Мутации генов при миелодиспластических синдромах
Добавил пользователь Валентин П. Обновлено: 08.01.2026
Миелодиспластический синдром — это несколько сходных гематологических заболеваний, которые развиваются, как правило в старшем возрасте (в основном старше 60 лет). Основным проявлением МДС является цитопения. В костном мозге при этом обнаруживаются диспластические изменения. В некоторых случаях миелодиспластический синдром может переходить в острый лейкоз.
Несколько причин лежит в основе миелодиспластического синдрома. И хотя конкретные факторы могут быть разными, все они приводят к генетическим повреждениям клетки, сложным хромосомным нарушениям или гиперметилированию ДНК. В конечном итоге это вызывает нарушение пролиферации миелоидных клеток (появлению миелобластных клеток) как костного мозга, так и крови. Это приводит к их неправильному функционированию и соответствующей клинической симптоматике.
При миелодиспластическом синдроме генетические исследования необходимы не только для выявления конкретных генетических аномалий, но и для мониторинга прогрессирования заболевания.
К сожалению, более чем половина пациентов с миелодиспластическим синдромом имеют нормальный кариотип., что делает мониторинг заболеваний у этих пациентов затруднительным.
Пациенты с нормальным кариотипом, но агрессивным заболеванием могут иметь отклонения, которые не могут быть идентифицированы FISH или обычным цитогенетическим методом (кариотипирование). Хромосомный микроматричный анализ является методом выбора для этих пациентов.
С помощью этого метода можно обнаружить делеции, дупликации, несбалансированные транслокации, а также потерю гетерозиготности, которая часто является результатом мутаций и последующего отбора мутантных опухолевых генов-супрессоров и онкогенов.
Сравнительная характеристика обычного цитогенетического метода, FISH и хромосомного микроматричного анализа.
В сочетании с обычной цитогенетикой, повышает точность диагностики. FISH является более чувствительным, чем обычный цитогенетический анализ при обнаружении клональных аномалий.
Тест выбора для больных миелодиспластическим синдромом с нормальным кариотипом Позволяет обнаруживать очень мелкие изменения, которые могут определять прогноз и тактику лечение пациентов с миелодиспластическим синдромом, которые имеют нормальный кариотип..
Острый миелоидный лейкоз
Обычная классификация острого миелоидного лейкоза с помощью цитогенетического анализа не позволяет точно определить прогноз заболевания и, соответственно выбрать правильную тактику лечения у более чем 60% пациентов. AML-профайлер, тест на основе молекулярный микроматричной технологии, позволяет дать более точную классификацию у половины этих пациентов и отнести их к группе с благоприятным и неблагоприятным прогнозом.
AML-профайлер заменяет 7 отдельных тестов на основе 3-х различных технологий: цитогенетики, анализа мутаций и анализа экспрессии.
С помощью AML-профайлера можно определить транслокации t(8, 21),t(15, 17), инверсии inv(16), мутации NPM1 (типа A, B и D), а также двойную мутацию CEBPA. Кроме того, определяются уровни экспрессии 2 прогностических генов, EVI1 и BAALC. Мутация гена FLT3 не входят в AML-профайлер и может быть выполнена в нашей лаборатории отдельно.
Мутации генов при миелодиспластических синдромах
Мутации генов при миелодиспластических синдромах
Мутация гена CSF1R (синоним FMS) выявляется в 12—20 % случаев миелодиспластических синдромов (МДС), особенно при вариантах с увеличенным числом бластных клеток. Этот ген в норме кодирует рецептор макрофагального колониестимулирующиего фактора. Мутантный ген часто связан с неблагоприятным прогнозом как в отношении трансформации миелодиспластических синдромов (МДС) в ОМЛ, так и низкой выживаемости больных.
Мутация гена FLT3 (представленная внутренней тандемной дупликацией) диагностируется приблизительно в 10 % случаев миелодиспластических синдромов (МДС) и ОМЛ с трехростковой дисплазией. Этот ген кодирует III класс рецептора тирозинкиназы, участвующий в пролиферации и дифференцировке стволовых клеток. Мутация гена FLТЗ ассоциирована с трансформацией миелодиспластических синдромов (МДС) в ОМЛ и низкой продолжительностью жизни больных.
Гиперэкспрессия гена С-KIT, ответственного за синтез рецептора стволово-клеточного фактора, особенно характерна для вариантов миелодиспластических синдромов (МДС) с увеличенным числом бластных клеток в костном мозге.
Гиперэкспрессия гена MPL, кодирующего рецептор тромбопоэтина, встречается преимущественно у больных миелодиспластическими синдромами (МДС) с увеличенным числом бластных клеток и ассоциирована с неблагоприятным прогнозом, включая высокую вероятность трансформации миелодиспластических синдромов (МДС) в ОМЛ.
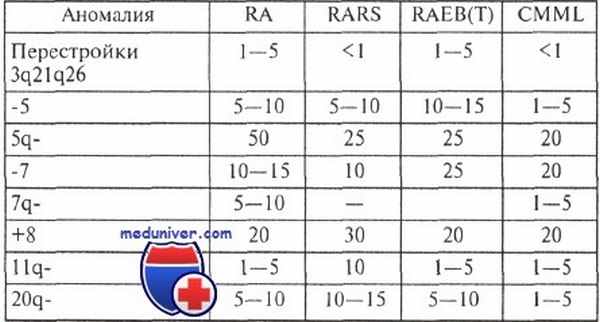
Частота (в процентах) характерных аномалий кариотипа при различных миелодиспластических синдромах
Точечная мутация гена GCSFR, вызывающая дефект рецептора G-CSF (гранулоцитарный колоние-стимулирующий фактор), является возможным объяснением развития миелодиспластических синдромов (МДС) и ОМЛ у больных с тяжелой врожденной нейтропенией (severe congenital neutropenia). Однако для возникновения миелодиспластических синдромов (МДС) или ОМЛ одной мутации гена GCSFR недостаточно. Развитие этих заболеваний ассоциировано с моносомией хромосомы 7 и мутацией гена RAS.
Мутации генов семейства RAS (наиболее часто гена NRAS) встречаются, по данным разных исследований, в 10—40 % случаев миелодиспластических синдромов (МДС), чаще при нарушениях миеломоноцитарной дифференцировки, например при хроническом миеломоноцитарном лейкозе. Мутация и гиперэкспрессия гена NRAS, участвующего в трансдукции сигнала с поверхности клетки, ассоциированы с повышенным риском развития ОМЛ и, возможно, являются одним из первых этапов лейкемической трансформации миелодиспластических синдромов.
Мутация гена опухолевой супрессии ТР53 (синоним Р53), кодирующего белок Р53, отмечается в 5— 25 % всех случаев миелодиспластических синдромов, особенно часто при вторичном характере МДС. Ген ТР53 в норме обеспечивает контроль целостности генома, и в случае повреждения ДНК либо происходит «арест» клеточного цикла в фазах G1 и G2, ли о активация апоптоза клеток. Существуют данные об увеличении частоты мутации гена ТР53 при «продвинутых» ФАБ-вариантах МДС — РАИБ и РАИБ-Т, а также при трансформации миелодиспластических синдромов (МДС) в ОМЛ.
Ген AML1 (синоним RUNX1) участвует в регуляции дифференцировки клеток миелоидного ростка кроветворения. Его мутация относится к одной из наиболее часто встречающихся мутаций при ОМЛ. При миелодиспластических синдромах мутация гена AML1 обнаруживается редко — в 5 % случаев.
Исследование Н. Harada и соавт. указывает на сопоставимую с данными предыдущей работы частоту мутации AML1 в случайной выборке больных МДС — 2,7 % и значительно более высокую — 46 % среди больных, подвергшихся воздействию малых доз облучения (выживших после атомной бомбардировки Хиросимы), а также — у 38 % больных вторичными миелодиспластическими синдромами и ОМЛ, ранее получавших терапию алкилирующими препаратами с локальным лучевым лечением и без такового лечения.
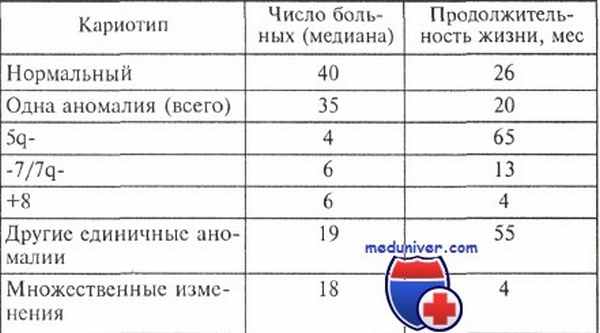
Продолжительность жизни больных миелодиспластическим синдромом с различными изменениями кариотипа
В патогенезе миелодиспластических синдромов задействовано множество других генов: реаранжировка гена MLL выявлена у 47 % больных, повышенная экспрессия гена WT1 — у 44 %, гена HFE — y 50 % [440], а также гена DLK1. Выявлены отсутствие или выраженное уменьшение экспрессии гена DCC.
При миелодиспластических синдромах (МДС), помимо мутаций генов, обнаружено гиперметилирование ДНК — присоединение метальной группы к цитозину. В настоящее время выявлено гиперметилирование четырех генов, регулирующих клеточный цикл: Р15INK4b, Р16INK4a, Р14ARF и гена ретинобластомы.
При миелодиспластических синдромах (МДС) наиболее изученным является ген р15INK4b. Его гиперметилирование отмечается у 26—63 % больных, причем не только в бластных клетках, но и в гранулоцитах крови. Гиперметилирование этого гена чаще определяется при вариантах МДС с увеличенным числом бластных клеток костного мозга (48 % больных), чем при вариантах с их нормальным числом (30 % больных).
Определенное внимание в патогенезе миелодиспластических синдромов уделяется повышенной активности теломеразы — рибонуклеиновому ферменту, содержащему фрагмент РНК, комплементарный ДНК теломеры (последовательности нуклеотидов, завершающих конец хромосом, которые стабилизируют их и предотвращают от разрушения). Повышенная активность теломеразы наряду с укорочением теломер может быть ранним маркером генетической нестабильности, приводящей в дальнейшем к возникновению хромосомных аберраций.
Точечная мутация митохондриальной ДНК и/или РНК может быть ранним этапом в патогенезе миелодиспластических синдромов. Мутации митохондриальной ДНК были выявлены в 40-60% случаев миелодиспластических синдромов. При рефрактерной анемии с кольцевыми сидеробластами выявлена мутация гена COII (митохондриальной ДНК) и генов 4MTIS1 и 4MTTD (митохондриальной тРНК).
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Мутации генов при миелодиспластических синдромах
Оценка роли молекулярно-генетических изменений в клиническом течении наследственных синдромов предрасположенности
к ОМЛ/МДС у детей
Инновационные подходы к терапии злокачественных лимфом у детей на основе современных методов геномной диагностики
Механизмы межклеточных взаимодействий и внутриклеточной сигнализации при иммунологических заболеваниях
Молекулярно-генетическая диагностика детей с синдромами предрасположенности к опухолевым заболеваниям
Особенности течения новой коронавирусной инфекции COVID-19 и формирования постинфекционного иммунитета у детей, получающих химио/иммуносупрессивную терапию, а также трансплантацию гемопоэтических стволовых клеток (ТГСК) в крупных гематологических клиниках России (преимущественно с диагнозом «острый миелобластный лейкоз»)
Оценка роли молекулярно-генетических изменений в клиническом течении наследственных синдромов предрасположенности к ОМЛ/МДС у детей
Развитие системы профессиональной психологической помощи семьям с детьми, успешно завершившими основной этап лечения онкологических, онкогематологических, гематологических заболеваний в специализированных клиниках России
Разработка и валидация метода диагностики нарушений гемостаза и тромбообразования в проточных камерах
Эффективность и безопасность сиролимуса у детей и подростков с ювенильной ангиофибромой основания черепа (ЮАОЧ)
к ОМЛ/МДС у детей
Н. С. Сметанина – Заместитель генерального директора-директор Управления по научно-аналитической работе с регионами, врач-гематолог НМИЦ ДГОИ им. Дмитрия Рогачева, д.м.н., профессор.
Д.В. Федорова – к.м.н., врач-гематолог НМИЦ ДГОИ им. Дмитрия Рогачева.
Врожденные синдромы с предрасположенностью к развитию первичного миелодиспластического синдрома/острого миелоидного лейкоза (СП МДС/ОМЛ) представляют собой клинически гетерогенную группу редких заболеваний с высоким риском развития онкогематологического заболевания, в основе которых лежит зародышевый (герминальный) молекулярно-генетический дефект.
Хорошо известно, что такие классические врожденные синдромы костномозговой недостаточности (СКМН), как анемия Фанкони, врожденный дискератоз, анемия Даймонда-Блекфена, анемия Швахмана-Даймонда и другие, имеют предрасположенность к развитию онкогематологических заболеваний. В последние годы накопились данные о новых наследственных СП МДС/ОМЛ, ассоциированных с герминальными мутациями в генах RUNX1, ANKRD24, ETV6, CEBPA, DDX41, GATA2, SRP7 и SAMD9/SAMD9L. С каждым годом перечень этих генов увеличивается. При последнем пересмотре классификации ВОЗ 2017 года, в 4ом издании эти синдромы были выделены в отдельные нозологические группы.
Часть синдромов предрасположенности к МДС/ОМЛ могут клинически не проявляться до дебюта собственно МДС/ОМЛ, а некоторые из них характеризуются наличием конституциональных особенностей, либо идиопатических моно-/панцитопений.
Несвоевременная диагностика СП МДС/ОМЛ может приводить к выбору неправильного лечения и резко ухудшать прогноз и качество жизни пациентов с этими синдромами. Более того, подходы к лечению онкогематологических заболеваний у пациентов с герминальными мутациями отличаются от лечения спорадически возникающих гемобластозов и требуют модификации стандартной терапии. В некоторых случаях возможно использование молекулярно-направленной терапии.
В случае генетически подтверждённого СП МДС/ОМЛ есть возможность провести преимплантационную/пренатальную диагностику для исключения повторного рождения ребенка с этим синдромом, а также эта информация крайне необходима при выборе родственного донора для проведения трансплантации гемопоэтических стволовых клеток (ТГСК).
1. Создание базы данных пациентов с синдромами предрасположенности к МДС/ОМЛ.
2. Исследование спектра гематологических и конституциональных проявлений у пациентов с СП МДС/ОМЛ.
3. Исследование спектра генетических причин (соматически/герминальных мутаций, хромосомных аберраций) при СП МДС/ОМЛ.
4. Провести анализ полученных результатов для получения клинико- генетических корреляций.
Длительность проекта: 01.12.2021 – 01.12.2025 гг.
По итогам проведенных исследований будет определена четкая взаимосвязь между установленными генетическими изменениями и клиническими проявлениями в данной группе пациентов, чтобы выработать сформулировать и обосновать единые принципы диагностики, лечения и реабилитации указанной категории пациентов.
Мутации генов при миелодиспластических синдромах
Мутации в гене эпигенетической регуляции ASXL1 часто (20,4%) встречаются при первичном миелофиброзе (ПМФ) независимо от наличия или отсутствия драйверной мутации.
Тройной негативный статус, наличие мутаций в генах JAK2, CALR, ASXL1 коррелируют с клинико-гематологическими проявлениями первичного миелофиброза - различные генетические нарушения способствуют формированию фенотипа опухолевой клетки. Появление мутаций TET2, DNMT3A, EZH2, ASXL1 и др. приводит к развитию более "продвинутых" форм МПН (миелопролиферативных новообразований), таких как ПМФ, и ухудшению прогноза. Дефекты гена ASXL1 ассоциированы с лейкоцитозом, бластозом, анемией, спленомегалией, конституциональными симптомами.
Синонимы русские
Миелофиброз, лейкемия, миелоидные злокачественные опухоли, миелодиспластический синдром, риск при трансплантации костного мозга, МПН, ПМФ.
Синонимы английские
PCR analysis of mutations in the gene ASXL1.
Метод исследования
Полимеразная цепная реакция в режиме реального времени.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
Название гена
OMIM
Локализация гена на хромосоме
Общая информация об исследовании
Ген ASXL1 кодирует ядерный белок, принадлежащий белковым комплексам, участвующим в эпигенетической регуляции экспрессии генов. Также он взаимодействует с компонентами комплекса PRC2 – EZH2 и SUZ12 в процессах, определяющих рост и дифференцировку организма. Мутации ASXL1 активируют ген HOXA, участвующий в возникновении и развитии лейкоза.
Из-за своей важной роли в регулировании синтеза белков, связанных с пролиферацией клеток, изменения в гене ASXL1 связаны с аномальным ростом клеток, характерным для онкологии. Наиболее часто данная патология обнаруживается у мужчин, у лиц старше 60 лет и у людей с наличием миелодиспластических синдромов (МДС) в роду.
Подавляющее большинство мутаций ASXL1 обнаружены при миелоидных злокачественных опухолях. Наиболее частой аномалией, которая составляет более половины всех мутаций ASXL1, представляет собой дублирование гуанинового нуклеотида. Мутации обычно гетерозиготные. "Поломка" ASXL1 встречается при миелопролиферативных новообразованиях (МПН), миелодиспластических синдромах, хроническом миеломоноцитарном лейкозе (ХММЛ) и острой миелоидной лейкемии (ОМЛ). Данная патология обнаруживается с частотой от нескольких процентов до более чем половины случаев в зависимости от заболевания. Чаще всего мутации встречаются при хроническом миеломоноцитарном лейкозе (45-50 %), при первичном миелофиброзе (30-35 %) и редко при полицитемии или эссенциальной тромбоцитемии. При остром миелоидном лейкозе, развившемся на фоне миелодиспластического синдрома или миелопролиферативного синдрома, они обнаруживаются у каждого третьего пациента. Известно, что при остром миелоидном лейкозе с нормальным кариотипом ASXL1-мутации являются взаимоисключающими с мутациями NPM1.
ASXL1 является вторым наиболее часто мутированным геном при миелодиспластических синдромах после TET2 и чаще встречается при рефрактерной анемии с избытком бластов (РАИБ), чем при других формах. Мутации ASXL1 также обнаруживаются в редких случаях ювенильной миеломоноцитарной лейкемии (ЮММЛ) и могут быть в первичных половых клетках (например, при синдроме Боринга - Опица, который приводит к смерти в раннем возрасте).
Анализ мутации ASXL1 - генетический тест, который обнаруживает аномалии в гене ASXL1 методом полимеразной цепной реакции. Мутации, как правило, связаны с агрессивным течением онкологического процесса, рефрактерностью к проводимому лечению и неблагоприятным прогнозом. Также положительное значение данного теста может быть показанием для исключения некоторых терапевтических препаратов в лечении острого миелоидного лейкоза, миелопролиферативных новообразований, миелодиспластических синдромов и хронического или ювенильного миеломоноцитарного лейкоза.
Мутации генов при миелодиспластических синдромах
Миелодиспластический синдром (МДС) сегодня является одной из самых сложных проблем гематологии. Лишь недавно лечение МДС вышло за рамки поддерживающей терапии, проводившейся с помощью облегчения симптомов. МДС является патологией старшей возрастной группы: 80 % случаев МДС приходится на лица старше 60 лет. МДС в детском возрасте встречается крайне редко. В европейских странах среди лиц 50—69 лет регистрируется 40 новых случаев МДС на 1 млн населения, а среди лиц 70 лет и старше — 150 новых случаев на 1 млн населения. Заболеваемость МДС в РФ в среднем составляет 3—4 случая на 100 тыс. населения в год и увеличивается с возрастом [1].
1.Рукавицын О. Гематология: национальное руководство / О. Рукавицын. – Москва: ГЭОТАР-Медиа, 2017. –С. 193-226.
2. Герминг У. Миелодиспластические синдромы: диагностика, прогноз, лечение / У. Герминг, Г. Коббе, Р. Хаас // Deutsches Ärzteblatt International. -2013. -№110(46). -90 с.
4. Иванага М. Риск миелодиспластических синдромов у людей, подвергшихся воздействию ионизирующего излучения: ретроспективное когортное исследование людей, переживших атомную бомбу Нагасаки / М. Иванга, М. Сода, Ю. Такасаки [и др.] // Журнал клинической онкологии. -2011. -№29(4). С. 34-42.
5. Каззола М. Экспрессия митохондриального ферритина в эритроидных клетках пациентов с сидеробластной анемией / М. Каззола, Р. Инверниззи, Дж. Бергамаши, С. Леви [и др.] // Журнал Кровь. -2003. -№101(5). –С. 1996-2000.
7. Бхатнагар Н. Транзиторный аномальный миелопоэз и острый миелоидный лейкоз при синдроме Дауна / Н. Бхатнагар, Л. Низери, О. Танстолл, П. Вьяс, И. Робертс // Текущие гематологические отчеты о злокачественных заболеваниях. -2016. -№11(5). –С. 33-41.
МДС – группа заболеваний со сложным патогенезом, который приводит к развитию диспластического кроветворения в сочетании с нормальным. Вначале симптомы обычно не проявляются. Позже симптомы могут включать чувство усталости, одышку, нарушения свертываемости крови, анемию или частые инфекции. Некоторые типы могут перерасти в острый миелоидный лейкоз [2].
Первичный (идиопатический) тип — 80—90 % случаев, вторичный (вследствие предшествующей химиотерапии и др. факторов) — 10—20 %. Большинство случаев МДС являются первичными — идиопатическими или de novo (с лат. — «вновь появившийся, новый»).
Вторичный МДС является значительно более неблагоприятным и резистентным к лечению типом МДС, обладающим заведомо более худшим прогнозом в сравнении с первичным МДС.
Признаки и симптомы неспецифичны и обычно связаны с цитопенией крови: анемия (хроническая усталость, одышка, ощущение холода, иногда боль в груди); нейтропения (повышенная восприимчивость к инфекции); тромбоцитопения (повышенная склонность к кровотечениям и экхимозам, а также к подкожным кровотечениям, приводящим к пурпуре или петехиям) [3].
Многие люди не имеют симптомов, и цитопения крови или другие проблемы выявляются при обычном анализе крови: нейтропения, анемия и тромбоцитопения; спленомегалия или редко гепатомегалия; аномальные гранулы в клетках, аномальная форма и размер ядер; хромосомная аномалия, включая хромосомные транслокации и аномальное количество хромосом
Хотя существует определенный риск развития острого миелоидного лейкоза, около 50% смертей происходит в результате кровотечения или инфекции. Однако лейкоз, возникающий в результате миелодисплазии, обычно не поддается лечению. На раннем этапе преобладает анемия. Большинство пациентов жалуются на постепенное наступление утомляемости и слабости, одышки и бледности, но, по крайней мере, у половины пациентов симптомы отсутствуют, и МДС обнаруживается лишь случайно при обычных анализах крови. Предшествующая химиотерапия или облучение являются важным фактором в истории болезни человека. Лихорадка и потеря веса должны указывать на миелопролиферативный, а не на миелодиспластический процесс. [3]
Некоторые люди в анамнезе подвергались химиотерапии (особенно алкилирующим агентам, таким как мелфалан, циклофосфамид, бусульфан и хлорамбуцил) или облучению (терапевтическому или случайному) или тому и другому (например, во время трансплантации стволовых клеток по поводу другого заболевания). Рабочие в некоторых отраслях промышленности, подвергающихся сильному воздействию углеводородов, таких как нефтяная промышленность, имеют несколько более высокий риск заражения этим заболеванием, чем население в целом. Воздействие ксилола и бензола было связано с миелодисплазией. Ветераны Вьетнама, подвергшиеся воздействию агент «оранж», рискуют заболеть МДС. Связь может существовать между развитием МДС «у людей, переживших атомную бомбу через 40-60 лет после облучения» (в данном случае имеется в виду людей, которые были в непосредственной близости от атомных бомб в Хиросиме и Нагасаки во время мировой войны. II). Дети с синдромом Дауна предрасположены к МДС, и семейный анамнез может указывать на наследственную форму сидеробластной анемии или анемии Фанкони [4].
МДС чаще всего развивается без видимой причины. Факторы риска включают воздействие агента, который, как известно, вызывает повреждение ДНК, такого как радиация, бензол и некоторые виды химиотерапии; о других факторах риска сообщалось непоследовательно. Доказать связь между предполагаемым воздействием и развитием МДС может быть сложно, но наличие генетических аномалий может предоставить некоторую подтверждающую информацию. Вторичный МДС может возникать как поздняя токсичность раковой терапии (МДС, ассоциированный с терапией, t-МДС). МДС после воздействия радиации или алкилирующих агентов, таких как бусульфан, нитрозомочевина или прокарбазин, обычно возникает через 3-7 лет после воздействия и часто демонстрирует потерю хромосомы 5 или 7. МДС после воздействия ингибиторов ДНК-топоизомеразы II возникает после более короткого латентного периода - всего 1–3 года и может иметь транслокацию 11q23. Другие ранее существовавшие заболевания костного мозга, такие как приобретенная апластическая анемия после иммуносупрессивного лечения и анемия Фанкони, могут перерасти в МДС.
Считается, что МДС возникает из-за мутаций в мультипотентных стволовых клетках костного мозга, но конкретные дефекты, ответственные за эти заболевания, остаются плохо изученными. Дифференциация клеток-предшественников крови нарушается, и в клетках костного мозга происходит значительное увеличение уровней апоптотической гибели клеток. Клональная экспансия аномальных клеток приводит к образованию клеток, утративших способность дифференцироваться. Если общий процент миелобластов костного мозга превышает определенный предел (20% для ВОЗ), то считается, что произошла трансформация в острый миелогенный лейкоз (ОМЛ). Прогрессирование МДС в ОМЛ - хороший пример многоэтапной теории канцерогенеза, в которой серия мутаций происходит в изначально нормальной клетке и превращает ее в раковую клетку.
Хотя признание лейкемической трансформации было исторически важным, значительная часть заболеваемости и смертности, связанных с МДС, является результатом не трансформации в ОМЛ, а, скорее, цитопений, наблюдаемых у всех пациентов с МДС. В то время как анемия является наиболее распространенной цитопенией у пациентов с МДС, учитывая доступность переливания крови, пациенты с МДС редко страдают от тяжелой анемии. Двумя наиболее серьезными осложнениями у пациентов с МДС в результате их цитопении являются кровотечение (из-за недостатка тромбоцитов) или инфекция (из-за недостатка лейкоцитов). Длительное переливание эритроцитов приводит к перегрузке железом.
Признание эпигенетических изменений в структуре ДНК при МДС объяснило успех двух (а именно гипометилирующих агентов 5-азацитидин и децитабин) из трех (третий - леналидомид) коммерчески доступных лекарств, одобренных Управлением по контролю за продуктами и лекарствами США для лечения МДС. Правильное метилирование ДНК имеет решающее значение для регуляции генов пролиферации, а потеря контроля метилирования ДНК может привести к неконтролируемому росту клеток и цитопении. Недавно одобренные ингибиторы ДНК-метилтрансферазы используют этот механизм, создавая более упорядоченный профиль метилирования ДНК в ядре гемопоэтических стволовых клеток, тем самым восстанавливая нормальные показатели крови и замедляя прогрессирование МДС до острого лейкоза [3].
Некоторые авторы предположили, что потеря митохондриальной функции с течением времени приводит к накоплению мутаций ДНК в гемопоэтических стволовых клетках, и это объясняет повышенную частоту МДС у пожилых пациентов. Исследователи указывают на накопление митохондриальных отложений железа в кольцевых сидеробластах как на доказательство митохондриальной дисфункции при МДС [5].
По крайней мере, с 1974 г. известно, что делеция в длинном плече хромосомы 5 связана с диспластическими аномалиями гемопоэтических стволовых клеток. К 2005 году леналидомид, химиотерапевтический препарат, был признан эффективным у пациентов с МДС с 5q-синдромом, а в декабре 2005 года FDA США одобрило этот препарат для этого показания. Пациенты с изолированным 5q-, низким риском IPSS и трансфузионной зависимостью лучше всего реагируют на леналидомид. Как правило, прогноз для этих пациентов благоприятный, средняя выживаемость составляет 63 месяца. Леналидомид имеет двойное действие, снижая количество злокачественных клонов у пациентов с 5q- и индуцируя лучшую дифференцировку здоровых эритроидных клеток, как это наблюдается у пациентов без делеции 5q [6].
Мутации в факторах сплайсинга были обнаружены в 40-80% случаев миелодиспластического синдрома, особенно у пациентов с кольцевидными сидеробластами.
Мутации в генах, кодирующих изоцитратдегидрогеназу 1 и 2 (IDH1 и IDH2), встречаются у 10–20% пациентов с миелодиспластическим синдромом и приводят к ухудшению прогноза при МДС низкого риска. Поскольку частота мутаций IDH1 / 2 увеличивается по мере увеличения злокачественности заболевания, эти данные вместе предполагают, что мутации IDH1 / 2 являются важными факторами прогрессирования МДС в более злокачественное состояние.
Преходящее миелопролиферативное заболевание - аномальная пролиферация клона доброкачественных мегакариобластов в печени и костном мозге. Заболевание ограничивается людьми с синдромом Дауна или генетическими изменениями, аналогичными таковым при синдроме Дауна, развивается во время беременности или вскоре после рождения и проходит в течение 3 месяцев, или примерно в 10% случаев прогрессирует до острого мегакариобластного лейкоза [7].
Читайте также: