин полинейропатии обычно проводится ряд лабораторных анализов, а также инструментальные методы исследования. К сожалению, до сих пор нет “золотого” стандарта диагностики, который с высокой точностью мог бы указывать на "ХВДП". Поэтому диагноз зачастую ставится на основании клинической картины и результатов исследования.
Ключевым инструментальным методом диагностики ХВДП, как и любой полинейропатии, является электронейромиография. Это исследование проводимости периферических нервов при помощи коротких электрических импульсов, передающихся по ходу нерва. Стимуляция приводит к сокращению мышцы, иннервируемой исследуемым нервом, которое регистрируется электродом.
Если данных для установки диагноза окажется недостаточно, могут дополнительно проводиться МРТ сплетений, анализ спинномозговой жидкости, УЗИ периферических нервов и в редких случаях биопсия нерва. К одним из критериев правильности установленного диагноза относится улучшение состояния, либо приостановление прогрессирования заболевания на фоне патогенетической терапии.
Какие варианты лечения ХВДП существуют?
До 80% пациентов с ХВДП имеют эффект от терапии, модулирующей работу иммунной системы. На основании крупных исследований в лечении ХВДП доказанную эффективность имеют следующие варианты лечения:
- глюкокортикостероидные препараты
- препараты внутривенного человеческого иммуноглобулина
- плазмаферез
Ни один из известных на сегодняшний день препаратов не излечивает ХВДП, только позволяет снизить активность заболевания, предупредить от дальнейшего ухудшения или обострения, а также уменьшить выраженность симптомов. Кроме того, ответ на лечение у разных людей может различаться.
Каждый способ лечения имеет свои плюсы и минусы, которые обговариваются врачом, учитывая все индивидуальные особенности?
Глюкокортикостероидные препараты (преднизолон, метилпреднизолон) назначаются в форме таблеток и инфузий. Вначале подбирается высокая доза из расчета массы тела, которая со временем постепенно снижается. Длительность приема и величина поддерживающей дозы зависят от тяжести симптоматики, скорости ее прогрессирования, а также ответа на лечение. Тем не менее, для оценки эффективности лечения, длительность терапии должна быть не менее 12 недель. Несмотря на доказанную эффективность и относительно низкую стоимость, лечение ГКС может быть сопряжено с рядом побочных явлений - набор веса, тошнота, бессонница, раздражительность, обострение язвенной болезни, повышение цифр артериального давления и уровня сахара крови, снижение плотности костной ткани. Поэтому наряду с основным препаратом назначается комплексная терапия для предупреждения развития вышеуказанных последствий лечения.
Аналогичными по эффективности глюкокортикостероидам являются препараты человеческого иммуноглобулина, однако последние гораздо реже сопряжены с развитием побочных эффектов, а потому более безопасны. Пожалуй, главным недостатком такого лечения является его высокая стоимость. Препараты человеческого иммуноглобулина получают путем очистки большого количества (>10,000 л) человеческой плазмы (>1000 доноров), что обуславливает их дороговизну. Лечение заключается в ежемесячном курсовом внутривенном введении препарата. Курс обычно занимает 4-5 дней. В дальнейшем частота введения препарата может варьироваться в зависимости от его эффективности. Важным моментом является выбор препарата. Необходимо обратить внимание на его основные характеристики: препарат должен подходить для проведения высокодозной внутривенной иммунотерапии, содержание IgG должно быть не менее 95%, количества IgA и IgM должны быть следовыми. При этом количество IgA должно быть четко обозначено в инструкции, так как именно с этим классом иммуноглобулинов ассоциировано развитие аллергических реакций.
Третьим вариантом лечения является высокообъемный плазмаферез. Данный способ терапии представляет собой забор плазмы с патогенными антителами через катетер и восполнение ее стерильными растворами, белковыми растворами и/или донорской плазмой. Процедура повторяется около 5 раз, обычно с интервалом через день. Эффект от такого лечения сохраняется на протяжении 3-4 недель. Учитывая его сложность, такой способ терапии не используется для длительного лечения и часто бывает полезен в случае стремительного и\или тяжелого обострения.
У некоторых больных, несмотря на грамотное лечение, заболевание всё равно может прогрессировать или не поддаваться контролю. В этих случаях назначаются иммунодепрессанты (микофенолата мофетил, азатиоприн, циклоспорин, циклофосфамид) или моноклональные антитела (ритуксимаб). Назначение данных препаратов должно исходить от врача, имеющего опыт их применения, учитывая все показания и противопоказания, с последующим тщательным контролем эффективности и безопасности терапии.
Нужно ли вносить какие-либо изменения в привычный образ жизни?
Да. Существуют ряд рекомендаций для больных с диагнозом ХВДП:
- избегать любых вирусных и бактериальных инфекций (респираторно-вирусных, энтеровирусных заболеваний и др.)
- больше двигаться - в случае выраженных двигательных нарушений может быть назначена физическая реабилитация, эрготерапия или поддерживающие средства для ходьбы – ортезы и пр.
- ограничить курение, употребление алкоголя, что оказывает негативное влияние на кровообращение, отягощая течение полинейропатии
- исключить прием нейротоксических препаратов, усугубляющий течение полинейропатии
- тщательный уход за стопами очень важен, особенно при наличии сопутствующего сахарного диабета. Необходимо ежедневно осматривать стопы на предмет порезов, мозолей, язв.
- придерживаться диеты с низким содержанием жиров, богатой злаками, фруктами и овощами.
- избегать длительного сдавления конечностей
Каковы прогнозы при данном заболевании?
В целом, продолжительность жизни не отличается от таковой у людей, не имеющих данное заболевание. Течение болезни может быть различным - протекать с частыми обострениями, иметь медленно прогрессирующее течение, либо достигнуть стойкой ремиссии с минимальными клиническими проявлениями. Крайне важным для прогноза является своевременное назначение лечения, тщательное наблюдение за пациентом и эффектами проводимой терапии.
Если у вас есть симптомы полинейропатии или вам поставлен диагноз "Полинейропатия" или "ХВДП", вы можете пройти комплексное обследование в Центре заболеваний периферической нервной системы ФГБНУ НЦН, где вам помогут уточнить диагноз, выявить причины поражения периферических нервов и назначат терапию с позиций доказательной медицины.
Сотрудники центра заболеваний периферической нервной системы консультируют пациентов амбулаторно в рамках ОМС и на коммерческой основе.
МРТ при хронической воспалительной демиелинизирующей полиневропатии (ХВДП)
ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, Москва, Россия
ГУ НИИ ОПиПФ РАМН, Москва
Российский национальный исследовательский медицинский университет им. Н.И. Пирогова, Москва
ООО Международный институт функциональной реконструктивной микрохирургии, Медицинский центр «Мотус», Ярославль, Россия
Хроническая дизиммунная гипертрофическая плексопатия как вариант атипичной хронической воспалительной демиелинизирующей полинейропатии
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. 2019;119(12): 94‑99
ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, Москва, Россия
Представлен клинический случай у ребенка 7 лет с постепенным развитием слабости в левой ноге, которая была расценена неврологом как поражение седалищного нерва неуточненного генеза. По результатам проведенного клинико-инструментального обследования верифицирован диагноз: фокальная форма хронической демиелинизирующей полинейропатии. Дизиммунный характер болезни подтвержден данными МРТ пояснично-крестцового сплетения с контрастным усилением невральных структур и ответом на терапию высокодозными внутривенными иммуноглобулинами в комбинации с кортикостероидами.
ФГБОУ ВО «Российский национальный исследовательский медицинский университет им. Н.И. Пирогова» Минздрава России, Москва, Россия
ГУ НИИ ОПиПФ РАМН, Москва
Российский национальный исследовательский медицинский университет им. Н.И. Пирогова, Москва
ООО Международный институт функциональной реконструктивной микрохирургии, Медицинский центр «Мотус», Ярославль, Россия
Термин «хроническая воспалительная демиелинизирующая полинейропатия» (ХВДП) объединяет группу иммуноопосредованных [1] состояний с частотой встречаемости у детей до 0,48 на 100 000 [2]. Традиционно выделяют типичные и атипичные формы ХВДП. Классическая клиническая картина ХВДП характеризуется симметричными чувствительными и двигательными нарушениями в дистальных и проксимальных отделах рук и ног, прогрессирующим или рецидивирующим течением в период 8 нед и более, повышением концентрации белка в цереброспинальной жидкости (ЦСЖ) и ответом на иммуномодулирующую терапию. Предложенные в 2000 г. клинические, электрофизиологические и лабораторные критерии ХВДП детского возраста имеют низкую чувствительность — до 77% [3, 4], в связи с чем эксперты приняли решение использовать критерии EFNS [5] для взрослых, чувствительность которых достигает 94% [4]. Независимо от возраста пациента диагностическую сложность представляют атипичные формы дизиммунных нейропатий и особенно фокальная ХВДП, проявляющаяся поражением одной или двух конечностей [6]. Симптомы и электрофизиологические изменения, выявляемые при атипичных формах ХВДП, не соответствуют принятым критериям диагноза, затрудняя диагностический поиск. Представлено наблюдение хронического дизиммунного поражения пояснично-крестцового сплетения у ребенка 7 лет как варианта атипичной ХВДП.
Клиническое наблюдение. Мальчик В., 2010 года рождения, с неотягощенным семейным анамнезом и без особенностей раннего развития. В возрасте 6,5 года мать впервые отметила, что ребенок без видимых причин начал прихрамывать на левую ногу. Неврологом в поликлинике было предположено поражение левого седалищного нерва, для уточнения генеза которого ребенок был направлен на УЗИ, которое не выявило изменений на доступном визуализации отрезке нерва.
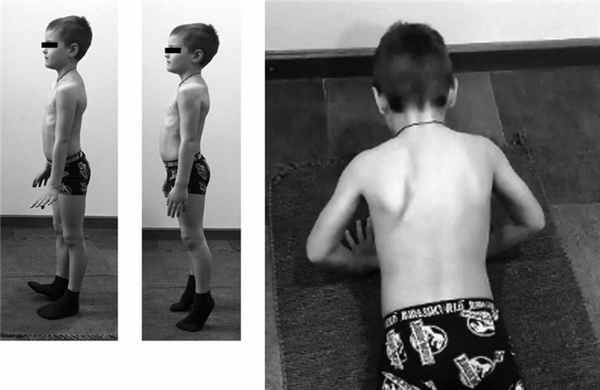
При осмотре обращали на себя внимание слабость (до плегии в мышцах тыльного сгибания стопы) и гипотрофия мышц, иннервируемых левым пояснично-крестцовым сплетением (четырехглавая мышца бедра, бицепс бедра, группа мышц, приводящих бедро, большая ягодичная мышца, передняя большеберцовая, икроножная, короткий разгибатель пальцев), а также слабость удерживающих лопатку слева мышц: широчайшей, зубчатой, надостной, подостной (рис. 1). Рис. 1. Пациент В., 7 лет. Клиническая оценка мышечной силы. а — отсутствие тыльного сгибания левой стопы; б — сохранность силы мышц подошвенного сгибания стопы; в — слабость мышц, удерживающих лопатку слева. Сухожильные рефлексы не вызывались только с левой ноги. Нарушения чувствительности ограничены зоной иннервации левого пояснично-крестцового сплетения. В остальном неврологический статус был без особенностей: менингеальных симптомов нет, движения глазных яблок в полном объеме, нистагма нет, сила мимической мускулатуры 5 баллов, лицо симметричное, сухожильные рефлексы, кроме ахиллова и коленного слева, в том числе, рефлекс с ости лопатки нормальной живости и симметричные, патологических стопных знаков нет. Координаторные пробы: пальценосовую пробу выполняет удовлетворительно.
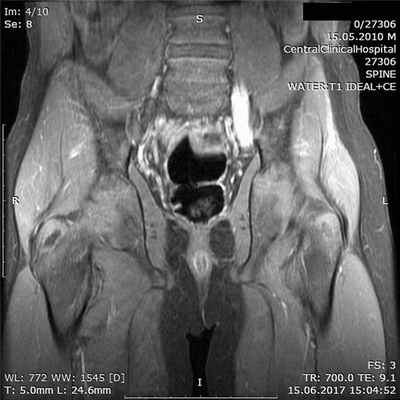
Результаты осмотра, УЗИ периферических нервов и ЭМГ констатировали поражение на уровне пояснично-крестцового сплетения. Для уточнения причины изменений проведено МРТ с контрастным усилением гадолинием, обнаружено увеличение нервных стволов и накопление контрастного вещества в пояснично-крестцовом сплетении (рис. 2). Рис. 2. Тот же пациент. МРТ пояснично-крестцового сплетения в режиме Т1 с контрастным усилением гадолинием. МРТ плечевого сплетения не выявило изменений.
В соответствии с алгоритмом обследования пациентов с подозрением на ХВДП проведено исследование ЦСЖ, которое не выявило белково-клеточной диссоциации; также не обнаружено антител к ганглиозидам периферических нервов в сыворотке крови.
На основании течения болезни (более 8 нед), данных МРТ — накопление контрастного вещества нервными стволами, результатов ЭМГ, указывающих на вовлечение пояснично-крестцового сплетения, предположено наличие дизиммунного процесса периферической нервной системы. Назначена комбинированная патогенетическая терапия 4 курсами с интервалами 1 мес: метилпреднизолон 3000 мг (суммарная доза на 1 курс) внутривенно и внутривенные иммуноглобулины в дозе 2 г на 1 кг массы тела в первый курс и 1 г на 1 кг массы тела в следующие 3 инфузии без последующей поддерживающей терапии.
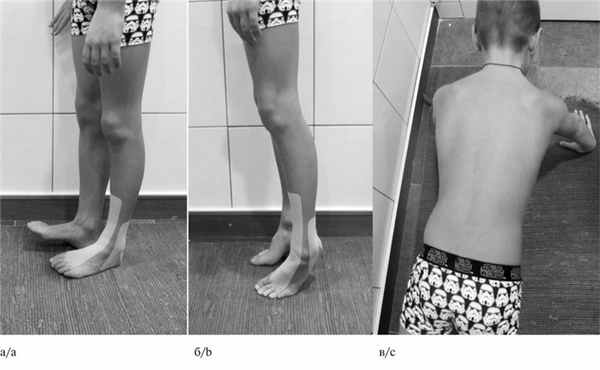
На фоне лечения отмечено значительное улучшение состояния ребенка в виде нарастания мышечной силы в левой ноге, мышцах, удерживающих левую лопатку, а также уменьшение выраженности мышечных гипотрофий (рис. 3). Рис. 3. Тот же пациент. Клиническая оценка мышечной силы на фоне иммуномодулирующей терапии через 4 мес. а — появилось тыльное сгибание стопы слева; б — сохранность силы мышц подошвенного сгибания стопы; в — регресс слабости мышц, удерживающих лопатку слева. Именно ответ на иммуномодулирующую терапию послужил фактом, подтверждающим диагноз фокальной формы ХВДП.
При очевидном улучшении состояния при повторном МРТ поясничного сплетения накопление контрастного вещества нервными стволами сохранялось (рис. 4). Рис. 4. Тот же пациент. МРТ пояснично-крестцового сплетения в режиме Т1 с контрастным усилением гадолинием на фоне иммуномодулирующей терапии через 4 мес.
Ответ на иммуномодулирующую терапию в представленном случае стал фактом, подтверждающим диагноз.
ХВДП вызывает в ряде случаев диагностические и дифференциально-диагностические сложности [7, 8].
Пациенты часто проходят долгий диагностический путь с исключением наследственных форм моторно-сенсорной полинейропатии, вторичных нейропатий на фоне болезней соединительной ткани, нервно-мышечной передачи и даже синдрома Гиейна—Барре [7, 8, 13].
Ретроспективное исследование ранее проведенной экспертной оценки случаев ХВДП из разных клиник показало, что только 37% пациентов имели достаточно данных для постановки диагноза [12]. Такая же ситуация имеет место и в педиатрической практике, когда неправильное толкование полученных данных, технические ошибки при проведении ЭМГ, недостаточный опыт исследователя, а также неполное обследование ребенка приводит к неправильной постановке диагноза [14].
С другой стороны, есть ограничения существующих электрофизиологических критериев ХВДП, которые хорошо известны и признаны [15]. Повышение белка в ЦСЖ у детей с ХВДП присутствует у большинства пациентов, но не у всех [2, 16], и у части больных не проводят исследование ЦСЖ [12]. МРТ-изменения в виде гипертрофии корешков конского хвоста, спинномозговых нервов плечевого и поясничного сплетений, сопровождаемые контрастным усилением, описываются во всех возрастных группах пациентов с ХВДП, и они включены в дополнительные критерии EFNS/PNS [5, 8, 17—19], но о том, как часто обнаруживаются изменения и на каком этапе болезни, данных в литературе мало. Так, пациенты, уже получающие патогенетическое лечение, в 100% случаев имели изменения в поясничном сплетении и были оценены ретроспективно [20]. Выявление характерных изменений для ХВДП при биопсии нерва, несомненно, подтверждает диагноз, но их отсутствие не исключает его [21].
Атипичные формы ХВДП требуют более полной оценки клинических данных в соответствии с получаемыми результатами инструментального и лабораторного обследования пациента [12], как и в описанном нами случае, когда нет соответствия полученных данных ни клиническим, ни электрофизиологическим критериям ХВДП. Положительный ответ на иммуномодулирующую терапию может служить подтверждением дизиммунного характера процесса [5], что было учтено у нашего больного.
Если типичные случаи ХВДП у детей хорошо представлены в литературе [22], то атипичные и тем более фокальные формы ХВДП обсуждаются мало. Большинство представленных случаев является результатом длительного наблюдения за пациентами, имевшими изменения в одной или двух конечностях, которые впоследствии генерализовались до типичной картины ХВДП [23, 24], в связи с чем обсуждается именно фокальное начало ХВДП, а не проявление клинической картины атипичной формы рассматриваемой патологии. Аналогичное мнение высказано в дискуссии с профессором J. Léger на 2 -м конгрессе Международной академии IG (Берн, Швейцария) в октябре 2018 г., на котором был представлен клинический случай 7-го наблюдения за пациентом с сенсорными изменениями только в левой руке с положительным ответом на введение иммуноглобулинов, расцененный как фокальная ХВДП. Экспертами рекомендовано наблюдение за такими больными даже в состоянии ремиссии.
Нами продолжается наблюдение за ребенком в виде клинического осмотра с частотой 1 раз в месяц с фото- и видеофиксацией результатов.
Представленный клинический случай является примером сложности трактовки неоднозначных результатов инструментальных методов обследования в сочетании с фокальным поражением конечности. Настороженность относительно атипичных форм ХВДП, в данном случае фокального варианта, позволяет уменьшить время постановки диагноза за счет сокращения дифференциально-диагностического ряда и начать патогенетическую терапию на ранних сроках, оправдывает использование ВВИГ ex juvantibus. Положительный ответ на иммуномодулирующую терапию следует рассматривать как дополнительный диагностический критерий в пользу дизиммунного процесса.
Информированное согласие. Матерью пациента подписано информированное согласие на публикацию данных сына.
Авторы заявляют об отсутствии конфликта интересов.
The authors declare no conflicts of interest.
Сведения об авторах
Как цитировать:
Хроническая воспалительная демиелинизирующая полинейропатия (ХВДП)
(хроническая приобретенная демиелинизирующая полинейропатия; хроническая рецидивирующая полинейропатия)
, MDCM, New York Presbyterian Hospital-Cornell Medical Center
Хроническая воспалительная демиелинизирующая полинейропатия представляет собой иммунноопосредованную полинейропатию, которая характеризуется развитием симметричной слабости проксимальных и дистальных мышц и прогрессированием симптомов в течение > 2 месяцев.
Симптомы хронической воспалительной демиелинизирующей полинейропатии (ХВДП) напоминают симптомы при синдроме Гийена-Барре Синдром Гийена-Барре (СГБ) Синдром Гийена – Барре – это острая, обычно быстро прогрессирующая воспалительная полинейропатия, характеризующаяся мышечной слабостью и умеренным выпадением дистальной чувствительности и самоограничивающимся. Прочитайте дополнительные сведения . Однако прогрессирование заболевания более 2 месяцев отличает ХВДП от СГБ, который характеризуется монофазным и самоограничивающимся течением. ХВДП развивается у 2-5% пациентов с первоначальным диагнозом синдрома Гийена-Барре.
Симптомы и признаки ХВДП
ХВДП обычно начинается незаметно и может медленно ухудшаться или протекать по типу с наличием периодов рецидивов и восстановления; восстановление между рецидивами может быть частичным или полным. У большинства пациентов превалируют вялые парезы, обычно мышц конечностей; характерно то, что более выражеными являются сенсорные нарушения (например, парестезии в области кистей и стоп). Выпадают глубокие сухожильные рефлексы.
У большинства пациентов вегетативная функция поражена меньше, чем при синдроме Гийена-Барре, кроме того, слабость может быть асимметричной и прогрессировать медленнее, чем при синдроме Гийена-Барре.
Диагностика ХВДП
Анализ спинномозговой жидкости (СМЖ), и нейрофизиологическык исследования
Обследования включают анализ ликвора и нейрофизиологические исследования. Результаты исследований сходны с изменениями при синдроме Гийена-Барре включая белково-клеточную диссоциацию (повышение уровня белка при нормальном содержании лейкоцитов) и демиелинизирующее поражение нервов, выявляемое при нейрофизиологическом исследовании.
В редких случаях проводится биопсия нервов, при которой также выявляется демиелинизация.
Лечение ХВДП
Внутривенное введение иммуноглобулина (ВВИГ)
ВВИГ, хотя и более дорогой, часто предлагается в первую очередь пациентам с хронической воспалительной демиелинизирующей полинейропатией по следующим причинам:
Он не имеет так много побочных эффектов в сравнении с длительным применением кортикостероидов.
Он проще в использовании, чем плазмаферез.
Тем не менее последние данные свидетельствуют о том, что прерывистое употребление кортикостероидов может привести к более длительным ремиссиям и имеет более низкий уровень серьезных побочных эффектов, чем ВВИГ. Пульсовая терапия кортикостероидами может проводится в следующих режимах:
Дексаметазон 40 мг перорально в день в течение 4 дней подряд каждый месяц в течение 6 циклов
Дексаметазон перорально один раз в неделю в течение 3 месяцев с ежемесячной корректировкой дозы в зависимости от клинического статуса пациента
Внутривенно метилпреднизолон 500 мг один раз в день в течение 4 дней подряд каждый месяц в течение 6 месяцев
У некоторых пациентов положительный результат может наблюдаться при комбинированном применении ВВИГ с кортикостероидами.
Плазмаферез также не имеет долгосрочных побочных эффектов, характерных для кортикостероидов, но он часто требует постоянной катетеризации, а также из-за значительного перемещения жидкости может вызвать гипотонию. Пациентам, которые не отвечают на терапию Ig в/в или имеют тяжелые заболевания, могут назначать обмен плазмы, но, поскольку данная процедура инвазивна и сопряжена с риском, её лучше использовать только для купирования тяжелого клинического ухудшения, а не в качестве длительного поддерживающего лечения.
Подкожный иммуноглобулин (ПКИГ) может быть столь же эффективным, как и ВВИГ.
В некоторых случаях необходимо применение иммуносупрессивных препаратов (например, азатиоприна), которые также могут уменьшить потребность в кортикостероидах.
Лечение может проводиться в течение длительного времени.
Ключевые моменты
Хотя симптомы хронической воспалительной демиелинизирующей полинейропатии напоминают симптомы при синдроме Гийена-Барре, эти два заболевания могут быть дифференцированы на основании того, как долго симптомы продолжают прогрессировать (то есть, > 2 месяцев в случае ХВДП).
Симптомы начинаются незаметно и могут медленно ухудшаться или развиваются по типу с наличием периодов рецидивов и восстановления.
Исследование СМЖ и результаты электродиагностических тестов аналогичны результатам при синдроме Гийена-Барре.
Следует проводить терапию ВВИГ и кортикостероидами, а в тяжелых случаях, рассмотреть проведение плазмафереза; иммуносупрессанты могут быть эффективными, а также могут снизить зависимость от кортикостероидов.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Читайте также: