Наследственные двигательные невропатии с предрасположенностью к параличам от сдавливания нерва
Добавил пользователь Валентин П. Обновлено: 27.01.2026
ГлавнаяHelixbook Детекция дупликации/делеции гена РМР22 при болезни Шарко-Мари-Тута 1А и наследственной нейропатии с подверженностью параличу от сдавления
Детекция дупликации/делеции гена РМР22 при болезни Шарко‑Мари‑Тута 1А и наследственной нейропатии с подверженностью параличу от сдавления
Молекулярно-генетическое исследование, позволяющее обнаружить в генетическом материале пациента определенные мутации, играющие основную роль в развитии некоторых наследственных заболеваний периферических нервов.
Синонимы русские
Исследование мутаций гена PMP22.
Синонимы английские
PMP22 (Duplication/Deletion) DNA Test, Charcot-Marie-Tooth disease type 1A (CMT1A), Hereditary Neuropathy with liability to Pressure Palsies (HNPP).
Метод исследования
Полимеразная цепная реакция (ПЦР).
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Наследственные моторно-сенсорные невропатии представляют собой группу врождённых заболеваний, общей чертой которых является поражение периферических нервов. В составе периферических нервов проходят сенсорные и моторные нервные волокна. Сенсорные нервные волокна проводят сигналы от болевых, тактильных и температурных рецепторов, а моторные – двигательные импульсы к мышцам. Поскольку при моторно-сенсорных невропатиях поражаются оба типа нервных волокон, эти заболевания проявляются как сенсорными нарушениями, такими как онемение и снижение чувствительности, так и моторными (мышечная слабость, потеря мышечной массы – атрофия мышц).
Передача сигнала в нервной системе подобна проведению электричества по проводам. При этом непосредственно проводник – нервное волокно – заключено в специальную изолирующую оболочку, состоящую из миелина (защитного вещества, которое покрывает нервы и способствует эффективной передаче нервных импульсов). Повреждение миелиновой оболочки приводит к замедлению проведения импульса по нерву. Основным компонентом миелина является периферический миелиновый белок 22 (англ. peripheral myelin protein 22, PMP22). Ген, кодирующий последовательность аминокислот этого белка, находится на коротком плече семнадцатой хромосомы. Соответственно, мутации в участке 17-й хромосомы, где расположен ген РМР22, приводят к нарушениям образования миелина и, как следствие, демиелинизации нервных волокон периферических нервов, что приводит к клиническим проявлениям полиневропатий.
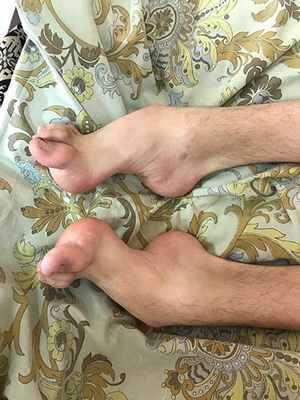
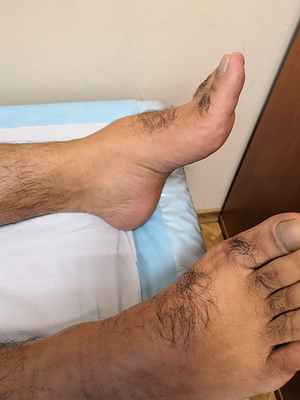
Болезнь Шарко - Мари - Тута типа 1А - наследственная моторно-сенсорная невропатия, обусловленная дупликацией (дублированием) гена PMP22 в 17-й хромосоме. В норме у человека должно быть две копии гена (по одной на каждой из двух хромосом), но в случае дупликации, то есть удвоения участка хромосомы, кодирующего этот белок, их становится три. Соответственно, возникает избыточный синтез белка PMP22, что приводит к увеличению числа слоев миелиновой оболочки с одновременным формированием демиелинизированных участков. В дальнейшем при морфологическом исследовании биоптата пораженного нерва находят рассеянную скудность миелина. Механизм демиелинизации при этом на данный момент не совсем ясен. Заболевание манифестирует в первые две декады жизни (чаще всего между 10 и 20 годами). Первым симптомом является слабость в ногах, приводящая к изменениям походки. В дальнейшем происходит атрофия мышц голеней, деформация стоп. Несколько позже в процесс вовлекаются мышцы кистей и предплечий. Сенсорные нарушения выражены меньше. Проявления заболевания у разных пациентов, даже внутри одной семьи, могут сильно различаться.
Наследственная невропатия с предрасположенностью к компрессионному параличу – также является заболеванием из группы наследственных полинейропатий, она связана с мутацией в 17-й хромосоме, однако в этом случае наблюдается делеция (то есть потеря) гена PMP22. Потеря одной копии гена уменьшает количество вырабатываемого белка PMP22, соответственно, наполовину. По данным различных исследований, белок PMP22 особенно важен для защиты нервных волокон от физического давления, он помогает им восстанавливать структуру после сжатия. Способность нервов восстановиться от нормальной, возникающей до нескольких раз в день компрессии (например, сидение в течение длительного времени) предохраняет конечности от постоянной потери ощущений. Недостаток белка PMP22 может влиять на структуру миелинового покрытия, нарушая передачу нервных импульсов. Кроме того, потеря этого белка, по-видимому, делает нервы менее способными к восстановлению после сжатия, что также прерывает нервную сигнализацию, вызывая признаки и симптомы данного заболевания. Первые симптомы появляются обычно во втором или третьем десятилетии жизни. Человека беспокоят безболезненные приступы онемения, мышечной слабости, как правило, возникающие после сдавления нерва. В половине случаев симптомы регрессируют в течение нескольких дней. Отмечается большая вариабельность клинических проявлений у разных пациентов.
Клинические проявления данных заболеваний имеют некоторые различия, но тем не менее не всегда позволяют с уверенностью провести дифференциальный диагноз. Электронейромиографические методики также позволяют определеить лишь группу полинейропатий, разграничивая поражение непосредственно нервного волокна и процессы демиелинизации. Появление молекулярно-генетического исследования для выявления мутации гена PMP22 позволило существенно повысить достоверность диагностики этих заболеваний.
Для чего используется исследование?
- Для выявления определенных мутаций гена PMP22 в 17-й хромосоме, специфичных для болезни Шарко - Мари - Тута типа 1А и наследственной невропатии с предрасположенностью к компрессионному параличу.
Когда назначается исследование?
- Характерная для данных заболеваний клиническая картина и снижение скорости проведения импульса по срединному нерву менее 38 метров в секунду по результатам стимуляционной электронейромиографии.
Что означают результаты?
Компонент
Референсные значения
Детекция дупликации/делеции гена РМP22
Дупликации/делеции гена РМP22 не обнаружено. Копий гена PMP22=2.
Это качественный анализ - он дает результат в виде информации о наличии/отсутствии исследуемой мутации.
Отсутствие мутации гена - это нормальный результат.
Обнаружение дупликации гена белка PMP22 характерно для болезни Шарко - Мари - Тута типа 1А, а делеции - для наследственной невропатии с предрасположенностью к параличу от сдавления.
Что может влиять на результат?
- При объеме крови меньше минимально необходимого провести исследование невозможно. Минимальный объем цельной крови для проведения исследования для взрослых пациентов - 6 мл, для детей - 1 мл. Требуемые объемы обеспечивают адекватное для проведения исследования количество ДНК, которая содержится исключительно в лейкоцитах. Дети имеют сравнительно большее нормальное количество лейкоцитов в единице объема крови, по сравнению со взрослыми, поэтому у них необходимо забирать меньше биоматериала.
Важные замечания
- Описанные заболевания относятся к полинейропатиям, обусловленным первичным поражением миелиновой оболочки (демиелинизирующий тип). Помимо этого, существуют полинейропатии с первичным поражением непосредственно нервного волокна, а также болезни, относящиеся к промежуточному варианту. Такое многообразие заболеваний, весьма сходных по клиническим проявлениям, не позволяет подобрать оптимальный алгоритм генетического обследования исключительно на основании жалоб пациента и данных осмотра врачом. В дифференциации типа полинейропатии ведущую роль играет электронейромиография – исследование, с помощью которого измеряют силу и скорость проведения сигнала по периферическим нервам (чаще всего по срединному нерву). Поэтому снижение скорости проведения импульса по данным электронейромиографии, характерное для демиелинизирующих полинейропатий, является необходимым условием для исследования мутации гена PMP2222.
- При выявлении мутации необходимо обследовать также близких родственников пациента (родители, братья и сестры).
Кто назначает исследование?
Литература
Неврология: национальное руководство. Под ред. Е. И. Гусева, А. Н. Коновалова, В. И. Скворцовой, А. Б. Гехт. - М. : ГЭОТАР-Медиа, 2009. С. 732-743.
Наследственные двигательные невропатии с предрасположенностью к параличам от сдавливания нерва (НCПС)
При наследственной двигательной невропатии со склонностью к параличам от сдавления (ННСПС), нервы становятся все более чувствительными к сдавлению и растяжению.
При ХВДП периферические нервы утрачивают миелиновую оболочку и способность нормально проводить импульсы. Заболевание носит аутосомно-доминантный характер. В 80% случаев его причиной становится утрата одной копии гена периферического миелинового белка 22 типа (PMP22), расположенного на коротком плече 17-й хромосомы. Для нормального функционирования нужны 2 копии гена.
Заболеваемость составляет 2-5 случаев на 100 000 человек.
Симптомы и признаки ННПС
Обычно симптомы наследственной моторной невропатии со склонностью к параличам от сдавления начинаются у подростков или молодых людей, но дебют возможен и в любом возрасте.
Часто развиваются паралич малоберцового нерва Паралич малоберцового нерва Единичные мононейропатии характеризуются нарушением чувствительности и слабостью в зоне иннервации пораженного нерва. Диагноз устанавливается по клинической картине, но его следует подтвердить. Прочитайте дополнительные сведения и карпальный туннельный синдром Синдром запястного канала Единичные мононейропатии характеризуются нарушением чувствительности и слабостью в зоне иннервации пораженного нерва. Диагноз устанавливается по клинической картине, но его следует подтвердить. Прочитайте дополнительные сведенияПосле эпизода паралича полное восстановление отмечается приблизительно у половины пациентов, у остальных сохраняются слабо выраженные симптомы.
Диагностика НКНП
Наследственную невропатию со склонностью к параличу от давления следует заподозрить при наличии любого из перечисленных ниже признаков:
Рецидивирующая демиелинизирующая полинейропатия
Множественная мононейропатия неизвестного происхождения
Симптомы, позволяющие заподозрить рецидивирующую демиелинизирующую полинейропатию (например, хроническая воспалительная демиелинизирующая полинейропатия [ХВДП]).
Карпальный синдром в семейном анамнезе
Нейрофизиологические исследования и генетическое тестирование помогают в диагностике, но биопсия требуется редко.
Лечение НКНП
Лечение наследственной невропатии со склонностью к параличам от сдавления включает в себя устранение или модификацию видов деятельности, которые вызывают симптомы. Использование ортезов для кистей и мягких накладок для локтей может уменьшить давление, предотвратить повторные повреждения и позволяет через некоторое время восстановить миелин.
В редких случаях показана операция.
Основные положения
Врожденная двигательная нейропатия со склонностью к параличам от сдавливания нервов(ННСПС), – это редкое, обычно аутосомно-доминантное заболевание.
Следует рассмотреть возможность ННСПС, если у пациентов наблюдаются необъяснимые периферические мононевропатии (например, паралич малоберцового или локтевого нерва, запястный туннельный синдром) или симптомы, согласующиеся с хронической воспалительной полинейропатией.
Диагностирование производится с помощью электродиагностического и генетического исследования.
Следует рекомендовать пациентам избегать тех видов деятельности (или изменить их), которые приводят к возникновению симптомов, и рекомендовать манжеты на запястье и/или налокотники при необходимости.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Наследственные полинейропатии
Наследственные полинейропатии
(информация для пациентов)
Что такое "Наследственные полинейропатии"?
Наследственные полинейропатии – это большая группа клинически разнообразных полинейропатий с возможным вовлечением различных органов и систем, в том числе и центральной нервной системы, которые развиваются в результате мутаций (поломок) в генах человека.
- "Наследственные заболевания развиваются с рождения или в раннем детстве"
Это не так - дебют наследственной болезни возможен и во взрослом возрасте, в том числе и после 50 лет. Более того, зачастую пациенты не могут точно назвать возраст начала заболевания, так как симптомы развиваются незаметно. - "Наследственная патология должна быть и у близких родственников. А если в роду ни у кого нет, значит и у меня тоже нет".
Это не так - наследственные заболевания передаются разными путями: по аутосомно-доминантному, аутосомно-рецессивному, Х-сцепленному типу, также возможен митохондриальный тип наследования и др. Часто в семье есть асимптомные носители мутантного гена без клинических признаков заболевания, а симптомы болезни могут быть только у одного из членов семьи. Поэтому отсутствие отягощенного семейного анамнеза не исключает наследственный генез заболевания.
- Наследственные моторно-сенсорные нейропатии (НМСН)
- Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС)
- Наследственные сенсорные и автономные нейропатии (НСАН)
- Болезнь Фабри
- Транстиретиновая семейная амилоидная нейропатия (ТТР-САП)
Как часто диагностируются наследственные полинейропатии среди населения?
Частота встречаемости всех форм НМСН варьирует от 10 до 40 случаев на 100 000 населения в различных популяциях. На НМСН 1 типа приходится до 70% всех случаев НМСН. Распространенность ННСПС 2-5 случая на 100 000 населения. Болезнь Фабри - 1 случай на 40 000 - 60 000 мужчин. По примерным подсчетам в США встречаемость ТТР-САП составляет 1 случай на 100 000 человек. Следует отметить, что настороженность относительно дебюта наследственной патологии во взрослом возрасте среди врачей разных специальностей достаточно низкая, поэтому некоторые цифры распространенности могут быть занижены.
- пациентов беспокоят болезненные спазмы мышц голеней (крампи), слабость и деформация стоп, изменение походки, затруднения при беге или подъеме по лестнице;
- постепенно слабость развивается в кистях, в результате чего появляются затруднения при застегивании пуговиц, открывании двери ключом и т.д. В целом руки вовлекаются не ранее чем через 10 лет после появления первых симптомов болезни;
- в меньшей степени беспокоит онемение кистей и стоп, часто эти изменения игнорируются пациентом.
- лучевой нерв на уровне плеча (спиральный канал), при этом развивается слабость разгибателей кисти, кисть "висит" как плеть, возникает онемение наружного края предплечья и кисти;
- малоберцовый нерв на уровне коленного сустава (фибулярный канал), при этом развивается слабость разгибателей стопы, стопа начинает "шлепать", возникает онемение наружного края голени и стопы;
- локтевой нерв на уровне локтевого сустава (кубитальный канал), при этом развивается слабость межкостных мышц кисти, мышцы - отводящих мизинец и других, возникает онемение 4 и 5 пальцев кисти;
- срединный нерв на уровне запястья (карпальный канал), при этом развивается слабость мышц возвышения большого пальца, возникает онемение 1-3 пальцев кисти, характерен болевой синдром;
- и т.д. с развитием соответствующей клинической картины поражения того или иного нерва.
Степень поражения нерва (защемления в канале) может быть различной - от незначительной, когда беспокоит только онемение и парестезии, до более выраженной, когда помимо чувствительных нарушений развивается и слабость мышц.
Развитию симптомов, как правило, предшествует незначительная травма, нахождение длительное время в неудобной статической поза (на четвереньках, на корточках, облокотившись на локоть и др.), ношение неудобной одежды, непривычная физическая нагрузка и прочее.
Мышечная слабость, как правило, сохраняется в течение нескольких дней или недель, а затем сила мышц постепенно восстанавливается (в течение нескольких недель или месяцев). Эпизоды "защемлений" нервов имеют тенденцию к повторению (всего у пациентов может быть от 1 до 10 таких эпизодов). Следует учитывать, что часто лечащие врачи не задумываются о том, что причиной рецидивирующих (повторяющихся) туннельных невропатий – является наследственная патология, что приводит к поздней диагностике и неоправданным медицинским вмешательствам (операциям по поводу туннельных невропатий).
Болезнь Фабри - редкое генетическое заболевание, обусловленное мутацией гена GLA, картированного на длинном плече хромосомы Хq 22.1 и контролирующего структуру фермента альфа-галактозидазы А. Нередко начало заболевания отмечается в подростковом возрасте. Тип наследования - сцепленное с Х-хромосомой.
- жгучая колющая боль в ладонях и стопах; острые приступы мучительной боли в кистях и стопах (кризы Фабри);
- нарушение потоотделения; непереносимость жары/холода;
- сыпь на коже (ангиокератомы);
- помутнение роговицы в виде завитка, которое не ослабляет зрение;
- шум в ушах, потеря слуха;
- желудочно-кишечные расстройства, диарея;
- кардиологические проявления (включая увеличенное сердце и нарушение ритма);
- нарушение функции почек, которое в конечном итоге приводит к терминальной стадии хронической почечной недостаточности;
- нарушение мозгового кровообращения (чаще ишемический инсульт, но может быть и внутримозговое кровоизлияние, или венозный тромбоз).
- анализ истории развития заболевания
- оценка неврологического статуса
- общий клинический и развернутый биохимический анализы крови;
- RW, анти-ВИЧ, НВsAg и анти-HCV;
- общий анализ мочи;
- анализ крови на уровни витаминов группы В, гомоцистеин;
- электрофорез белков сыворотки и мочи с иммунофиксацией; ;
- анализ цереброспинальной жидкости (ликвор) и другие.
- уровень фитановой кислоты в крови методом хроматомасс-спектрометрии (повышение при болезни Рефсума);
- определение активности лизосомного фермента альфа-галактозидазы А, в том числе с помощью флюориметрического метода ("сухое" пятно) - исключение болезни Фабри;
- биопсия икроножного нерва, малых слюнных желез и абдоминального жира – для выявления отложения амилоида, характерного для ТТР-САП).
- Однако окончательный диагноз той или иной наследственной полинейропатии может быть подтвержден только по результатам генетического анализа. Оптимальным является генетическое обследование по следующему алгоритму:
- при убедительной клинико-анамнестической и параклинической картине в пользу той или иной наследственной полинейропатии проводится прицельная генетическая диагностика одного гена, в том числе с использованием скрининг тестов методом "сухого пятна";
- если подозревается наследственный генез полинейропатии, тип её не определен целесообразно проведение более широкого генетического анализа - секвенирования нового поколения (NGS).
- при болезни Фабри - это фермент-заместительная терапия рекомбинантными препаратами альфа-галактозидазы А
- при ТТР-САП – стабилизация молекулы транстиретина (Тафамидис), трансплантация печени;
- при порфирийной полинейропатии - введение аргината гема;
- при болезни Рефсума – диетотерапия с ограничением поступления фитановой кислоты (исключение потребления зеленых овощей, говядины, рыбы (тунец, пикша, треска)), плазмаферез.
При некоторых наследственных нейропатиях, для которых характерно поражение органов и систем, важно мониторировать функцию сердечно-сосудистой системы и других органов, наблюдаться у профильных специалистов.
- рекомендуется вести здоровый образ жизни, правильно и сбалансировано питаться, полностью отказаться от приема алкоголя, воздерживаться от вегетарианства;
- исключить, ограничить прием лекарственных препаратов с нейротоксическим действием;
- проводится коррекция ортопедических нарушений: подбор стелек, стоподержателей, коленоупорников, использование дополнительной опоры, кинезиотейпирование;
- регулярно, в поддерживающем режиме проводятся реабилитационные мероприятия: миостимуляция, массаж, растяжка, ЛФК.
Прием "нейрометаболических" препаратов и витаминов группы В не целесообразно, в связи с отсутствием доказательной базы.
.
Какой прогноз данного заболевания?
Прогноз определяется типом наследственной полинейропатии и многими сопутствующими факторами. Так, при НМСН 1 и 2 типа при позднем дебюте крайне замедленное развитие мышечной слабости приводит к тому, что больные приспосабливаются к своему дефекту и, несмотря на слабость и похудание кистей и стоп, могут выполнять ручную работу, свободно передвигаться. Даже при многолетнем течении сохраняется способность к самостоятельной ходьбе. Наследственная нейропатия со склонностью к параличам от сдавления также имеет относительно благоприятный прогноз при соблюдении рекомендаций по ограничению длительных статических поз и снижению потенциальных провокаторов компрессий нервов в повседневной жизни. НМСН III, V типов вызывают тяжелый двигательный дефект и в большинстве случаев приводит к выраженной инвалидности. При ранней диагностике наследственных нейропатий, ассоциированных с метаболическим дефектом (ТТР-САП, болезнь Рефсума, порфирия, болезнь Фабри) и назначении необходимой терапии возможно пролонгировать наступление инвалидизации и улучшить качество жизни.
В ФГБНУ НЦН проводят консультации сотрудники Центра заболеваний периферической нервной системы, сотрудники 5 неврологического отделения, профилем которого является диагностика и лечение наследственных заболеваний нервной системы. Консультации пациентов проводятся амбулаторно в рамках ОМС и на коммерческой основе.
На базе ФГБНУ НЦН проводится весь перечень необходимых для уточнения диагноза обследований, разработаны подходы к комплексной восстановительной терапии при наследственных полинейропатиях.
Наследственные двигательные невропатии с предрасположенностью к параличам от сдавливания нерва
Facebook Если у вас не работает этот способ авторизации, сконвертируйте свой аккаунт по ссылке ВКонтакте Google RAMBLER&Co ID
Авторизуясь в LiveJournal с помощью стороннего сервиса вы принимаете условия Пользовательского соглашения LiveJournal
Наследственная невропатия со склонностью к параличам от сдавления

… наследственные невропатии относятся к числу достаточно распространенных заболеваний периферической нервной системы и встречаются в общей популяции с частотой примерно 1:2500. Большинство наследственных невропатий характеризуется неуклонно прогрессирующим течением. В то же время имеются формы фенотипически представленные рецидивирующими мононевропатиями с острым или подостым началом. Одним из заболеваний, входящих в эту группу, является ткак называемая наследственная невропатия со склонностью к параличам от сдавления (ННПС).
Наследственная невропатия со склонностью к параличам от сдавления – редкое аутосомно-доминантное заболевание, начинающееся на 2-3* десятилетии жизни и характеризующееся повышенной чувствительностью периферических нервов к сдавлению, что приводит повторяющимся эпизодам компрессионных невропатий (* однако описано как более раннее начало – в 4 года, так и более позднее – в 67 лет и 73 года).
В большинстве случаев ген заболевания обнаруживается на коротком плече 17-й хромосомы (р11.2 – р12), в области гена, кодирующего РМР 22. В отличие от НМСН – IА (наследственная моторно-сенсорная невропатия типа IА) для данного варианта невропатии характерно не удвоение участка того гена, а делеция. Но описаны случаи невропатии, имеющие иную генетическую основу. Частота заболевания у мужчин и женщин, по данным одних авторов, примерно одинакова, других – соотносится как 4:3, при этом у мужчин отмечается более ранний дебют болезни.
Факторами, способствующими развитию параличей, служат мелкие травмы и незначительные кратковременные эпизоды сдавления нервов. Так, они могут возникать после работы за письменным толом с порой на локти (повреждение локтевого нерва), при сидении в позе «нога на ногу», стоянии на коленях, на корточках (паралич малоберцового нерва). Для каждого нерва существуют свои предрасполагающие к сдавлению позиции. К сдавлению лучевого нерва предрасполагают положение руки на спинке скамьи, кошение травы, сон на надувном матраце. Компрессия плечевого сплетения возникает при закладывании руки за голову во время сна, при сне на одном боку, при наркозе, ношении тяжелого чемодана или рюкзака, длительной игре на скрипке либо виолончели. Компрессия затылочного нерва нередко возникает при сне на полу, тройничного - при опоре одной стороны лица о твердый предмет. Примерами двигательной активности и физических факторов, которые способны вызвать эпизоды онемения, парестезий и слабости, может также служить выкапывание клубней, использование ножниц, вязание, длительный разговор по телефону в одной позе, продолжительные малярные работы, характерная поза при рисовании, ходьба более 1 часа, быстрая потеря массы тела, использование компьютерной «мышки» и т.д. Описано повреждение периферических нервов на фоне беременности, родов, наложения гипса и т.д. Все перечисленные провоцирующие факторы не являются специфичными для возникновения компрессионных невропатий при ННПС, они однотипны для всех компрессионных невропатий (в частности, для нетравматических туннельных синдромов). В ряде случаев у новорожденных парезы от сдавления в качестве первого проявления ННПС расцениваются как акушерский паралич.
■ семейный анамнез болезни;
■ наличие рецидивирующих параличей периферических нервов;
■ электрофизиологические изменения как пораженных, так и клинически интактных нервов;
■ морфологические изменения периферических нервов с наличием томакул.
В большинстве случаев имеется смешанный сенсомоторный дефицит, гораздо реже наблюдаются изолированные чувствительнее или двигательные расстройства. Патоморфологически выявляются сегментарная демиелинизация и ремиелинизация с локальным утолщением миелиновой оболочки с нарушением ее внутренней структуры, что придает нервному волокну вид связки сосисок.
1. Классический вариант с типичными эпизодами рецидивирующих мононевропатий (описан выше).
2. Медленно прогрессирующая сенсомоторная невропатия, напоминающая хроническую приобретенную демиелинизирющую полинейропатию. Некоторые авторы отмечали на фоне генерализованной невропатии преимущественное вовлечение перонеальных нервов. Этот вариант можно дифференцировать с приобретенными демиелинизирующими невропатиями только на основании исследования биоптата нервов и ДНК-диагнстики.
3. Прогрессирующая сенсорная невропатия. Описано несколько ее разновидностей: а) наличие транзиторных симптомов онемения и парестезий; б) наличие хронического синдрома карпального канала с вовлечением только чувствительных волокон; в) по типу прогрессирующей сенсорной невропатии.
4. Множественная мононевропатия. Данную форму ННПС необходимо дифференцировать с мультифокальной моторной (аутоиммунной) невропатией, характеризующейся сочетанным поражением периферических нервов и ядер нервных клеток. Для мультифокальной моторной невропатии характерно преимущественное болевое поражение двигательных нервов конечностей с наличием множественных блоков проведения, выявляемых при электромиографии.
5. Олигосимптомный вариант. Может имитировать любой вид туннельной невропатии, радикулопатию, паралич Белла.
6. Асимптомный вариант. Его наличие предполагается примерно у 40% и более больных ННПС.
На ЭНМГ выявляется замедление или блокада проведения в одной или нескольких зонах компрессии, но нередко имеются и более распространенные изменения, которые обнаруживают и при исследовании интактных нервов.
1.Наличие диффузных электрофизиологических изменений у всех носителей делеции 17р11.2 старше 15 лет вне зависимости от наличия или отсутствия клинических признаков: билатеральное увеличение дистальной моторной латенции, коррелирующее с замедлением проведения импульса по чувствительным волокнам срединного нерва на ладонно-запястном уровне, а также изменение хотябы одного показателя (дистальной латенции или скорости проведения) при исследовании малоберцового нерва.
2.Частое сопутствующее локальное снижение проведения возбуждения по локтевому нерву на локтевом сегменте.
3.Незначительное снижение скорости проведения по двигательным волокнам остальных периферических нервов.
4.Снижение амплитуды чувствительных потенциалов, особенно по нервам верхних конечностей.
Специфической терапии не существует. К сожалению, на сегодняшний день медицина не располагает эффективными способами лечения наследственных невропатий. В случае диагностированной ННПС больным следует рекомендовать соответствующую коррекцию стиля жизни, физических нагрузок и профессиональной ориентации, что имеет решающее значение для профилактики осложнений этого заболевания.
Литература: 1) Руководство для врачей «Болезни нервной системы» под редакцией члена-корреспондента РАМН, профессора Н.Н. Яхно, профессора Д.Р. Штульмана. Изд. 2-ое, переработанное, том 1; Москва; «Медицина», 2001. 2) Статья «Особая форма демиелинизирующей полиневропатии: наследственная невропатия с предрасположенностью к параличам от сдавления» Н.Г. Савицкая, И.А. Иванова-Смоленская, С.Н. Иллариошкин, С.С. Никитин (НИИ неврологии РАМН, Москва), 2002.
Наследственная нейропатия с подверженностью параличу от сдавления. Анализ числа копий гена РМР22
Ген РМР22 (PERIPHERAL MYELIN PROTEIN 22) расположен на хромосоме 17 в регионе 17p12. Мутации в этом гене приводят также к развитию таких заболеваний как болезнь Шарко-мари-Тута типы 1А и 1Е, синдром Руси-Леви, нейропатия демиелинизирующая воспалительная и болезнь Дежерина-Сота.
Заболевание возникает в возрасте от 8 до 64 лет (средний возраст - 26 лет) и характеризуется рецидивирующими парезами периферических нервов, возникающими остро после небольших травм или сдавлений. У 40% больных парезы возникают после интенсивных физических нагрузок, а у 10% при пробуждении. Продолжительность двигательных нарушений колеблется от одного дня до нескольких месяцев. У 50% больных в патологический процесс вовлекаются два и более периферических нерва, патологический процесс часто бывает асимметричным. В редких случаях в процесс вовлекаются черепно-мозговые нервы, особенно бульбарная группа, что проявляется парезом голосовых связок или лицевой мускулатуры. У 62% больных сухожильные рефлексы с рук и ног не изменяются, у 30-35% больных выявляется их умеренное снижение, и лишь у 12% больных сухожильные рефлексы исчезают. Характерно возникновение расстройств чувствительности, особенно выраженное в период существования пареза. В литературе это заболевание впервые было описано как «болезнь сборщиков картофеля», поскольку было замечено в семье фермеров, у которых частичный паралич ног развивался после длительной работы в положении на коленях.
В большинстве случаев пациенты с данной формой нейропатии восстанавливаются за несколько дней или недель, но могут наблюдаться рецидивы с тенденцией к сохранению парезов в течение более длительного периода. Несмотря на доминантный характер наследования, больные с ННПС сравнительно редко попадают в поле зрения врача-генетика, поскольку у многих носителей заболевание протекает бессимптомно, и семейный характер заболевания часто выявляется только при специальном неврологическом исследовании. Описаны больные с наличием сколиоза и деформации стоп по типу полых, а также с глухотой. На электромиограмме выявляется умеренное увеличение латенции по срединному нерву, снижение скоростей проведения по сенсорным и моторным нервам.
При морфологическом изучении биоптата периферических нервов выявляются изменения сходные с таковыми при наследственной моторно- сенсорной нейропатии 1 типа. Характерно возникновение очагов демиелинизации и образование томакул - колбасовидных утолщений миелиновой оболочки периферических нервов. Их наличие отмечено как в моторных, так и сенсорных нервах. Считается, что томакулы формируются в период раннего развития ребенка и отражают нарушение взаимодействия аксона и миелина.
Литература
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
типичная клиническая картина.
Литература
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
Читайте также: