Наследственные нарушения обмена аминокислот. Фенилпировиноградная олигофрения
Добавил пользователь Алексей Ф. Обновлено: 20.01.2026
Фенилкетонурия xe "Фенилкетонурия" (ФКУ, фенилпировиноградная xe "Олигофрения фенилпировиноградная" олигофрения) — наследственное заболевание обмена веществ, обусловленное дефицитом одного из ферментов обмена фенилаланина, сопровождающееся нарушением гидроксилирования аминокислоты фенилаланина в тирозин. В результате в организме больного ребёнка происходит постепенное накопление фенилаланина и его метаболитов, оказывающих токсическое действие на ЦНС с дальнейшей задержкой психического развития. Болезнь впервые описана в 1934 г. А. Феллингом.
Частота. Выявлены значительные этнические и географические различия в частоте разных мутаций при наиболее распространённой классической ФКУ. Частота классической ФКУсоставляет 1 на 4500 в Ирландии, 1 на 6000–10 000 в России, 1 на 16 000–20 000 среди белого населения США, 1 на 12 000 в Италии, 1 на 16 000 в Швейцарии и значительно снижена у афроамериканцев (1 на 50 000), китайцев и японцев, евреев ашкенази. Необычно редко наблюдают ФКУ в Финляндии (реже 1 на 100 000), к 1995 г. было выявлено четверо больных, среди полинезийцев ФКУ не выявлена. В Татарстане частота классической формы ФКУ составляет 1 на 6000 новорождённых (вне зависимости от этнической принадлежности).
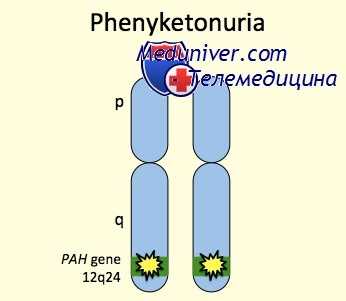
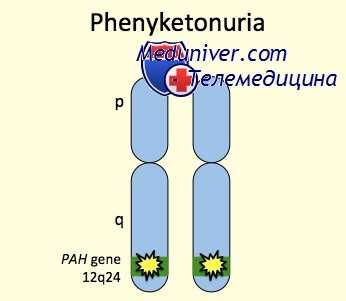
Генетические аспекты. ФКУ развивается при дефектах генов следующих ферментов • ГТФ циклогидролаза 1 (233910, GCH1, 600225, 14q22.1–q22.2) • Фенилаланин гидроксилаза (261600, PAH, PKU1, 12q24.1) • Дигидробиоптеридин редуктаза (261630, QDPR, DHPR, 4p15.31) • Дигидробиоптерин синтетаза (261640, PTS, 11q22.3–q23.3).
Патоморфология — обнаружены изменения в структуре миелиновых волокон у нелеченых больных.
Клиническая картина Дети с ФКУ рождаются без клинических признаков болезни. Однако фенилаланин, поступающий с первых дней жизни с молоком матери или обычной молочной смесью, способствует проявлению заболевания • Неврологические и психические расстройства •• Умственная отсталость (олигофрения, идиотия или имбецильность, глубокая психическая инвалидность). При отсутствии лечения коэффициент интеллектуального развития за каждые 10 нед снижается на 5 пунктов) •• Повышенная возбудимость в детстве •• Специфическая походка •• Специфические осанка и поза при сидении •• Необычное положение конечностей (поза портного) •• Стереотипные движения •• Повышение сухожильных рефлексов •• Судороги •• Дефектное формирование миелина • Раннее закрытие большого родничка • Микроцефалия • Изменения кожи •• Гипопигментация •• Сухость •• Экзема •• Дерматит •• Склеродермия • Волосы гипопигментированы • Рвота в периоде новорождённости • Светлые радужки, катаракта • Специфический «мышиный» запах тела и мочи.
Методы исследования.
• Рентгенологическое исследование: мозговые кальцификаты.
• Лабораторные исследования •• Дефицит гидроксилазы фенилаланина (ФКУ-1), дигидроптеридинредуктазы (ФКУ-2) или дигидробиоптерин синтетазы (ФКУ-3) •• Гиперфенилаланинемия. Нормальная концентрация фенилаланина крови 58 ± 15 мкмоль/л — у взрослых, 60 ± 13 мкмоль/л — у подростков, 62 ± 18 мкмоль/л — у детей. У новорождённых верхний предел нормы составляет 120 мкмоль/л (2 мг/100 мл) •• При нелеченой классической ФКУ содержание фенилаланина крови повышается до 2,4 ммоль/л •• Фенилпировиноградная ацидемия •• Повышение в моче содержания o-гидроксифенилуксусной, фенилпировиноградной и фенилуксусной кислот и фенилацетилглютамина.
• Специальные исследования: метод массового неонатального скрининга — обязательное определение содержания фенилаланина в образцах крови у всех новорождённых, полученных на 4–5-й день жизни в родильном доме.
• Молекулярная генетика: известно более 200 различных мутаций гена фенилаланингидроксилазы. Большинство из них сцеплено с определёнными гаплотипами полиморфизма длины рестрикционных фрагментов (RFLP) и числа танедемных повторов (VNTR). Один гаплотип может быть ассоциирован более чем с одной мутацией, следовательно, на основании исследования гаплотипа нельзя сделать определённого вывода о локализации мутации. Главная мутация для славянских народов — R408W/HP2/VNTR3. Исследование, проведённое в Татарстане, показало существенное различие в частоте этой мутации среди больных ФКУ русской (78%) и татарской (37%) национальностей. Интересно, что среди больных ФКУ татарской национальности часто (40%) отмечают мутации, характерные для средиземноморских, в т.ч. тюркских популяций (R261Q и др.), и не выявлено мутаций, характерных для восточных народов.
ЛЕЧЕНИЕ
Режим амбулаторный, госпитализация показана для коррекции диеты в случае нестабильной концентрации фенилаланина плазмы.
Диета с резким ограничением содержания фенилаланина вводится с момента подтверждения диагноза классической ФКУ. Учитывая высокое содержание фенилаланина в белке, полностью исключают продукты животного происхождения (мясо, птицу, рыбу, яйца, грибы, молоко и продукты, приготовленные из них, колбасы, хлебобулочные изделия, крупы, бобовые, орехи, шоколад и др.), содержание растительных белков тщательно нормируют с учётом массы тела и возраста ребёнка. Дефицит пищевых белков и микроэлементов, возникающий вследствие длительного применения ограничительной диеты, компенсируют назначением специальных продуктов питания — смесей аминокислот или белковых гидролизатов с низким содержанием фенилаланина. Безфенилаланиновые препараты вводят с фруктовыми и овощными соками, пюре, супами. Учитывая высокую стоимость этих препаратов, лечение проводят за счёт государства под наблюдением врачей специализированного центра • При атипичных формах ФКУ (2 и 3), а также при несвоевременном ограничении продуктов в пищевом рационе даже строгое соблюдение диеты с низким содержанием фенилаланина не приводит к предупреждению тяжёлых неврологических нарушений. Назначение диеты, обогащённой тетрагидробиоптерином, также не приводит к клиническому улучшению у таких больных.
Лекарственная терапия.
• Препараты выбора •• Ноотропные препараты, например пирацетам •• Иглорефлексотерапия •• Для компенсации белковой и витаминной недостаточности при назначении ограничительной диеты используют белковые гидролизаты и аминокислотные смеси, обогащённые микроэлементами и витаминами: «Лофеналак», «Фенил-40», «Фенил-100», Афенилак, «Аналог SP», «Эро-Бэби» (для детей до года), «Максамайд», «Максамум», «П-АМ универсальный», «ФенилФри», «Тетрафен», «Фенил-400» (для детей после года), а также такие диетические продукты, как «PKU-1mix», «PKU-1» «PKU-2» (Германия). Дозировки рассчитывают с учётом содержания естественного белка в рационе, массы тела и возраста ребёнка •• В резистентных к лечению случаях отмечают некоторый эффект от применения препаратов леводопы.
Генотерапия. Из трёх основных мероприятий, требуемых для генотерапии при фенилкетонурии, два выполнены: получена кДНК, обеспечивающая экспрессию гидроксилазы фенилаланина человека, разработана гидроксилазадефицитная животная модель. Однако векторы для эффективной передачи гена in vivo требуют дальнейшей разработки. Ретровирусные векторы, несмотря на то, что достаточно эффективны in vitro, имеют низкую эффективность передачи in vivo. Рекомбинантные аденовирусные векторы, хотя и полностью успешны на короткое время, не сохраняются в организме больше нескольких недель из-за иммунного ответа.
Течение и прогноз. Своевременно начатое диетическое лечение позволяет избежать развития клинических проявлений классической ФКУ. Необходимо проводить лечение до полового созревания, а по индивидуальным показаниям и дольше. Т.к. женщина, больная ФКУ, не может выносить здоровый плод, показано проведение специального лечения, начатого перед зачатием и продолжающегося до момента родов с целью исключить поражение плода фенилаланином плазмы крови матери.
Беременность. Повышенное содержание фенилаланина в плазме крови матери приводит к разнообразным врождённым заболеваниям плода, спектр зависит от выраженности и продолжительности повышения содержание фенилаланина • ВПС при высокой концентрации фенилаланина • Аномалии развития головного мозга, внутриутробная и послеродовая задержка роста, изменения внешности (широкая переносица с вывернутыми ноздрями) при средней концентрации фенилаланина в I триместре беременности • Неврологические симптомы при среднем повышении концентрации фенилаланина в течение всей беременности • Женщинам с классической ФКУ рекомендуют диету с низким содержанием фенилаланина, чтобы достичь концентрации фенилаланина плазмы менее 360 мкмоль/л ещё до зачатия и поддерживать эту концентрацию в течение всей беременности.
Сокращение. ФКУ — фенилкетонурия.
МКБ-10 • E70.0 Классическая фенилкетонурия
Наследственные нарушения обмена аминокислот. Фенилпировиноградная олигофрения
Генетика фенилкетонурии. Наследование
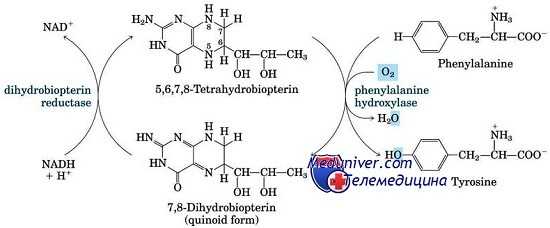
Аномалии, приводящие к увеличению уровня фенилаланина крови, чаще всего недостаточность фенилаланингидроксилаза (ФАГ) или фенилкетонурия (ФКУ), иллюстрируют почти все принципы биохимической генетики, относящиеся к дефектам ферментов. Все генетические аномалии метаболизма фенилаланина — следствие мутаций со снижением функции в гене, кодирующем ФАГ, или в генах, необходимых для синтеза или восстановления ее кофактора, ВН4.
Классическую фенилкетонурию (ФКУ) по праву считают образцовым представителем врожденных ошибок метаболизма. Это аутосомно-рецессивное заболевание распада фенилаланина, вызванное мутациями в гене, кодирующем ФАГ, фермент, преобразующий фенилаланин в тирозин. Открытие фенилкетонурии (ФКУ) Фелингом в 1934 г. впервые продемонстрировало генетический дефект как причину умственной отсталости.
Из-за неспособности к утилизации фенилаланина пациенты с фенилкетонурией (ФКУ) накапливают эту аминокислоту в жидкостях тела. Гиперфенилаланинемия повреждает формирующуюся в раннем детстве ЦНС и создает помехи функционированию зрелого мозга. Небольшая часть фенилаланина метаболизируется по альтернативным путям, производя повышенные количества фенилпировиноградной кислоты (кетокислота, по которой названа болезнь) и других метаболитов, выделяющихся с мочой.
Любопытно, что хотя ферментный дефект известен уже десятилетия, точный патогенетический механизм, каким образом увеличение фенилаланина повреждает мозг, все еще неизвестен. Важно, что развитие неврологического ущерба, вызванного метаболическим блоком при классической ФКУ, может в основном предупреждаться изменениями диеты, предохраняющими от накопления фенилаланина. Лечение фенилкетонурии (ФКУ) стало образцом для лечения многих метаболических болезней, исходы которых могут улучшаться за счет предотвращения накопления субстрата фермента и его производных.
Скрининг новорожденных на фенилкетонурию (ФКУ)
Широко используется популяционный скрининг новорожденных на фенилкетонурию (ФКУ). Фенилкетонурия (ФКУ) — образец генетических болезней, для которых оправдан массовый неонатальный скрининг; заболевание сравнительно часто встречается в ряде популяций (до 1 на 2900 живых новорожденных). Лечение, начатое в начале жизни, весьма эффективно; без лечения неизбежно развивается тяжелая умственная отсталость. Скрининг-тесты выполняют через несколько дней после рождения.
Капельку крови, полученную при проколе пятки, наносят на бумажный фильтр, высушивают и отправляют в централизованную лабораторию для оценки уровня фенилаланина в крови и соотношения фенилаланин/ тирозин. В прошлом образцы собирали перед выпиской ребенка из роддома. Тенденция к ранней выписке матери и новорожденного после родов изменила эту практику. Тест предпочтительно не делать до возраста 24 ч, поскольку уровень фенилаланина при фенилкетонурии (ФКУ) повышается только после рождения. Положительные результаты теста должны быть быстро подтверждены, поскольку задержка начала лечения более 4 нед после родов не позволяет избежать влияния на интеллектуальное состояние пациентов с фенилкетонурией (ФКУ).
Различные формы фенилкетонурии и гиперфенилаланинемия
Поскольку фенилкетонурия (ФКУ) связана с выраженной недостаточностью активности фенилаланингидроксилазы (ФАГ) (менее 1% по сравнению с контролем), мутантная ФАГ, имеющая остаточную активность, вызывает менее тяжелые фенотипические проявления, так называемую гиперфенилаланинемию и атипичную фенилкетонурию (ФКУ).
Гиперфенилаланинемию, отличную от фенилкетонурии (ФКУ), диагностируют, если концентрация фенилаланина в плазме ниже 1 ммоль/л на фоне нормальной диеты. Эта степень гиперфенилаланинемии только в 10 раз выше нормы и значительно ниже, чем концентрации, обнаруживаемые при классической фенилкетонурии (ФКУ) (>1 ммоль/л). Умеренное увеличение фенилаланина при гиперфенилаланинемии не способно повреждать функции мозга и может даже быть благоприятным, если увеличение небольшое (
Атипичная фенилкетонурия (ФКУ) — категория, включающая пациентов с промежуточным уровнем фенилаланина между классической ФКУ и гиперфенилаланинемией; такие пациенты требуют некоторого ограничения фенилаланина в диете, но меньшего, чем для пациентов с классической фенилкетонурии (ФКУ). Комплекс из этих трех клинических фенотипов с мутациями в гене ФАГ — пример клинической гетерогенности.
Гиперфенилаланинемии: аллельная и локусная гетерогенность при фенилкетонурии (ФКУ)
Молекулярные дефекты в гене фенилаланингидроксилазы. У пациентов с гиперфенилаланинемией, включая классическую фенилкетонурию (ФКУ), атипичную фенилкетонурию (ФКУ) и доброкачественные гиперфенилаланинемии, обнаружена поразительная степень аллельной гетерогенности в локусе фенилаланингидроксилазы (ФАГ) (более 400 различных мутаций по всему миру).
Подавляющее большинство аллелей фенилаланингидроксилазы (ФАГ) — достаточно редкие мутации, нарушающие ферментные свойства фенилаланингидроксилазы (ФАГ) и приводящие к гиперфенилаланинемии, хотя также обнаружены и доброкачественные полиморфизмы или менее частые доброкачественные варианты.
В популяциях европейского происхождения около двух третей известных мутантных хромосом представлены шестью мутациями. Шесть других мутаций ответственны за чуть более 80% мутаций фенилаланингидроксилазы (ФАГ) в азиатских популяциях. Остальные патогенные мутации встречаются реже. Чтобы сделать эту информацию широкодоступной, международным консорциумом разработана база данных мутаций в гене фенилаланингидроксилазы (ФАГ).
Во всех популяциях существует выраженная генетическая гетерогенность фенилаланингидроксилазы (ФАГ). Благодаря высокой степени аллельной гетерогенности в локусе, большинство пациентов с фенилкетонурией (ФКУ) во многих популяциях — компаундные гетерозиготы (т.е. у них присутствуют два разных патогенных аллеля), что полностью соответствует наблюдаемой ферментативной и фенотипической гетерогенности при нарушениях фенилаланингидроксилазы (ФАГ).
Сначала казалось, что знание генотипа фенилаланингидроксилазы (ФАГ) надежно предсказывает детали фенотипа; это ожидание оправдалось не полностью, хотя обнаружена определенная корреляция между генотипом ФАГ и биохимическим фенотипом.
В общих чертах мутации, которые полностью подавляют или резко снижают активность фенилаланингидроксилазы (ФАГ), вызывают классическую фенилкетонурию (ФКУ), тогда как мутации, приводящие к достаточно большой остаточной активности фермента, связаны с легкими фенотипами.
Тем не менее некоторые мутации фенилаланингидроксилазы (ФАГ) у гомозиготных пациентов определяют весь спектр фенотипов, от классической фенилкетонурии (ФКУ) до доброкачественной гиперфенилаланинемии.
Таким образом, стало очевидно, что в формировании фенотипа, наблюдаемого при специфическом генотипе, участвуют другие неопознанные биологические факторы, несомненно, включая гены-модификаторы. Это наблюдение, признанное в настоящее время общей характеристикой множества моногенных болезней, указывает на то, что даже моногенные болезни, подобные фенилкетонурии (ФКУ), — генетически не простые заболевания.
Дефекты в метаболизме тетрагидробиоптерина при фенилкетонурии (ФКУ)
Первоначально считали, что все дети с наследственной гиперфенилаланинемией имеют первичную недостаточность фенилаланингидроксилазы (ФАГ). Сейчас ясно, что примерно у 1-3% пациентов ген ФАГ нормален, а их гиперфенилаланинемия — результат генетического дефекта в одном из нескольких других генов, задействованных в синтезе или регенерации кофактора ФАГ, ВН4. Ассоциация одного фенотипа, например гиперфенилаланинемии, с мутациями в разных генах — пример локусной гетерогенности.
Как показывают мутации в генах, кодирующих белок фенилаланингидроксилазы (ФАГ) и метаболизм его кофактора биоптерина, белки, закодированные генами, демонстрирующими локусную гетерогенность, обычно входят в одну цепочку биохимических реакций. Пациенты с недостаточностью ВН4 сначала были выявлены из-за того, что, несмотря на успешное поддержание в диете низкой концентрации фенилаланина, у них рано развивались глубокие неврологические проблемы.
Плохие результаты частично объясняются необходимостью кофактора ВН4 для активности двух других ферментов, тирозингидроксилазы и триптофангидроксилазы. Обе этих гидроксилазы критичны для синтеза моноаминовых нейротрансмиттеров, таких как дегидроксифенилаланин, норэпинефрин, эпинефрин и серотонин. Пациенты с недостаточностью ВН4 имеют нарушение или в его биосинтезе из ГТФ, или в регенерации ВН4. Подобно классической фенилкетонурии (ФКУ), нарушение наследуется по аутосомно-рецессивному типу.
Очень важно отличать пациентов с дефектами в метаболизме ВН4 от больных с мутациями в фенилаланингидроксилазы (ФАГ), поскольку их лечение заметно разнится. Во-первых, так как белковая структура фенилаланингидроксилазы (ФАГ) у больных с нарушениями ВН4 нормальная, ее активность может восстанавливаться, если этим пациентам давать большие дозы ВН4, что приводит к снижению уровня фенилаланина плазмы. Следовательно, степень ограничения фенилаланина в диете пациентов с дефектами в метаболизме ВН4 может быть значительно уменьшена, а некоторые пациенты могут перейти на нормальную диету (т.е. без ограничения фенилаланина).
Во-вторых, необходимо также постараться нормализовать уровень нейротрансмиттеров в мозге этих пациентов, назначая продукты тирозингидроксилазы и триптофангидроксилазы: L-dopa и 5-гидрокситриптофан соответственно. По этим соображениям всем новорожденным с гиперфенилаланинемией показано обследование для определения аномалий в метаболизме ВН4.
Реакция на тетрагидробиоптерин при мутациях в гене ФАГ при фенилкетонурии (ФКУ)
У большинства пациентов с мутациями в гене фенилаланингидроксилазы (ФАГ), а не в метаболизме ВН4, отмечено отчетливое уменьшение уровня фенилаланина крови на фоне перорального приема больших доз кофактора фенилаланингидроксилазы (ФАГ) ВН4. Лучше отвечают на такое лечение пациенты со значимой остаточной активностью фенилаланингидроксилазы (ФАГ) (т.е. пациенты с атипичной фенилкетонурией (ФКУ) и гиперфенилаланинемией), но также поддается лечению небольшое число пациентов даже с классической фенилкетонурией (ФКУ). В то же время наличие остаточной активности ФАГ не дает гарантии влияния на уровень фенилаланина плазмы при назначении ВН4.
Наиболее вероятно, что степень ответной реакции на ВН4 зависит от специфических свойств каждого мутантного белка фенилаланингидроксилазы (ФАГ), отражающих лежащую в основе мутаций ФАГ аллельную гетерогенность. Показано, что введение в диету ВН4 оказывает лечебный эффект через несколько механизмов, вызванных повышением количества нормального кофактора, входящего в контакт с мутантным.
Эти механизмы включают стабилизацию мутантного фермента, защиту фермента от разложения клеткой, увеличение поступления кофактора к ферменту, имеющего низкое сродство с ВН4, и другие полезные эффекты в кинетических и каталитических свойствах фермента. Обеспечение повышенного количества кофактора — общая стратегия, применяемая в лечении многих врожденных ошибок метаболизма.
Материнская фенилкетонурия
Обычно успешное лечение фенилкетонурии (ФКУ) позволяет больным гомозиготам вести полноценную жизнь и иметь практически нормальные перспективы деторождения. В прошлом диету с низким содержанием фенилаланина прекращали у большинства пациентов с ФКУ в среднем детстве, основываясь на предположении (ошибочном, как установлено в настоящее время), что функционирование зрелой нервной системы не нарушается при возврате гиперфенилаланине-мии. Впоследствии было обнаружено, что почти все потомство женщин с фенилкетонурией (ФКУ), не получавших лечения, аномально; большинство этих детей с задержкой умственного развития, многие имеют микроцефалию, задержку роста и пороки развития, особенно сердца.
Как предсказывают принципы менделирующего наследования, все эти дети — гетерозиготы. Таким образом, задержка их развития вызвана не собственной генетической конституцией, а высокотератогенным эффектом высоких уровней фенилаланина в материнской крови. Соответственно необходимо, чтобы женщины с фенилкетонурией (ФКУ), планирующие беременность, начинали соблюдать диету с низким содержанием фенилаланина еще до зачатия.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Фоллинга (Folling) - синонимы, авторы, клиника
Синонимы синдрома Фоллинга. Слабоумие (Foiling). Фенилкетонурия. Фенилурия. Фенилпировиноградная олигофрения (Foiling). Фенилпировиноградная имбецильность. Альфа-кетонокислотная олигофрения. Альфа-кетонокислотное слабоумие.
Определение синдрома Фоллинга. Этиологически самостоятельная форма олигофрении, вызванная наследственным расстройством аминокислотного обмена. Относится к ферментопатиям.
Автор. Foiling Ivar Asbjorn — современный норвежский врач, Осло. Синдром был впервые описан в 1934 г.
Симптоматология синдрома Фоллинга:
1. Тяжелая олигофрения (идиотия или имбецильность).
2. Задержка общего физического развития, малый рост, инфантилизм.
3. Общая недостаточность пигментации (светлые волосы, светло-голубые глаза) (не обязательный признак), светочувствительность, склонность к фотодерматозам, гипергидроз (в результате его больные издают характерный мышиный запах). Вторичная экзематизация кожи и склонность к развитию интертриго.
4. Расстройства движений и тонуса: ригидность, гипер- или дискинезии (хорео-атетотические и миоклонические гиперкинезы). При стоянии и ходьбе все суставы слегка согнуты (поза питекантропа). Повышение рефлексов. Эпилептиформные припадки.
5. Биохимия крови: гипофосфатемия, уменьшение щелочных резервов, гипогликемия натощак, патологическая реакция Staub—Traugott.
6. Выделение с мочой повышенных количеств фенил-альфа-кетоновой кислоты [выявляемая при помощи хлористого железа (FeCl)3 насыщенно зеленая окраска]. Однако степень выделения этой кислоты не соответствует степени слабоумия.
7. Часто обнаруживают другие пороки развития: spina bifida, заячья губа, расщепленное небо, брахицефалия, пороки развития головного мозга, ногтей и т. д.
8. Иногда у братьев и сестер больных выявлют обменные нарушения, но без слабоумия.
Этиология и патогенез синдрома Фоллинга. Наблюдают семейные случаи заболевания, а также кровное родство родителей. По-видимому, наследование носит рецессивный характер. Речь идет об обменном расстройстве по типу «врожденной ошибки обмена». При этом ферментативная система печени характеризуется патологическим состоянием фенилаланингидроксилазы, принимающей участие в нормальном построении фенилаланина. В результате наличия больших количеств фенилаланина и фенил-альфа-кетоновой кислоты частично инактивируются и другие ферменты («компенсаторное торможение»). В результате патологического образования фенилаланина в крови и тканевых жидкостях (спинномозговая жидкость) его наряду с другими аминокислотами обнаруживают в моче. Часть его в почках окисляется до фенил-альфа-кетоновой кислоты.
Дифференциальный диагноз. S. Abderhalden—Fanconi (см.). Олигофрении, не связанные с обменными нарушениями. S. Bessman—Baldwin (см.).
Наследственные нарушения обмена аминокислот представляют собой наиболее изученную группу генетически детерминированных энзимопатий. Они обусловлены рецессивными мутациями генов, локализованных в аутосомах. При большинстве заболеваний известны молекулярные механизмы, приводящие к формированию патологии.
В основе патогенеза заболеваний этой группы лежит избирательное снижение активности фермента, участвующего в метаболизме той или иной аминокислоты. В результате энзиматического дефекта аминокислоты не утилизируются в организме, а в тканях и биологических жидкостях накапливаются недоокисленные продукты нарушенного метаболизма, обладающие токсическим действием на ткани и органы, в первую очередь на нервную систему.
Большинство нарушений обмена аминокислот проявляется в первые недели и месяцы жизни диспентическим синдромом, неврологическими симптомами и изменениями кожи.
Совершенствование методов диагностики позволило установить частоту этой группы заболеваний и определить их значение и удельный вес в структуре патологии раннего возраста. Частота наследственных нарушений обмена аминокислот колеблется от 1 : 10 000 до 1 : 100 000 новорожденных. Частота гетерозиготных носителей патологического гена составляет в общей популяции 1 : 100 — 1 : 400.
В настоящее время апробирована и внедряется в практику лечебных учреждений этапная биохимическая система диагностики наследственных нарушений обмена аминокислот. Первый этап обследования с помощью качественных проб и полуколичественных методов ставит своей целью выявление группы детей с повышенным содержанием аминокислот в крови и увеличенной экскрецией их с мочой (тотальный скрининг). Эти дети подлежат динамическому наблюдению. Им проводят количественное биохимическое исследование крови и мочи (второй этап), целью которого является идентификация патологии.
Усилия, направленные на раннюю диагностику, оправданы возможностью патогенетической терапии, основным принципом которой является «разгрузка» дефектной ферментной системы посредством исключения из рациона аминокислоты, не метаболизирующейся в организме.
В профилактике заболеваний, обусловленных нарушением обмена аминокислот, большое значение имеют медико-генетическое консультирование, выявление гетерозиготных носителей патологического гена и антенатальная диагностика. Гетерозиготные носители определяются с помощью нагрузочных тестов, выявляющих слабость той или иной ферментной системы. При гетерози-готности мутантного гена активность детерминируемого им фермента, хотя и обеспечивает нормальную жизнедеятельность организма, но ниже, чем у гомозигот по нормальному гену.

Фенилпировиноградная олигофрения (ФПО)
Наследственное заболевание, обусловленное нарушением обмена фенилаланина, описано A. Foiling в 1934 г. Встречается с частотой 1 : 10 000 новорожденных.
В основе патогенеза заболевания лежит дефект (отсутствие или инактивация) фермента фенилаланин-4-гидроксидазы в печени, катализирующего реакцию гидроксилирования фенилаланина в тирозин. Накопление в организме фенилаланина и его патологических производных приводит к вторичным нарушениям обмена веществ. Накопление в мозговой ткани кетокислот блокирует ферментный метаболизм мозга. Недостаточное усвоение фенилаланина приводит также к дефициту тирозина и нарушению синтеза меланина и катехоламинов.
Патоморфологическое исследование выявляет демиелинизацию и глиозное перерождение центральной нервной системы.
Признаки заболевания появляются в период новорожденности или несколько позже. Характерен внешний вид больных: светлые волосы, голубые глаза, отсутствие пигментации кожи. Часто наблюдаются экссудативный диатез, дерматиты, экземы. От больных исходит специфический «затхлый мышиный» запах. Дети вялые, адинамичные или, наоборот, беспокойные, беспричинно кричат, тревожно спят, плохо сосут, часто срыгивают.
Основной клинический симптом болезни — задержка психического развития, которая может быть выражена в различной степени. У некоторых детей на фоне снижения общего уровня психического развития отмечаются периоды улучшения: появляется улыбка, гуление, слежение за предметом. Спустя короткий промежуток времени эти функции снова угасают. Часто наблюдаются развернутые или малые судорожные припадки, которые появляются обычно во втором полугодии жизни. Двигательные нарушения на ранней стадии характеризуются снижением мышечного тонуса. В более старшем возрасте у некоторых больных отмечаются мышечная дистония и гипертония. Сухожильные рефлексы высокие с расширенными зонами, клонусы стоп. Затем могут присоединяться координаторные нарушения, гиперкпнезы. Дети поздно начинают сидеть, ходить. Походка спастико-атактическая.
Изменения со стороны черепномозговых нервов включают косоглазие, нистагм. Отмечается нарушение зрительно-моторной координации.
В плазме крови больных повышен уровень фенилаланина (до 0,15 г/л и выше) и кетокислот. Встречаются также менее тяжелые формы заболевания, обусловленные патогенетически иными нарушениями метаболизма фенилаланина.
Скринирующими тестами на ФПО являются проба Феллинга и тест с 2,4-динитрофенилгидразином. Следует отметить, что эти тесты могут быть положительными и при других нарушениях обмена аминокислот, а также при отсутствии клинических признаков заболевания. Более специфическим является тест Гатри.
Дифференциальный диагноз ФПО у детей раннего возраста проводят с другими нарушениями аминокислот, последствиями асфиксии, внутричерепной родовой травмы и инфекционных поражений центральной нервной системы. ФПО дифференцируется от транзиторной фенилкетонурпи и нарушения обмена других аминокислот с помощью хроматографии. Широкое применение получили белковые гидролизаты, такие, как лофенолак, кетонил, гипофенат, цимогран, минафен, а также смеси альфа-аминокислот. Диетический рацион считается правильным, если уровень фенилаланина в крови не превышает 0,02— 0,06 г/л.
Гетерозиготные носители патологического гена выявляются с помощью нагрузочных тестов с фенилаланипом. Частота их в популяции составляет в среднем 1:50—1:70. Предпринимаются попытки антенатальной диагностики ФПО путем определения концентрации фенилаланина в амнотической жидкости.
- Вернуться в оглавление раздела "Неврология."
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Фенилкетонурия ( Болезнь Феллинга , Фенилпировиноградная олигофрения )
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко - врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
Диагностика
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Читайте также:
- Выявление метициллинрезистентных стафилококков. Определение чувствительности к антибиотикам у анаэробных бактерий.
- Принципы операций при врожденной и травматической катаракте. Тактика
- Паралич третьего черепного нерва
- Лихорадка Денге . Эпидемиология лихорадки денге. Патогенез лихорадки денге. Диагностика, лечение и профилактика лихорадки денге.
- Диагностика мастоидита. Атипичное течение мастоидита