Мышечная дистрофия с офтальмоплегией. Митохондриальные миопатии
Добавил пользователь Skiper Обновлено: 27.01.2026
Окулофарингеальная мышечная дистрофия (ОФМД) - заболевание, которое в мире встречается в двух генетических вариантах: аутосомно-рецессивном и аутосомно-доминантном. Эти варианты болезни являются аллельными и обусловлены различными мутациями в одном гене. Клинические варианты описаны в зависимости от типа наследования. Несмотря на то что, согласно литературным данным, ОФМД относится к более поздним формам мышечных дистрофий (4 – 5 –е десятилетие), имеется клинический пример раннего дебюта ОФМД у пациента в возрасте 23 года, с установленным диагнозом в 31 год.
При длительном течении заболевания - заметная гипотрофия нескольких групп мышц: лица, плечевого пояса, в т.ч. спины, конечностей. Часто появляются бронхо-легочные осложнения.
Средний возраст манифестации: 49 ± 1,42 лет
Особенность окулофарингеальной миодистрофии - наличие в ядрах нитевидных трубочек диаметром 8,5 нм. Аутосомно-доминантный и аутосомно-рецессивный варианты болезни обусловлены мутациями в одном и том же гене - РАВР2 (полиаденилсвязывающем протеине-2; OMIM 602 279), локализованном в области 14q11.2-q13. Основной тип мутаций - короткая экспансия тринуклеотидного повтора GCG в кодирующей части гена. В норме число повторов не превышает 6, однако у 2 % здоровых людей число повторов может достигать 7, что расценивается как проявление нормального полиморфизма. У больных с окулофарингеальной миопатией число повторов увеличено до 8–13.
Электрофореграмма сиквенса GCG – повторов
в 1 экзоне гена PABPN1
Результаты электрофореза ДНК на ДНК-анализаторе ABIPRISM 310
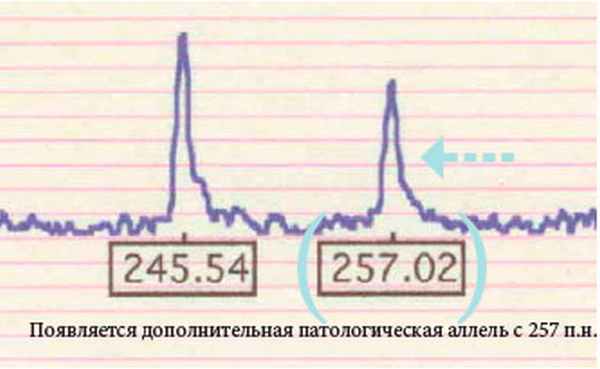
Тяжесть проявления заболевания зависит от количества повторов. Антиципация, обусловленная увеличением количества повторов, не характерна. Возникновение аутосомно-рецессивного варианта обусловлено гомозиготностью по GCG7-повтору, который является примером аллели-модификатора. Наиболее тяжелый фенотип наблюдался у компаунд-гетерозигот GCG9/GCG7, а также гомозигот по GCG9-повторам. Патогенетически белок РАВР2 является высоконсервативным и содержится в ядре, где участвует в полиаденилировании мРНК. GCG-повторы кодируют включение полиаланинового тракта вблизи N-конца мутантного белка. Считается, что образующиеся в ядре нитевидные структуры представлены удлиненными нитями мутантного белка. Заболевание особенно часто встречается у франко-язычных канадцев и в латиноамериканских семьях на юго-западе США. Оно описано также в большой еврейской семье восточноевропейского происхождения. На сегодняшний день для профилактики ОФМД возможна дородовая диагностика с использованием методов ДНК-анализа.
ДНК-диагностика ОФМД в 8% ПААГ:
(М - маркер PUC19/MspI)
1, 4 - генотип 6/6 GCG-повторов (здоровые).
2, 3 - генотип 6/10 GCG-повторов (больные).
Опасность окулофарингеальной миодистрофии состоит в прогрессировании с нарастающей дисфагией и требует применения методов паллиативной неврологии. Паллиативная неврология предусматривает зондовое питание или наложение стомы. Эффективного лечения на данный момент нет. Описаны методики рассечения перстнеглоточной мышцы для улучшения глотания, но не предотвращения аспирации. Если птоз мешает зрению, используют специальные скотчевые наклейки на веки, проволочные держатели век, которые крепятся к оправе очков, либо, если нет выраженной слабости мимических мышц, прибегают к хирургическому лечению.
Аутосомно-доминантный вариант заболевания (впервыеописан Victor и соавтор. в 1962 году у 9 членов одной семьи из трех поколений ). Первые симптомы возникают на 4–5-м десятилетиях жизни и в большинстве случаев характеризуются сочетанием дисфагии с прогрессирующим птозом верхних век. По мере прогрессирования заболевания отмечается распространение симптомов мышечной слабости на мышцы плечевого и тазового поясов. Также писана (Satoyoshi & Kinoshita, 1977) семья с аутосомно-доминантной сегрегацией окулофарингеальной миопатии, характеризующейся значительной генерализацией процесса по мере течения болезни. У наблюдаемых больных мышечная слабость распространялась на мышцы лица, шеи, дистальных отделов конечностей, а также анального сфинктера. Описаны единичные больные с наличием пигментной дегенерации сетчатки. Большинство авторов эту форму аутосомно-доминантной миопатии относят к довольно редким, возникающим в зрелом возрасте и медленнотекущим заболеваниям, считают, что клинически болезнь проявляет себя как локальная миопатия. Поражаются мышцы, осуществляющие движения глазных яблок, и мышца, поднимающая верхнее веко. Фарингеальные расстройства обусловлены включением в процесс констрикторов глотки, что затрудняет глотание. Типична симметричность процесса. Начальные признаки болезни появляются в возрасте 30–40 лет: двустороннее опущение верхнего века при ограничении движения глазных яблок. Как правило, на диплопию больные не жалуются. Объяснение этому находят в медленном и симметричном развитии парезов глазодвигательных мышц. Значительно утяжеляют заболевание и ухудшают прогноз фарингеальные симптомы. Начинаясь с дисфагии, они имеют тенденцию к нарушению функций (афагии). Следует иметь в виду существование окулярной миопатии, при которой фарингеальные расстройства не выражены. Этот вариант миопатии рассматривается одними исследователями как самостоятельное заболевание, другими- как дебют окулофарингеальной миопатии.
Аутосомно-рецессивный вариант заболевания (впервые описан Fried и соавт. в 1975 году у двух сестер, родившихся от кровнородственного брака). Для этой формы болезни характерно более раннее начало и вовлечение в процесс дистальных групп мышц конечностей. При (ЭМГ) электромиографическом обследовании больных с ОФМД определяют первично-мышечный характер поражения. При морфологическом исследовании выявляют нитевидные образования в ядрах скелетных мышц. Эти нити имеют ветвящуюся трубчатую структуру и иногда поперечно исчерчены. Наряду с этим отмечаются атрофические изменения в мышечных волокнах 1-го типа. При электронном микроскопировании обнаруживается увеличение размеров митохондрий с наличием в них крестовидных включений. Также могут быть обнаружены вакуоли, при электронной микроскопии в них видны обрывки мембран, скопления гликогена и другие неспецифичные остатки лизосомного происхождения.
Распространенность ОФМД у якутов в 10 раз выше распространенности описанной в мире. ОФМД получила накопление на территории улусов центральной Якутии и вилюйских улусов, особенно в Усть-Алданском улусе и городе Якутске.Выявлены мажорные мутации, характерные для якутской популяции: экспансия (GCG)10 в гене PÁBPN1 для окулофарингеальной миодистрофии. Распространенность аутосомно-доминантной окулофарингеальной миодистрофии в PC (Я) среди якутов составляет 11,1 на 100 тыс. населения с преимущественным накоплением в центральной и вилюйской группах улусов Якутии. Единственная молекулярно-генетическая причина всех случаев ОФМД в якутской популяции - мутация (GCG)m в 1 экзоне гена PABPN1. Выявленный единственный гаплотип является гаплотипом основателя.
Публикации в СМИ
Миопатии — обобщённое название множества заболеваний мышечной системы (главным образом, скелетной мускулатуры), обусловленных нарушением сократительной способности мышечных волокон и проявляющихся мышечной слабостью, уменьшением объёма активных движений, снижением тонуса, атрофией, иногда псевдогипертрофией. Точная диагностика миопатий сложна, во многих случаях необходима биопсия мышц, практически во всех случаях обязателен углублённый семейный анамнез. Классификация миопатий запутана, многие нозологические единицы имеют эпонимические, слишком общие и частично совпадающие наименования. Обычно выделяют первичные (наследственные) и вторичные миопатии. Первые включают в себя врождённые структурные миопатии (см. ниже) и прогрессирующие мышечные дистрофии (см. Дистрофии мышечные, Дистрофия мышечная Дюшенна). Вторые объединяют поражения мышц при различные системных заболеваниях (коллагенозы, эндокринные нарушения и пр., см. также Миопатии метаболические).
Генетические аспекты. Типы, гены и дефект: • недостаточность карнитин пальмитоилтрансферазы II, 255110, CPT2, 600650, 1p32; • недостаточность сукцинат дегидрогеназы, SDH1, 185470, 1p22.1 qter; • недостаточность фосфоглицерат мутазы, PGAM2, PGAMM, 261670, 7p13 p12.3; • Броди, 601003, ATP2A1, SERCA1, 108730, 16p12; • врождённая доброкачественная, 158810, ген коллагена COL6A1, 120220, 21q22.3; • врождённая доброкачественная, 158810, ген коллагена COL6A3, 120250, 2q37; • дистальная, MPD1, 160500, 14q; • миотубулярная (центронуклеарная) X сцепленная, MTM1, MTMX, 310400, Xq28; • Миоши, 254130, LGMD2B, 253601 (тазово плечевая дистрофия мышечная), 2p13.3 p13.1; • немалиновая, тип 1, 161800, NEM1, 191030 (тропомиозин 3, TPM3), 1q22 q23; • немалиновая, тип 2, 256030, NEM2, 191030 (тропомиозин 3, TPM3), 1q22 q23; • с тельцами включения (), IBM2, 601073, 9p1 q1; • 125660, ген десмина DES, 2q35.
Некоторые формы врождённых миопатий
• Немалиновая — непрогрессирующая мышечная слабость, чаще поражающая проксимальные мышцы. В мышечных волокнах характерные палочковидные и нитеобразные стержни (элементы Z-линий). Синонимы: миопатия врождённая непрогрессирующая, миопатия нитеобразная.
• Центронуклеарная (миотубулярная) — медленно прогрессирующие мышечная слабость и атрофия, начинающиеся в детском возрасте; ядра большинства мышечных волокон локализованы преимущественно в центральной части, нежели по периферии мышечных волокон.
• Болезнь центрального стержня — медленно прогрессирующая слабость мышц; при биопсии в сердцевине мышечных волокон отсутствуют митохондрии и элементы саркоплазматической сети, активность окислительных ферментов, фосфорилазы, АТФазы; миофибриллы расположены в виде компактных образований.
• Митохондриальные — разнообразная группа редко встречающихся заболеваний, обусловленных дефектами митохондриального и/или ядерного генома. На первый план выступают прогрессирующие мышечная слабость и утомляемость, часты миокардиодистрофии, симптоматика со стороны ЦНС (задержка психомоторного развития, деменция, интермиттирующая кома, судорожные припадки), а также симптомы поражения различных органов и систем. Диагностика затруднена, обязательно включает биопсию мышц.
• Митохондриальные энцефаломиопатии — разнородная группа заболеваний с общим морфологическим признаком — нарушением структуры митохондрий в мышцах и ЦНС, но различными молекулярными дефектами в виде нарушений транспорта субстратов из цитозоля в митохондрии, утилизации субстратов, структуры ферментов цикла Кребса, сопряжения окисления и фосфорилирования.
Клиническая картина • Сходная для всех врождённых миопатий • Дети рождаются с низким мышечным тонусом и слабостью лицевой мускулатуры • Заболевание обычно не прогрессирует или регрессируют с возрастом • Иногда развивается дыхательная недостаточность • В подростковом периоде, как правило, развивается сколиоз. Диагноз устанавливают по результатам биопсии мышц.
Лечение • В первые месяцы жизни ребёнка могут потребоваться борьба с дыхательными расстройствами и питание через зонд • Позже возникает необходимость в ортопедической коррекции, лечебной гимнастике, физиотерапии и рациональном трудоустройстве.
МКБ-10 • G71.2 Врождённые миопатии • G71.3 Митохондриальная миопатия, не классифицированная в других рубриках • G72 Другие миопатии
OMIM • 160560, 252010 Митохондриальная миопатия с гигантскими митохондриями • 251945 Митохондриальная миопатия с дефектами транспорта белка в митохондриях • 251950 Митохондриальная миопатия и молочнокислый ацидоз • 160550 Митохондриальная миопатия с ранней катарактой • 161800, 256030 Миопатия немалиновая • 191030 Миопатия немалиновая • 160500, 160300, 254130 Миопатия поздняя дистальная • 160150, 255200, 310400 Миопатия центронуклеарная • 117000 Болезнь центрального стержня • 160200 Миопатия врождённая с внутриядерными включениями кристаллина E • 160570 Миопатия с отложением гликопротеинов и гликозаминогликанов • 254940 Синдром Карей–Файнман–Зитера • 254950 Миопатия грануловакуолярная дольковая с миотонией • 254960 Миопатия вследствие дефекта малат-аспартатного шунта • 255100 Миопатия с нарушением метаболизма липидов • 255160 Миопатия с лизисом миофибрилл типа I • 255300 Миопатия врождённая Бюттена–Тёрнера • 255310 Миопатия врождённая с диспропорцией волокон • 255320 Миопатия врождённая многоцентровая с наружной офтальмоплегией.
Код вставки на сайт
Миопатии
Миопатии — обобщённое название множества заболеваний мышечной системы (главным образом, скелетной мускулатуры), обусловленных нарушением сократительной способности мышечных волокон и проявляющихся мышечной слабостью, уменьшением объёма активных движений, снижением тонуса, атрофией, иногда псевдогипертрофией. Точная диагностика миопатий сложна, во многих случаях необходима биопсия мышц, практически во всех случаях обязателен углублённый семейный анамнез. Классификация миопатий запутана, многие нозологические единицы имеют эпонимические, слишком общие и частично совпадающие наименования. Обычно выделяют первичные (наследственные) и вторичные миопатии. Первые включают в себя врождённые структурные миопатии (см. ниже) и прогрессирующие мышечные дистрофии (см. Дистрофии мышечные, Дистрофия мышечная Дюшенна). Вторые объединяют поражения мышц при различные системных заболеваниях (коллагенозы, эндокринные нарушения и пр., см. также Миопатии метаболические).
Генетические аспекты. Типы, гены и дефект: • недостаточность карнитин пальмитоилтрансферазы II, 255110, CPT2, 600650, 1p32; • недостаточность сукцинат дегидрогеназы, SDH1, 185470, 1p22.1 qter; • недостаточность фосфоглицерат мутазы, PGAM2, PGAMM, 261670, 7p13 p12.3; • Броди, 601003, ATP2A1, SERCA1, 108730, 16p12; • врождённая доброкачественная, 158810, ген коллагена COL6A1, 120220, 21q22.3; • врождённая доброкачественная, 158810, ген коллагена COL6A3, 120250, 2q37; • дистальная, MPD1, 160500, 14q; • миотубулярная (центронуклеарная) X сцепленная, MTM1, MTMX, 310400, Xq28; • Миоши, 254130, LGMD2B, 253601 (тазово плечевая дистрофия мышечная), 2p13.3 p13.1; • немалиновая, тип 1, 161800, NEM1, 191030 (тропомиозин 3, TPM3), 1q22 q23; • немалиновая, тип 2, 256030, NEM2, 191030 (тропомиозин 3, TPM3), 1q22 q23; • с тельцами включения (), IBM2, 601073, 9p1 q1; • 125660, ген десмина DES, 2q35.
Некоторые формы врождённых миопатий
• Немалиновая — непрогрессирующая мышечная слабость, чаще поражающая проксимальные мышцы. В мышечных волокнах характерные палочковидные и нитеобразные стержни (элементы Z-линий). Синонимы: миопатия врождённая непрогрессирующая, миопатия нитеобразная.
• Центронуклеарная (миотубулярная) — медленно прогрессирующие мышечная слабость и атрофия, начинающиеся в детском возрасте; ядра большинства мышечных волокон локализованы преимущественно в центральной части, нежели по периферии мышечных волокон.
• Болезнь центрального стержня — медленно прогрессирующая слабость мышц; при биопсии в сердцевине мышечных волокон отсутствуют митохондрии и элементы саркоплазматической сети, активность окислительных ферментов, фосфорилазы, АТФазы; миофибриллы расположены в виде компактных образований.
• Митохондриальные — разнообразная группа редко встречающихся заболеваний, обусловленных дефектами митохондриального и/или ядерного генома. На первый план выступают прогрессирующие мышечная слабость и утомляемость, часты миокардиодистрофии, симптоматика со стороны ЦНС (задержка психомоторного развития, деменция, интермиттирующая кома, судорожные припадки), а также симптомы поражения различных органов и систем. Диагностика затруднена, обязательно включает биопсию мышц.
• Митохондриальные энцефаломиопатии — разнородная группа заболеваний с общим морфологическим признаком — нарушением структуры митохондрий в мышцах и ЦНС, но различными молекулярными дефектами в виде нарушений транспорта субстратов из цитозоля в митохондрии, утилизации субстратов, структуры ферментов цикла Кребса, сопряжения окисления и фосфорилирования.
Клиническая картина • Сходная для всех врождённых миопатий • Дети рождаются с низким мышечным тонусом и слабостью лицевой мускулатуры • Заболевание обычно не прогрессирует или регрессируют с возрастом • Иногда развивается дыхательная недостаточность • В подростковом периоде, как правило, развивается сколиоз. Диагноз устанавливают по результатам биопсии мышц.
Лечение • В первые месяцы жизни ребёнка могут потребоваться борьба с дыхательными расстройствами и питание через зонд • Позже возникает необходимость в ортопедической коррекции, лечебной гимнастике, физиотерапии и рациональном трудоустройстве.
МКБ-10 • G71.2 Врождённые миопатии • G71.3 Митохондриальная миопатия, не классифицированная в других рубриках • G72 Другие миопатии
OMIM • 160560, 252010 Митохондриальная миопатия с гигантскими митохондриями • 251945 Митохондриальная миопатия с дефектами транспорта белка в митохондриях • 251950 Митохондриальная миопатия и молочнокислый ацидоз • 160550 Митохондриальная миопатия с ранней катарактой • 161800, 256030 Миопатия немалиновая • 191030 Миопатия немалиновая • 160500, 160300, 254130 Миопатия поздняя дистальная • 160150, 255200, 310400 Миопатия центронуклеарная • 117000 Болезнь центрального стержня • 160200 Миопатия врождённая с внутриядерными включениями кристаллина E • 160570 Миопатия с отложением гликопротеинов и гликозаминогликанов • 254940 Синдром Карей–Файнман–Зитера • 254950 Миопатия грануловакуолярная дольковая с миотонией • 254960 Миопатия вследствие дефекта малат-аспартатного шунта • 255100 Миопатия с нарушением метаболизма липидов • 255160 Миопатия с лизисом миофибрилл типа I • 255300 Миопатия врождённая Бюттена–Тёрнера • 255310 Миопатия врождённая с диспропорцией волокон • 255320 Миопатия врождённая многоцентровая с наружной офтальмоплегией.
Мышечная дистрофия с офтальмоплегией. Митохондриальные миопатии
Мышечная дистрофия с офтальмоплегией. Митохондриальные миопатии
Мышечная дистрофия с офтальмоплегией описана у новорожденных Е. К. Hurwitz и др. в 1969 г., тип наследования — аутосомно-рецессивный.
При гистологическом исследовании биопсированной мышцы обнаруживают изменения, характерные для первичного миодистрофического процесса: центрально расположенные ядра, их вакуолизацию, гиалиновую дегенерацию и атрофию мышечных волокон.
Симптомы поражения мышц появляются в первые месяцы жизни или пепосредствено после рождения. Ограничен объем движения глазных яблок, иногда вплоть до полной офтальмоплегии. Мышечный тонус скелетных мышц снижен, дети вяло сосут, крик тихий. В дальнейшем функция мышц несколько улучшается, но офтальмоплегия и слабость проксимальных групп мышц конечностей остаются постоянными симптомами. Течение заболевания — медленно прогрессирующее.
Митохондриальные миопатии
К врожденным миопатиям относится также группа так называемых митохондриальных миопатии, клинически отличающихся от выше описанных главным образом прогредиентностью течения.
Мегаконеальная миопатия описана G. Shy и N. Gonatas в 1964 г., наследуется аутосомно-рецессивно. Гистохимически и при электронной микроскопии в мышцах выявляются митохондрии, увеличенные до 1—2\i в диаметре, особенно в волокнах I гистохимического типа. Каких-либо других изменений, указывающих на дистрофический процесс в скелетной мышце, не обнаруживают.
Заболевание быстро прогрессирует и в течение нескольких лет приводит к почти полной обездвиженности больных. Угасают сухожильные рефлексы, развиваются мышечные атрофии и деформации скелета. Сердечная мышца остается интактной.
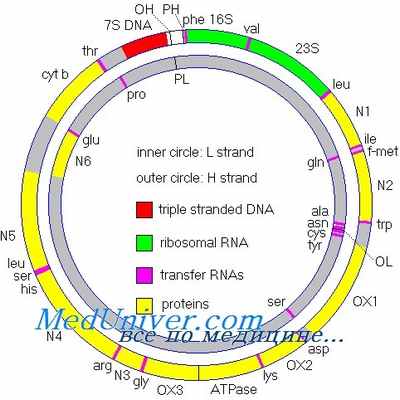
Структура ДНК митохондрий (мтДНК)
Мегаконеальная миопатия с гиперметаболизмом описана A. Ernster и др. в 1959 г., наследуется аутосомно-рецессивно. Гистологически определяются умеренные дегенеративные изменения в мышечных волокнах. При электронной микроскопии выявляется большое количество митохондрий различной величины с необычными зигзагообразными или шаровидными включениями и измененной внутренней структурой. В митохондриях повышены общее содержание белка, и активность цитохромоксидазы. В изолированных митохондриях определяется гиперметаболизм вследствие нарушения фосфатных акцепторов и повышения активности эндогенной магнийзависимой АТФ. Помимо мышечных нарушений у детей отмечаются гипотрофия несмотря на повышенный аппетит и жажда без полиурии.
Плеокониальная миопатия описана G. Shy и др. в 1966 г., наследуется аутосомно-рецессивно. При цитохимическом исследовании мышцы выявляется большое количество митохондрий нормального размера, нарушающих фибриллярную архитектонику мышечной клетки. Встречаются также волокна с центрально расположенными ядрами, умеренно выражено разрастание соединительной ткани.
На фоне имеющейся мышечной слабости у больных эпизодически наблюдаются приступы жажды, рвоты, профузного пота. Во время приступа мышечная слабость усиливается вплоть до вялой тетраплегии с отсутствием сухожильных рефлексов. Клинически заболевание напоминает периодический нормокалиемический паралич, но отличается отсутствием терапевтического эффекта при введении солей натрия. Течение прогредиентное, однако с возрастом темп прогрессирования несколько замедляется.
При всех врожденных миопатиях изменения на ЭМГ указывают на первично-мышечный характер патологии. Заболевания этой группы следует дифференцировать от спинальной амиотрофии Верднига — Гоффмана, травмы шейного отдела позвоночника в родах, врожденных миелодисплазий; болезней, обусловленных нарушением обмена аминокислот и липидов; травматических невритов и инфекционных поражений нервной системы. Дифференциальный диагноз проводится на основании гистологического исследования биопсированных мышц. Лечение аналогично таковому при болезни Верднига — Гоффмана.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Митохондриальные заболевания, комплексная диагностика: митохондриальная ДНК, ч. м. (Mitochondrial Diseases, multiplex mutations detection assay)
Митохондриальные заболевания клинически представляют собой гетерогенную группу состояний, развивающихся в связи с нарушением работы ферментов дыхательной цепи митохондрий. Нарушения могут быть связаны как с мутациями, появляющимися в геноме человека, так и с мутациями в митохондриальной ДНК (мтДНК). Наиболее часто наблюдаются повреждения мтДНК, которые характеризуются протяженными делециями и дупликациями, а также точечными мутациями (чаще всего 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C).
Митохондриальные заболевания могут манифестировать в любом возрасте. При некоторых митохондриальных заболеваниях поражается только один орган (наследственная оптическая нейропатия Лебера), однако в большинстве данная группа состояний имеет системные проявления, чаще всего связанные с поражением нервной и мышечной системы. Довольно часто клинические проявления митохондриальных заболеваний могут подпадать под специфические синдромы: MELAS-синдром (митохондриальная миопатия, энцефалопатия, лактат-ацидоз, инсультоподобные эпизоды), MERRF-синдром (миоклоническая эпилепсия, ассоциированная с порванными красными волокнами), прогрессирующая наружная офтальмоплегия, синдром Кернса-Сейра, синдром Лея, нейрогенная слабость с атаксией и пигментным ретинитом. Однако довольно много клинических случаев митохондриальных заболеваний не подпадают ни под одну из перечисленных категорий, имея перемежающийся спектр проявлений и симптомов, что возможно объясняется гетероплазмией мутаций в мтДНК (вариация количества «мутантных» митохондрий в одной клетке).
Частыми клиническими проявлениями митохондриальных заболеваний являются блефароптоз, офтальмоплегия, проксимальная миопатия, кардиомиопатия, нейросенсорная тугоухость, атрофия глазного нерва, пигментная ретинопатия, энцефалопатия, судороги, деменция, мигрень, инсультоподобные эпизоды, атаксия, спастичность.
Мутации в мтДНК могут передаваться по материнской линии. При наличии мутаций в мтДНК мужчины риск передачи аберрации детям отсутствует. Делеции в мтДНК чаще всего возникают de novo (единственный случай в семье), тогда как точечные аберрации и дупликации часто передаются по материнской линии.
Специальной подготовки не требуется. Взятие крови желательно проводить не ранее 4 часов после приема пищи.
- подозрение на митохондриальную патологию;
- дифференциальный диагноз синдрома Девика (оптическая нейропатия);
- дифференциальный диагноз миопатии;
- дифференциальный диагноз энцефалопатии, судорожного синдрома.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Делеций и дупликаций митохондриальной ДНК, а также мутаций 3243A>G, 3460G>A, 8344A>G, 11778G>A, 14484T>C обнаружено не было.
В связи с тем, что одна и та же мутация может вызывать различные клинические проявления у разных пациентов, иногда сложно определить точно корреляцию генотипа и фенотипа. Однако считается, что делеции и дупликации мтДНК чаще всего встречаются при синдроме Кернса-Сейра, прогрессирующей офтальмоплегии и синдроме Пирсона. Мутация 3243A>G характерна для синдрома MELAS, 11778G>A, 14484T>C, 3460G>A - для наследственной оптической нейропатии Лебера Отрицательный результат исследования не исключает диагноз митохондриального заболевания в связи с возможностью наличия менее распространенных точечных мутаций, а также появления генетических аберрации в участках геномной ДНК, кодирующих митохондриальные белки.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации. ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
При миопатии Дюшенна нарушается поглощение митоходриями ионов кальция
Ученые из Марийского государственного университета, Института теоретической и экспериментальной биофизики и Института биофизики клетки показали на мышах, что при мышечной дистрофии Дюшенна происходят структурные перестройки энергетических станций клетки — митохондрий. В результате нарушается транспортировка ионов кальция, которые участвуют в регуляции мышечного сокращения. Этот механизм, вероятно, способствует развитию болезни, но он же может послужить мишенью для ее терапии. Работа выполнена в рамках Президентской программы исследовательских проектов Российского научного фонда. Статья с итогами работы опубликована в журнале Biochimica et Biophysica Acta — Molecular Basis of Disease, кратко о них рассказывается в пресс-релизе РНФ.
Миопатия (или миодистрофия) Дюшенна — тяжелое генетическое заболевание, которым болеют, в основном, мальчики: примерно один из пяти тысяч. В Москве ежегодно рождается примерно 10 детей с таким нарушением, а в России — около 100. Болезнь дает о себе знать еще в раннем детстве и постепенно прогрессирует. Это проявляется в снижении плотности костей, ограничении подвижности суставов. У страдающих этим заболеванием плохая осанка, увеличенные икры, нарастающая слабость, нарушение сердечного ритма, они чаще остальных ломают руки и ноги. Уже в подростковом возрасте человек теряет способность к самостоятельному передвижению, а после возникает сердечная и дыхательная недостаточность. Большинство пациентов умирают в 15–25 лет.
Причина заболевания кроется в мутации гена белка дистрофина. Он отвечает за связь мышечных волокон с внеклеточным матриксом, окружающим клетки. Из-за нарушения в структуре соответствующего гена патологически изменяется структура и функции белка. Развитие болезни Дюшенна также тесно связано с нарушением работы митохондрий — энергетических станций клетки.
«Отечественными учеными регулярно проводятся исследования, направленные на клиническую оценку митохондриальных изменений при миодистрофиях. Удалось выявить устойчивые пути патоморфологических изменений, в том числе связанных с митохондриями. Известно, что эти органеллы не только обеспечивают клетку энергией, но и регулируют содержание кальция и, следовательно, работу основных двигательных белков мышцы», — рассказывает Михаил Дубинин, кандидат биологических наук, доцент кафедры биохимии, клеточной биологии и микробиологии Марийского государственного университета (Йошкар-Ола).
Ученые исследовали состояние структур митохондрий, связанных с транспортом кальция в митохондриях скелетных мышц. Для этого использовали мышей с точечной мутацией в гене дистрофина, а также здоровых животных в качестве контроля. Сначала проводили тест на струне: мышь висит на жесткой проволоке, держась за нее передними лапами, ее задача — провисеть 30 секунд. В зависимости от результата прохождения теста ученые делали вывод о ее физической силе и выносливости. Для изучения структуры митохондрий готовили препараты мышечной ткани бедра и фиксировали их в специальном растворе. Затем образцы обезвоживали концентрированными спиртами и помещали в капсулу со смолой. Ультратонкие срезы мышечной ткани наблюдали под электронным микроскопом. На следующем этапе исследовали, как митохондрии поглощают, высвобождают и удерживают ионы кальция. Оказалось, что при развитии мышечной дистрофии Дюшенна в этих органеллах меняются структура и роль систем, ответственных за транспорт ионов кальция. С одной стороны, снижается эффективность накопления кальция, с другой — митохондрии намного хуже удерживают ионы.

Картинка: Михаил Дубинин проводит тест на струне. Источник: Михаил Дубинин
В экспериментах исследователи оценивали скорость притока кальция в митохондрии по времени поглощения ионов органеллами. Оказалось, что у животного с мутацией эта скорость снижена более чем в два раза по сравнению со здоровыми. Кроме того, митохондрии больных мышей способны поглощать гораздо меньше кальция, чем здоровые животные. Это чревато нарушением проницаемости мембраны митохондрии, из-за чего может начаться высвобождение веществ, запускающих программу клеточной гибели.
«Мы обнаружили перестройки в кальций-транспортирующих системах митохондрий. Из-за них ион плохо аккумулируется в этих органеллах. Это, в свою очередь, способствует значительному увеличению уровня свободного внутриклеточного кальция в мышечной ткани и драматически ускоряет развитие болезни, — говорит Михаил Дубинин. — Также мы полагаем, что, воздействуя на кальций-транспортирующие системы митохондрий, возможно корректировать протекание этого нервно-мышечного заболевания. Такой метод позволит разработать новые подходы к терапии болезни Дюшенна».
Читайте также: