Наследственные нейропатии
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Полинейропатии (ПНП) относятся к числу самых распространенных болезней периферической нервной системы. Это группа заболеваний, характеризующаяся множественным и диффузным поражением корешков, сплетений и периферических нервов.
В зависимости от того, какие нервные волокна в большей степени вовлекаются в патологический процесс, возникают следующие жалобы:
чувствительные симптомы: пациента беспокоят онемение, покалывание, жжение, "ползанье мурашек" в кистях и/или стопах; неустойчивость при ходьбе, усиливающаяся в темноте и при закрывании глаз;
двигательные симптомы: развивается неловкость, слабость в кистях и/или стопах; похудание мышц рук и ног; нарушение мелкой моторики (сложности при застегивании пуговиц и молнии, завязывании шнурков и т.д.); затруднения при ходьбе ("шлепанье" стоп, трудности при подъеме и спуске с лестницы и т.д.);
вегетативные симптомы: колебание цифр артериального давления, "перебои" в работе сердца, запоры или диарея, сухость кожи или повышенная потливость, снижение либидо, нарушение эрекции.
При полинейропатии может происходить как изолированное поражение определенного типа нервных волокон, так и их сочетание, что приводит к появлению разнообразной клинической картины.
По скорости развития выделяют:
острые полинейропатии развиваются менее чем за четыре недели: наиболее часто это синдром Гийена-Барре;
подострой полинейропатию называют, если болезнь развивается за 4-8 недель;
хроническое течение, когда болезнь развивается более двух месяцев (8 недель). Хроническое течение более характерно при алкоголизме, некомпенсированном сахарном диабете, вегетарианстве, или просто неправильном питании, приводящем к дефициту витаминов группы В, наследственных причинах, воспалительных процессах с вовлечением аутоиммунных механизмов, поражающих преимущественно миелиновую оболочку нервов и др..
Кроме вышеуказанных причиной даннного заболевания могут быть: применение нейротоксичных препаратов, проведение химиотрерапии интоксикация солями тяжелых металлов, некоторые хронические соматические заболевания с явлениями недостаточности печени, почек, заболевания эндокринных органов, например, щитовидной железы.
Также полинейропатия возникает при аутоиммунных болезнях, как поражающих преимущественно периферические нервы (синдром Гийена-Барре, мультифокальная моторная невропатия, хроническая воспалительная демиелинизирующая полинейропатия, парапротеинемическая полинейропатия), так и системных болезнях соединительной ткани (ревматоидный артрит, болезнь Съегрена, системная красная волчанка).
В развитии полинейропатии этиологическую роль часто имеют инфекционные факторы как бактериальной природы (боррелиоз, ботулизм, дифтерия,сифилис, Вич-инфекция), так и вирусной( грипп, цитомегаловирус, герпес и др.). Особое место в группе полинейропатий занимают наследственные нейродегенеративные заболевания, такие как транстиретиновая семейная амилоидная полинейропатия, полинейропатия при порфирии, наследственная нейропатия со склонностью к параличам от сдавления.
Важно
Для того, чтобы определить тактику лечения и верно составить прогноз принципиально важно правильно установить причину заболевания.
Естественно начинаться это должно с тщательного сбора анамнеза пациента и подробного неврологического осмотра. Врач-невролог анализирует полученные в процессе консультации сведения и, опираясь на свой опыт и знания, обнаружив симптомы полинейропатии, назначает обследование.
- ЭНМГ (электронейромиография)-метод является важнейшим не только для определения повреждения периферического нерва, но и для уточнения характера повреждения (нарушение миелиновой оболочки, блока проведения импульса или аксональное повреждение).
- УЗИ периферических нервов.
- МРТ нервных сплетений с контрастированием.
- Проведение биопсии (исследование нерва морфологическими методами)
- Выполнение люмбальной пункции.
Для диагностики и уточнения причин проводят лабораторные исследования:
- клинический и биохимический анализ крови;
- определяется уровень витаминов группы В, гомоцистеин крови: определяют наличие антител к наиболее распространенным инфекциям- ВИЧ, сифилис, гепатиты В и С, и др по показаниям);
- если имеются основания подозревать наследствненную причину заболеваний проводится комплекс генетических исследований;
- определяется белковый состав сыворотки крови с иммунофиксацией freelite;
- проводится анализ спинномозговой жидкости.
Для исключения паранеопластических синдромов выполняется онкоскрининг:
- тщательно обследуют молочные железы (УЗИ и маммография);
- легкие (КТ);
- желудочно-кишечный тракт (выполняют эндоскопические исследования); УЗИ органов брюшной полости, малого таза, половых органов;
- при необходимости ПЭТ-КТ.
Лечение
Лечение полинейропатии включает в себя максимально возможное устранение причины, приведшей к её развитию.
- Так, если имеется диабетическая полинейропатия, то необходимо строго и постоянно контролировать сахар крови, находиться под наблюдением врача-эндокринолога, чтобы не допускать декомпенсации основного заболевания.
- Если полинейропатия развилась на почве алкоголизма, надо полностью отказаться от приема алкоголя.Необходимо наладить питание и провести лечение витаминами при развившемся их дефиците.
- Лечение полинейропатий, в основе которых лежат аутоиммунные процессы называют патогенетической терапией. При этом лечение также индивидуализировано и подбирается в зависимости от типа заболевания, с учетом всех нюансов механизма болезни и индивидуальных особенностей пациента. Это лечение высокими дозами глюкокортикоидных гормонов, использование в лечении цитостатиков и препаратов моноклональных антител, проведение высокодозной внутривенной иммунотерапии препаратами иммуноглобулина человеческого класса G и высокообъемного программного плазмафереза.
Для некоторых наследственных полинейропатий также разработана терапия с учетом патогенеза заболевания:
- при болезнь Фабри применяется заместительная терапия ферментами;
- порфирийная полинейропатия назначается аргинат гема, исключается применение у пациента порфириногенных лекарственных препаратов;
- при болезни Рефсума рекомендуется диета, при которой исключают зеленые овощи, содержащие хлорофилл, а накопление фитановой кислоты удается устранить с помощью сеансов плазмафереза.
- широко используется симптоматическая терапия для лечения неприятных ощущений, вызванных полинейропатией, таких как онемение, жжение, ползание мурашек, боли.
- врачи используют в своей практике анестетики, анальгетики, антиконвульсанты.
- применяются методы психотерапии, иглорефлексотерапии, БОС (биологическая обратная связь).
- назначают также лечение физическими факторами, а именно: массаж, физиотерапия, электронейромиостимуляция, механо- и роботизированная терапия, которые позволяют снизить количество и дозы лекарственных препаратов, купировать симптомы и достичь эффектов в более быстрые сроки.
Автор статьи: Бутцев Вячеслав Александрович (врач - невролог)
Оформите заявку на сайте, мы свяжемся с вами в ближайшее время и ответим на все интересующие вопросы.
Наследственные полинейропатии
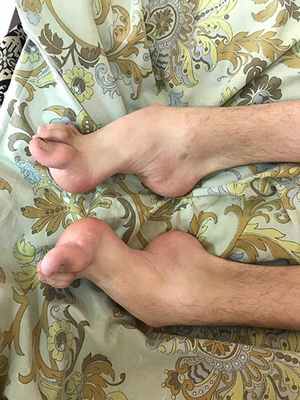
Наследственные полинейропатии
(информация для пациентов)
Что такое "Наследственные полинейропатии"?
Наследственные полинейропатии – это большая группа клинически разнообразных полинейропатий с возможным вовлечением различных органов и систем, в том числе и центральной нервной системы, которые развиваются в результате мутаций (поломок) в генах человека.
- "Наследственные заболевания развиваются с рождения или в раннем детстве"
Это не так - дебют наследственной болезни возможен и во взрослом возрасте, в том числе и после 50 лет. Более того, зачастую пациенты не могут точно назвать возраст начала заболевания, так как симптомы развиваются незаметно. - "Наследственная патология должна быть и у близких родственников. А если в роду ни у кого нет, значит и у меня тоже нет".
Это не так - наследственные заболевания передаются разными путями: по аутосомно-доминантному, аутосомно-рецессивному, Х-сцепленному типу, также возможен митохондриальный тип наследования и др. Часто в семье есть асимптомные носители мутантного гена без клинических признаков заболевания, а симптомы болезни могут быть только у одного из членов семьи. Поэтому отсутствие отягощенного семейного анамнеза не исключает наследственный генез заболевания.
- Наследственные моторно-сенсорные нейропатии (НМСН)
- Наследственная нейропатия со склонностью к параличам от сдавления (ННСПС)
- Наследственные сенсорные и автономные нейропатии (НСАН)
- Болезнь Фабри
- Транстиретиновая семейная амилоидная нейропатия (ТТР-САП)
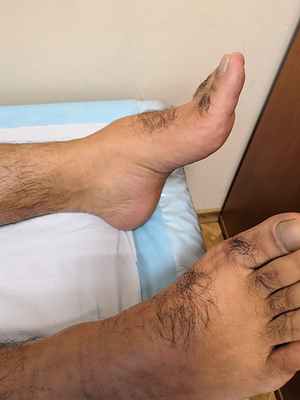
Как часто диагностируются наследственные полинейропатии среди населения?
Частота встречаемости всех форм НМСН варьирует от 10 до 40 случаев на 100 000 населения в различных популяциях. На НМСН 1 типа приходится до 70% всех случаев НМСН. Распространенность ННСПС 2-5 случая на 100 000 населения. Болезнь Фабри - 1 случай на 40 000 - 60 000 мужчин. По примерным подсчетам в США встречаемость ТТР-САП составляет 1 случай на 100 000 человек. Следует отметить, что настороженность относительно дебюта наследственной патологии во взрослом возрасте среди врачей разных специальностей достаточно низкая, поэтому некоторые цифры распространенности могут быть занижены.
- пациентов беспокоят болезненные спазмы мышц голеней (крампи), слабость и деформация стоп, изменение походки, затруднения при беге или подъеме по лестнице;
- постепенно слабость развивается в кистях, в результате чего появляются затруднения при застегивании пуговиц, открывании двери ключом и т.д. В целом руки вовлекаются не ранее чем через 10 лет после появления первых симптомов болезни;
- в меньшей степени беспокоит онемение кистей и стоп, часто эти изменения игнорируются пациентом.
- лучевой нерв на уровне плеча (спиральный канал), при этом развивается слабость разгибателей кисти, кисть "висит" как плеть, возникает онемение наружного края предплечья и кисти;
- малоберцовый нерв на уровне коленного сустава (фибулярный канал), при этом развивается слабость разгибателей стопы, стопа начинает "шлепать", возникает онемение наружного края голени и стопы;
- локтевой нерв на уровне локтевого сустава (кубитальный канал), при этом развивается слабость межкостных мышц кисти, мышцы - отводящих мизинец и других, возникает онемение 4 и 5 пальцев кисти;
- срединный нерв на уровне запястья (карпальный канал), при этом развивается слабость мышц возвышения большого пальца, возникает онемение 1-3 пальцев кисти, характерен болевой синдром;
- и т.д. с развитием соответствующей клинической картины поражения того или иного нерва.
Степень поражения нерва (защемления в канале) может быть различной - от незначительной, когда беспокоит только онемение и парестезии, до более выраженной, когда помимо чувствительных нарушений развивается и слабость мышц.
Развитию симптомов, как правило, предшествует незначительная травма, нахождение длительное время в неудобной статической поза (на четвереньках, на корточках, облокотившись на локоть и др.), ношение неудобной одежды, непривычная физическая нагрузка и прочее.
Мышечная слабость, как правило, сохраняется в течение нескольких дней или недель, а затем сила мышц постепенно восстанавливается (в течение нескольких недель или месяцев). Эпизоды "защемлений" нервов имеют тенденцию к повторению (всего у пациентов может быть от 1 до 10 таких эпизодов). Следует учитывать, что часто лечащие врачи не задумываются о том, что причиной рецидивирующих (повторяющихся) туннельных невропатий – является наследственная патология, что приводит к поздней диагностике и неоправданным медицинским вмешательствам (операциям по поводу туннельных невропатий).
Болезнь Фабри - редкое генетическое заболевание, обусловленное мутацией гена GLA, картированного на длинном плече хромосомы Хq 22.1 и контролирующего структуру фермента альфа-галактозидазы А. Нередко начало заболевания отмечается в подростковом возрасте. Тип наследования - сцепленное с Х-хромосомой.
- жгучая колющая боль в ладонях и стопах; острые приступы мучительной боли в кистях и стопах (кризы Фабри);
- нарушение потоотделения; непереносимость жары/холода;
- сыпь на коже (ангиокератомы);
- помутнение роговицы в виде завитка, которое не ослабляет зрение;
- шум в ушах, потеря слуха;
- желудочно-кишечные расстройства, диарея;
- кардиологические проявления (включая увеличенное сердце и нарушение ритма);
- нарушение функции почек, которое в конечном итоге приводит к терминальной стадии хронической почечной недостаточности;
- нарушение мозгового кровообращения (чаще ишемический инсульт, но может быть и внутримозговое кровоизлияние, или венозный тромбоз).
- анализ истории развития заболевания
- оценка неврологического статуса
- общий клинический и развернутый биохимический анализы крови;
- RW, анти-ВИЧ, НВsAg и анти-HCV;
- общий анализ мочи;
- анализ крови на уровни витаминов группы В, гомоцистеин;
- электрофорез белков сыворотки и мочи с иммунофиксацией; ;
- анализ цереброспинальной жидкости (ликвор) и другие.
- уровень фитановой кислоты в крови методом хроматомасс-спектрометрии (повышение при болезни Рефсума);
- определение активности лизосомного фермента альфа-галактозидазы А, в том числе с помощью флюориметрического метода ("сухое" пятно) - исключение болезни Фабри;
- биопсия икроножного нерва, малых слюнных желез и абдоминального жира – для выявления отложения амилоида, характерного для ТТР-САП).
- Однако окончательный диагноз той или иной наследственной полинейропатии может быть подтвержден только по результатам генетического анализа. Оптимальным является генетическое обследование по следующему алгоритму:
- при убедительной клинико-анамнестической и параклинической картине в пользу той или иной наследственной полинейропатии проводится прицельная генетическая диагностика одного гена, в том числе с использованием скрининг тестов методом "сухого пятна";
- если подозревается наследственный генез полинейропатии, тип её не определен целесообразно проведение более широкого генетического анализа - секвенирования нового поколения (NGS).
- при болезни Фабри - это фермент-заместительная терапия рекомбинантными препаратами альфа-галактозидазы А
- при ТТР-САП – стабилизация молекулы транстиретина (Тафамидис), трансплантация печени;
- при порфирийной полинейропатии - введение аргината гема;
- при болезни Рефсума – диетотерапия с ограничением поступления фитановой кислоты (исключение потребления зеленых овощей, говядины, рыбы (тунец, пикша, треска)), плазмаферез.
При некоторых наследственных нейропатиях, для которых характерно поражение органов и систем, важно мониторировать функцию сердечно-сосудистой системы и других органов, наблюдаться у профильных специалистов.
- рекомендуется вести здоровый образ жизни, правильно и сбалансировано питаться, полностью отказаться от приема алкоголя, воздерживаться от вегетарианства;
- исключить, ограничить прием лекарственных препаратов с нейротоксическим действием;
- проводится коррекция ортопедических нарушений: подбор стелек, стоподержателей, коленоупорников, использование дополнительной опоры, кинезиотейпирование;
- регулярно, в поддерживающем режиме проводятся реабилитационные мероприятия: миостимуляция, массаж, растяжка, ЛФК.
Прием "нейрометаболических" препаратов и витаминов группы В не целесообразно, в связи с отсутствием доказательной базы.
.
Какой прогноз данного заболевания?
Прогноз определяется типом наследственной полинейропатии и многими сопутствующими факторами. Так, при НМСН 1 и 2 типа при позднем дебюте крайне замедленное развитие мышечной слабости приводит к тому, что больные приспосабливаются к своему дефекту и, несмотря на слабость и похудание кистей и стоп, могут выполнять ручную работу, свободно передвигаться. Даже при многолетнем течении сохраняется способность к самостоятельной ходьбе. Наследственная нейропатия со склонностью к параличам от сдавления также имеет относительно благоприятный прогноз при соблюдении рекомендаций по ограничению длительных статических поз и снижению потенциальных провокаторов компрессий нервов в повседневной жизни. НМСН III, V типов вызывают тяжелый двигательный дефект и в большинстве случаев приводит к выраженной инвалидности. При ранней диагностике наследственных нейропатий, ассоциированных с метаболическим дефектом (ТТР-САП, болезнь Рефсума, порфирия, болезнь Фабри) и назначении необходимой терапии возможно пролонгировать наступление инвалидизации и улучшить качество жизни.
В ФГБНУ НЦН проводят консультации сотрудники Центра заболеваний периферической нервной системы, сотрудники 5 неврологического отделения, профилем которого является диагностика и лечение наследственных заболеваний нервной системы. Консультации пациентов проводятся амбулаторно в рамках ОМС и на коммерческой основе.
На базе ФГБНУ НЦН проводится весь перечень необходимых для уточнения диагноза обследований, разработаны подходы к комплексной восстановительной терапии при наследственных полинейропатиях.
Наследственные нейропатии
Наследственные нейропатии представлены гетерогенной группой заболеваний, которым присуще несколько общих признаков. Во-первых, генетическая детерминированность, во-вторых — объемное поражение периферических нервных волокон. Данный вид патологии является изменчивым, каждый конкретный случай отличается от других особенностями наследования, клиническими проявлениями, тяжестью, прогнозом и т. д. Наследственные нейропатии встречаются достаточно часто. Среди всех форм полинейропатий на их долю приходится больше половины.
Клинические проявления
Заболевание развивается в результате мутации в определенных генах, которых насчитывается большое количество. В зависимости от конкретных генетических нарушений и их локализации, у пациентов с наследственной нейропатией могут развиваться следующие симптомы:
- слабость мышц нижних конечностей;
- паралич голосовых связок;
- задержка моторного развития;
- затруднение передвижения;
- сниженный порог температурной и болевой чувствительности;
- сильная боль в области нижних конечностей;
- нарушение поверхностной чувствительности;
- полная потеря чувствительности;
- деформация стоп;
- образование трофических язв на ногах;
- отечность, онемение нижних конечностей.
Для каждой формы заболевания характерен определенный набор нарушений, которые будут иметь различную степень выраженности и тяжесть протекания.
Классификация наследственных нейропатий
Наследственная нейропатия включает в себя несколько подвидов исходя из клинического течения:
- моторная,
- сенсорная,
- моторно-сенсорная,
- сенсорно-вегетативная.
Кроме того, наследственные нейропатии отличаются между собой и по другим признакам.
- Характер наследования: аутосомно-доминантный, аутосомно-рецессивный, Х-сцепленный доминантный.
- Первые симптомы могут проявляться уже с рождения, а при отдельных формах только после 70 лет.
- Скорость проведения нервного импульса по периферическим нервным волокнам может быть снижена незначительно или отсутствовать вовсе.
Разнообразие форм наследственной нейропатии, а также значительная вариабельность течения заболевания может приводить к трудностям установления точного диагноза. Современные методы диагностики и большой практический опыт врача помогут избежать этой проблемы и достоверно выявить болезнь.
Диагностика и лечение наследственной нейропатии
Как правило, пациенты с наследственной нейропатией обращаются к неврологу при появлении определенных жалоб (слабость в ногах, боли, онемение). Для того чтобы точно подтвердить диагноз, специалист назначает комплексное обследование.
- Определение мутаций в специфических генах. Данный вид исследования можно пройти в медико-генетическом центре «Геномед».
- Скорость проведения импульса по нервным волокнам.
- Анализы крови.
- Миография.
В зависимости от конкретной клинической картины врач может назначить другие методы диагностики, которые помогут получить необходимую информацию.
Лечение наследственной нейропатии в настоящее время не разработано. Пациентам назначается поддерживающая терапия, которая помогает сгладить основные симптомы и улучшить качество жизни. Точный план лечения подбирается индивидуально.
Наследственные нейропатии
Наследственные нейропатии включают в себя различные врожденные дегенеративные периферические нейропатии (например, болезнь Шарко-Мари-Тута).
Наследственные невропатии классифицируются как
Двигательные и сенсорные
Сенсорные и вегетативные
Наследственные невропатии могут быть первичными или вторичными по отношению к другим наследственным заболеваниям, включая болезнь Рефсума Классическая болезнь Рефсума Пероксисомы являются внутриклеточными органеллами, которые содержат ферменты для бета-окисления. Эти ферменты выполняют такие же функции, что и имеющиеся в митохондриях, за исключением того. Прочитайте дополнительные сведения , порфириновую болезнь Обзор порфирий (Overview of Porphyrias) Порфирии - это редкие нарушения при которых аномалии метаболизма гемоглобина обусловлены генетической или приобретенной недостаточностью ферментов биосинтеза гема, что приводит к накоплению. Прочитайте дополнительные сведения и болезнь Фабри Болезнь Фабри Болезнь Фабри – это сфинголипидоз, наследственное нарушение метаболизма, вызванное дефицитом α-галактозидазы А, который вызывает появление ангиокератом, акропарестезий, помутнение роговицы. Прочитайте дополнительные сведенияСенсорно-моторные нейропатии
Выделяют 3 типа (CMT1, CMT2, и CMT3) моторной и сенсорной нейропатий, каждый из которых начинается в детстве. Реже встречающиеся типы дебютируют с рождения и приводят к более тяжелой инвалидизации.
Обычно аутосомно-доминантные, рецессивные или X-сцепленные заболевания CMT1 и CMT2 (разновидности болезни Шарко–Мари–Тута, или перонеальной амиотрофии) являются наиболее частыми. Заболевание I типа связано с дупликацией гена (запасного гена) периферического миелинового белка 22 типа (PMP22), расположенного на коротком плече 17-й хромосомы; он составляет 70–80% случаев СМТ1.
CMT1 и CMT2 характеризуются слабостью и атрофией, преимущественно малоберцовых и дистальных мышц ног. Часто в семейном анамнезе пациента имеются данные о нейропатии. Данные анамнеза заболевания могут варьировать: у некоторых пациентов заболевание протекает бессимптомно и у них отмечается только замедление скорости проведения по нервам (при исследовании скорости распространения возбуждения); у других пациентов заболевание проявляется более тяжело.
Наследственная моторно-сенсорная нейропатия 1 типа (болезнь Шарко-Мари-Тута) начинается в среднем детском возрасте со слабости в стопах и медленно прогрессирующей дистальной амиотрофии («ноги аиста»). Характерные амиотрофии кистей развиваются позднее. Вибрационная, температурная и болевая чувствительность нарушаются по типу «перчаток» и «носков». Выпадают глубокие сухожильные рефлексы. Иногда единственными признаками, которые выявляются у членов семьи – носителей заболевания, – это деформации стоп (высокий свод вплоть до «полой» стопы) и молоткообразная деформация пальцев на ногах. Скорость проведения нервных импульсов замедлена, а дистальные латентности удлинены. Имеет место сегментарная демиелинизация и ремиелинизация. Иногда удается пальпировать утолщенные периферические нервы. Заболевание прогрессирует медленно и не уменьшает продолжительность жизни. При одном из подтипов у мужчин симптомы более выражены, а у женщин проявляются слабо или вовсе отсутствуют.
Генетика СМТ2 менее понятна. Этот тип составляет около 25% всех случаев амиотрофии ШМТ; патогенные мутации с множеством подтипов были выявлены только у небольшого количества пациентов, вероятно, у 25%. CMT2A является наиболее распространенным фенотипом CMT2, чаще всего из-за мутации в гене, который кодирует митохондриальный гибридный белок митофусин-2 (MFN2). CMT2A обычно является аутосомно-доминантным и аксональным заболеванием. Болезнь прогрессирует медленно, слабость обычно развивается в более позднем возрасте. Скорость проведения возбуждения относительно нормальна, однако амплитуда потенциалов действия чувствительных нервов снижена, определяются полифазные потенциалы действия мышц. При биопсии выявляется аксональная (валлеровская) дегенерация.
ШМТ 3 (также известная как болезнь Дежерина–Сотта) – редкая врожденная гипомиелинизирующая нейропатия, аутосомное доминантное или рецессивное заболевание с мутацией в нескольких генах, включая PMP22, MPZ, EGR2 которое начинается в детстве с прогрессирующей слабости, потери чувствительности и отсутствия глубоких сухожильных рефлексов. Вначале напоминает болезнь Шарко–Мари–Тута, но мышечная слабость прогрессирует быстрее. Демиелинизация и ремиелинизация приводят к утолщению периферических нервов и формированию «луковиц», что определяется при биопсии.
Сенсорно-вегетативные нейропатии
Наследственные сенсорно-вегетативные нейропатии встречаются редко. Описано семь основных типов заболевания.
Утрата болевой и температурной чувствительности в дистальных отделах выражена больше, чем нарушение вибрационной чувствительности и чувства локализации. Основное осложнение – мутиляция стопы из-за снижения болевой чувствительности, что сопровождается риском инфицирования и остеомиелита Остеомиелит Остеомиелит – воспалительное деструктивное заболевание кости, вызываемое бактериями, микобактериями или грибами. К его симптомам относятся боли и болезненность в области пораженной кости, сопровождающиеся. Прочитайте дополнительные сведенияДиагностика наследственной нейропатии
Характерное распределение мышечной слабости, деформации стопы и наследственный характер заболевания позволяют заподозрить наследственную нейропатию, которая должна быть подтверждена при нейрофизиологическом исследовании.
Возможен генетический анализ.
Лечение наследственных нейропатий
Фиксация помогает при коррекции отвислой стопы; ортопедическое хирургическое вмешательство может помочь стабилизировать стопу.
Могут помочь физиотерапия (увеличить мышечную силу) и эрготерапия; консультация по выбору профессии может помочь молодым людям подготовиться к профессиональной деятельности, несмотря на прогрессирование заболевания.
Ключевые моменты
Наследственные нейропатии могут поражать двигательные и сенсорные нервы, чувствительные нервы, чувствительные и вегетативные нервы или только двигательные нервы.
Выделяют 3 основных типа моторных и сенсорных нейропатий, которые различают по степени тяжести и скорости прогрессирования; почти все они начинаются в детстве.
Следует использовать фиксаторы для коррекции отвислой стопы, а также рекомендованы физическая и трудотерапия для того, чтобы помочь пациентам сохранить функцию; иногда требуется проведение ортопедического хирургического вмешательства.
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Наследственная моторно-сенсорная нейропатия (болезнь Шарко-Мари-Тута) тип I, дупликация на хромосоме 17 в области гена РМР22 м.
К настоящему времени картировано более 40 локусов, отвечающих за наследственные моторно-сенсорные нейропатии, идентифицировано более двадцати генов, мутации в которых приводят к развитию клинического фенотипа НМСН.
Мутации в гене РМР22 приводят также к развитию болезни Дежерина-Сотта, болезни Русси-Леви, воспалительной демиелинизирующей нейропатии, нейропатии с параличом от сдавления.
Болезнь Шарко-Мари-Тута (ШМТ), или невральная амиотрофия Шарко-Мари, известная и как наследственная моторно-сенсорная невропатия (НМСН) - обширная группа генетически гетерогенных заболеваний периферических нервов, характеризующихся симптомами прогрессирующей полинейропатии с преимущественным поражением мышц дистальных отделов конечностей. НМСН являются не только самыми частыми среди наследственных заболеваний периферической нервной системы, но и одними из самых частых наследственных заболеваний человека.
Возникновение заболевания в большинстве случаев обусловлено избыточной экспрессией периферического белка миелина (РМР22)вследствие дупликации гена, который составляет от 2% до 5% миелиновых белков периферических нервов. Возникновение признаков заболевания при наличии точковых мутаций в гене РМР22 связано с нарушением процессов деградации шванновских клеток и их включения в компактный миелин. Заболевание возникает в 1-2 десятилетии жизни. У 75% больных первые признаки выявляются до 10 летнего возраста, а у остальных 25% - до 20 лет. Первыми в патологический процесс вовлекаются сгибатели стоп, что клинически проявляется их гипотрофией и нарушением походки в виде степпажа. По мере прогрессирования заболевания возникает деформация стоп в виде фридрейховой, полой или эквино-варусной и голени приобретают вид перевернутых бутылок. Поражение дистальных отделов рук возникает, как правило, спустя несколько месяцев или лет. Первыми поражаются межкостные мышцы кистей и мышцы гипотенара. По мере прогрессирования заболевания кисть приобретает вид «когтистой лапы» или «обезьяньей лапы». В области пораженных мышц рук и ног обнаруживаются расстройства поверхностной и глубокой чувствительности. В 56% случаев у больных отмечается сенситивно-мозжечковая атаксия и интенционный тремор кистей. Сухожильные рефлексы в начальной стадии болезни снижаются, и по мере прогрессирования заболевания быстро угасают. Характерным симптомом этой формы заболевания является определяемое пальпаторно утолщение нервных стволов. Наиболее часто этот симптом можно отметить в ушном и локтевом нервах. Вовлечение в процесс проксимальных мышц рук и ног не характерно и наблюдается лишь у 10% больных в пожилом возрасте. Течение заболевания медленно прогрессирующее, не приводящее к тяжелой инвалидизации. Больные до конца жизни сохраняют способность к самообслуживанию и самостоятельно передвигаются. Описано несколько больных, с клиническими проявлениями периферической нейропатии в сочетании с умственной отсталостью, дизморфическими чертами строения лица и патологией зрения, у которых делеция в области короткого плеча 17 хромосомы была более протяженная и могла быть определена при использовании цитогенетических методов. В настоящее время к этому варианту наследственных моторно-сенсорных нейропатий относят болезни Русси-Леви и Дежерина-Сотта, которые до недавнего времени выделяли в самостоятельные нозологические формы.
Электронейромиографические признаки поражения периферических нервов возникают задолго до появления первых клинических симптомов. Наличие этих признаков можно отметить, начиная с двухлетнего возраста, а у гомозигот по мутации (при наличии четырех копий РМР 22 гена) - с годовалого возраста. Основными электромиографическими признаками являются: резкое снижение скоростей проведения импульса по периферическим нервам, которое в среднем составляет 17-20 м/сек и колеблется от 5 до 34 м/сек; снижение амплитуды М-ответа; удлинение дистальной латенции и F-волны; отсутствие или резкое снижение амплитуды сенсорного потенциала.
В биоптате периферических нервов определяются специфические луковицеподобные утолщения миелиновой оболочки периферических нервов, образованные отростками шванновских клеток и базальной мембраны, чередующиеся с участками де- и ремиелинизации.
Литература
Специальной подготовки к исследованию не требуется.
Обязательны к заполнению:
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Литература
Типичная клиническая картина. Обязательным условием является снижение скоростей проведения импульса по периферическим нервам.
Литература
Интерпретация результатов исследования содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
другие формы НМСН, сопровождающиеся снижением скоростей проведения импульса по периферическим нервам.
Читайте также:
- Сесамоидит
- Патологическое заживление переломов. Ложный сустав - псевдоартроз
- Глюкагон и глюконеогенез. Регуляция секреции глюкагона
- Абсансные припадки - абсансы. Разновидности абсансов
- Легочная гипертензия при дефекте межпредсердной перегородки (ДМПП). Эуфиллин и нитроглицерин при легочной гипертензии