Оптиконевромиелит на МРТ
Добавил пользователь Morpheus Обновлено: 07.01.2026
Оптиконевромиелит является демиелинизирующим заболеванием, которое преимущественно поражает глаза и спинной мозг, но может затрагивать и другие структуры центральной нервной системы (ЦНС), содержащие аквапорин-4.
Расстройство из спектра оптиконевромиелита вызывает острый неврит оптического нерва Неврит зрительного нерва Неврит зрительного нерва - это воспаление зрительного нерва. Симптомы воспаления зрительного нерва обычно носят односторонний характер, характерна боль и полная или частичная потеря зрения. Прочитайте дополнительные сведения , иногда двусторонний, плюс демиелинизацию шейных или грудных отделов спинного мозга. Ранее его считали разновидностью рассеянного склероза Рассеянный склероз (РС) Рассеянный склероз (РС) характеризуется появлением в головном и спинном мозге диссеминированных очагов демиелинизации. Характерные симптомы включают зрительные и глазодвигательные нарушения. Прочитайте дополнительные сведенияПри невромиелите зрительного нерва соспектральным расстройством мишенью иммунной системы становится аквапорин 4 - протеин, который присутствует в астроцитах головного мозга, и, в частности, в спинном мозге и зрительных нервах, а также, возможно, и в других органах-мишенях. Астроциты повреждаются вследствие аутоиммунно-опосредованных реакциях воспаления, а также при демиелинизации.
Симптомы и признаки заболевания СОНМ
Симптомы расстройства из спектра оптиконевромиелита включают потерю зрения, мышечные спазмы, пара- или тетрапарез и недержание.
Специфические характерные проявления включают:
Тяжелый двусторонний неврит зрительного нерва, который включает хиазму зрительных нервов, приводящую к потере зрения выше или ниже горизонтали (высотный дефект поля зрения) или потере остроты зрения (20/200 или хуже)
Синдром полного повреждения спинного мозга, особенно с пароксизмальными тоническими спазмами
Синдром area postrema, вызывающий трудноизлечимую икоту или тошноту и рвоту (area postrema является структурой, контролирующей рвоту и расположенной на дне 4-го желудочка)
Острый поперечный миелит, распространяющийся на 3 и более смежных сегмента спинного мозга
Диагностика NMOSD (заболеваний оптиконейромиелитного спектра)
МРТ головного и спинного мозга
Исследование зрительных вызванных потенциалов
Приведенные ниже особенности помогают отличить зрительный нейромиелит от рассеянного склероза (РС):
Оптиконевромиелит затрагивает несколько (обычно ≥ 3) соседних спинальных сегментов спинного мозга, тогда как РС обычно поражает одиночный сегмент.
По данным МРТ поражения белого вещества головного мозга при оптиконевромиелите не характерны, в отличие от РС.
На результатах МРТ морфология и локализация поражений отличаются от таковых результатов при РС.
Зрительные вызванные потенциалы могут способствовать дифференцированию оптиконейромиелита от других нейропатий зрительного нерва. Обнаруженные при нейромиелите зрительного нерва со спектральным расстройством находки включают в себя сниженную амплитуду или пролонгированную латентность. Этот тест также полезен для выявления клинически незаметных повреждений, существующих еще до появления симптомов.
Можно выполнить анализы для измерения антител к IgG, специфичных для оптиконевромиелита со спектральным расстройством (антитела к аквапорину 4 [также именуемые NMO-IgG]) для его дифференциации от РС. Антитела к MOG (миелиновому олигодендроцитарному гликопротеину) идентифицируют подгруппу пациентов с нейромиелитом зрительного нерва, которые, по-видимому, имеют другие клинические характеристики: у них наблюдается меньше осложнений и лучшее восстановление, чем у пациентов с антителами AQP4 или при отсутствии антител.
Справочные материалы по диагностике
1. Wingerchuk DM, Banwell B, Bennett JL, et al: International consensus diagnostic criteria for neuromyelitis optica spectrum disorders. Neurology 85 (2):177–189, 2015. doi: 10.1212/WNL.0000000000001729 Epub 2015 Jun 19
Лечение ОНМ
Глюкокортикоиды и иммуномодулирующее или иммуносупрессивное лечение
Оптиконевромиелит неизлечим. Тем не менее, лечение может предотвратить, замедлить или уменьшить степень выраженности обострений.
Экулизумаб, ингибитор комплемента С5, недавно был одобрен для выработки антител к аквапорину-4 при нейромиелите зрительного нерва со спектральным расстройством. Побочные эффекты включают респираторные инфекции, головную боль и пневмонию и могут быть значительными; таким образом, пациенты должны находиться под пристальным наблюдением ( 1 Справочные материалы по лечению Оптиконевромиелит является демиелинизирующим заболеванием, которое преимущественно поражает глаза и спинной мозг, но может затрагивать и другие структуры центральной нервной системы (ЦНС), содержащие. Прочитайте дополнительные сведения ). Поскольку у одного пациента развился менингококковый сепсис, вакцинация против менингококка требуется до начала терапии.
Сатрализумаб (моноклональное антитело, против рецептора интерлейкина-6) и инебилизумаб (моноклональное антитело, против CD19 на В-клетках) были недавно одобрены для лечения аквапорин-4 антител-положительного нейромиелита. Пациентов следует тщательно контролировать на наличие инфекций, таких как инфекции мочевыводящих путей и респираторные инфекции.
Натализумаб и финголимод показали свою неэффективность и могут оказать вредное воздействие.
Симптоматическая терапия Симптоматическая терапияСправочные материалы по лечению
1. Pittock SJ, Berthele A, Fujihara K, et al: Eculizumab in aquaporin-4-positive neuromyelitis optica spectrum disorder. N Engl J Med 381 (7):614–625, 2019. doi: 10.1056/NEJMoa1900866 Epub 2019 May 3
Основные положения
Нейромиелит зрительного нерва со спектральным расстройством является следствием демиелинизации, при которой, как правило, имеются антитела к аквапорину-4 или миелиновому гликопротеину олигодендроцитов.
Типичные симптомы включают в себя потерю зрения, мышечные спазмы, пара- или тетрапарез и инконтиненцию.
Для диагностики оптиконевромиелита со спектральным расстройством используется МРТ головного и спинного мозга, а также исследование зрительных вызванных потенциалов.
Лечение включает кортикостероиды и иммуномодулирующую или иммуносупрессивную терапию (например, экулизумаб, ритуксимаб).
Авторское право © 2022 Merck & Co., Inc., Rahway, NJ, США и ее аффилированные лица. Все права сохранены.
Алгоритм диагностики оптиконевромиелита (болезнь Девика) Текст научной статьи по специальности «Клиническая медицина»
Похожие темы научных работ по клинической медицине , автор научной работы — Симанив Тарас Олегович, Васильев Алексей Владимирович, Воробьёва Анна Александровна, Захарова Мария Николаевна, Переседова Анастасия Вячеславовна
Текст научной работы на тему «Алгоритм диагностики оптиконевромиелита (болезнь Девика)»
Алгоритм диагностики оптиконевромиелита (болезнь Девика)
Т.О. Симанив, А.В. Васильев, А.А. Воробьёва,
М.Н. Захарова, А.В. Переседова
Демиелинизирующие заболевания центральной нервной системы (ЦНС) - актуальная медико-социальная проблема, так как нозологии данной группы являются второй по частоте причиной инвалидизации лиц молодого возраста после черепно-мозговой травмы. Проведение дифференциальной диагностики данной группы заболеваний является важнейшей задачей неврологов, особенно на ранних стадиях заболевания, поскольку подходы к терапии рассеянного склероза (РС), оптиконевромиелита (ОМ), острого рассеянного энцефаломиелита (ОРЭМ) принципиально отличаются. Сложность постановки диагноза РС подтверждается данными исследований: в 10% случаев аутопсии РС не выявляется, а около 20% пациентов, направляемых на консультацию с диагнозом “рассеянный склероз”, имеют другие заболевания.
Оптиконевромиелит - вторая по частоте форма воспалительного демиелинизирующего заболевания после РС. Заболевание преимущественно распространено среди лиц африканского и азиатского происхождения, выходцев из стран Латинской Америки, коренных индейцев Северной Америки, индусов, где встречаемость РС невысока. В европейских странах и США доля ОМ среди остальных демиелинизирующих заболеваний ЦНС составляет 1-5%. Распространенность ОМ среди представителей европеоидной расы по данным различных эпидемиологических исследований составляет 0,3-4,4 на 100 000 населения. Однако многие случаи ОМ нередко ошибочно трактуются как РС, рецидивирующий поперечный миелит либо рецидивирующий ретробульбарный неврит.
Возраст начала заболевания - от детского до пожилого со снижением частоты встречаемости после 50 лет. Средний возраст первой атаки заболевания - 35-41 год при рецидивирующем типе течения заболевания и 29 лет при монофазном течении. Женщины болеют гораздо чаще
6-е неврологическое отделение ФГБУ “Научный центр неврологии” РАМН, Москва.
Тарас Олегович Симанив - аспирант.
Алексей Владимирович Васильев - канд. мед. наук, науч. сотр.
Анна Александровна Воробьёва - аспирант.
Мария Николаевна Захарова - докт. мед. наук, вед. науч. сотр.
Анастасия Вячеславовна Переседова - докт. мед. наук, и.о. рук. отделения.
(85% случаев), чем мужчины, и обычно частота заболевания у женщин выше, чем при РС. При монофазном типе течения соотношение мужчин и женщин приближается к 1 : 1.
Представления об ОМ как одном из вариантов РС сохранялись вплоть до 90-х годов прошлого века, когда дальнейшие клинические и гистопатологические исследования изменили концепцию и место ОМ среди расширяющегося спектра аутоиммунных расстройств ЦНС. Новым этапом в развитии и понимании патогенеза ОМ явилось открытие в 2004 г. V. Lennon et al. сывороточных аутоантител NMO-IgG (neuromyelitis optica - Immunoglobulin G), которые в совокупности с наличием признаков острого миелита и оптического неврита отличали ОМ от РС и других демиелинизи-рующих заболеваний. В последующие годы было показано, что NMO-IgG селективно связывается с аквапорином-4, одним из основных белков водных каналов ЦНС, локализующихся в ножках астроцитов, образующих гематоэнцефалический барьер (ГЭБ). Таким образом, в настоящее время ОМ - единственное демиелинизирующее заболевание, при котором выявлен специфический биомаркер.
В основе ОМ лежит аутоиммунный процесс, начинающийся с активной продукции специфических антител на периферии, с последующим проникновением их через ГЭБ и связыванием с белком аквапорином-4, который расположен на мембране астроцитарных ножек, участвующих вместе с капиллярами в формировании ГЭБ. Это приводит к развитию воспалительной реакции, активации системы комплемента, повышенной продукции воспалительных цитокинов (IL-17, IL-8, гранулоцитарный колониестимулирующий фактор). Нарушение клеточных механизмов транспорта воды, повреждение ГЭБ и активная инфильтрация периваскулярного пространства клеточными элементами крови (нейтрофилами, эозинофилами) способствуют развитию демиелинизации, сосудистой гиперплазии с гиали-низацией стенок капилляров, некрозу олигодендроцитов и нейронов и образованию специфических полостей как в белом, так и в сером веществе спинного мозга. Большое значение имеют В-клеточно-индуцированные реакции, что не характерно для типичного течения РС, но наблюдается при тяжелых его формах.
В отличие от РС, в основе развития которого лежат преимущественно клеточные механизмы, ОМ является ауто-
иммунной астроцитопатией/каналопатией, которая имеет гуморально-опосредованный механизм.
Другими патогенетическими механизмами при ОМ являются: 1) активация клеточного иммунитета, что приводит к инфильтрации гранулоцитов и гибели астроцитов с потерей аквапорина-4, олигодендроцитов и демиелинизации, гибели нейрональных клеток; 2) нарушение транспорта и утилизации глутамата. При действии NMO-IgG происходит уменьшение связанных с аквапорином-4 транспортеров возбуждающих аминокислот EAAT2, что нарушает транспорт глутамата. Также в пораженных астроцитах снижается синтез глутаминсинтетазы, превращающей глутамат в глутамин, и нарушается утилизация последнего. Таким образом, происходит высвобождение глутамата из астроцитов, его внеклеточное накопление с последующей гибелью олигодендроцитов по типу эксайтотоксичности.
Клиническая картина характеризуется сочетанием синдромов оптического неврита и/или продольно-поперечного миелита. Поражения зрительных нервов и спинного мозга в некоторых случаях могут возникать одновременно, но чаще - с временным интервалом, который может составлять месяцы, годы и даже десятилетия. Симптоматика нарастает в течение нескольких дней, а регрессирует неделями и месяцами, зачастую с сохранением остаточного неврологического дефицита. С каждой последующей атакой неврологический дефицит накапливается и приводит к тяжелейшим нарушениям со стороны двигательной, чувствительной, зрительной и вегетативной систем. Для оценки степени выраженности неврологического дефицита, как и при РС, используется расширенная шкала инвалидиза-ции EDSS.
При поражении зрительного нерва при ОМ имеет место следующее:
• в 80% случаев поражение предшествует миелиту;
• поражение может быть одно- либо двусторонним;
• возникновение неврита зрительного нерва одновременно с двух сторон или последовательно с одной и другой стороны в короткий срок свидетельствует о высокой вероятности наличия ОМ;
• поражение зрительных нервов обычно тяжелое, в редких случаях - асимптомное;
• болевой синдром в области орбит;
• позитивные зрительные феномены (мерцающие огни, пятна или линии);
• односторонняя слепота развивается в 60% случаев при рецидивирующем и в 22% случаев у пациентов с монофазным течением заболевания;
• при офтальмоскопии обнаруживается нормальная картина глазного дна либо небольшая стушеванность дисков зрительных нервов и отек, в хронических случаях - атрофия и бледность дисков;
• при периметрии - квадрантная, триквадрантная или битемпоральная гемианопсия, периферические скотомы, утрата цветочувствительности.
Вовлечение спинного мозга при ОМ обычно проявляется миелитом протяженностью три и более вертебральных сегмента. Поражение является обычно более тяжелым в сравнении с РС, с острым (на протяжении часов/дней) развитием симметричных грубых двигательных, чувствительных и тазовых нарушений. При поражении спинного мозга (миелит) при ОМ имеет место следующее:
• в отличие от наблюдаемого при типичном РС поражение является обычно более тяжелым;
• острое развитие симметричных грубых двигательных (пара- и тетрапарезы), чувствительных и сфинктерных нарушений (“полный поперечный миелит”);
• радикулярные боли, пароксизмальные тонические спазмы и симптом Лермитта развиваются в 1/3 случаев при ремиттирующем течении заболевания, но крайне редки или не встречаются при монофазном течении заболевания;
• у 77-88% больных после атаки миелита наблюдается частичное восстановление двигательных функций; полный регресс нехарактерен;
• в 80% случаев миелит локализуется в грудном отделе (как правило у пациентов, положительных по антителам к ак-вапорину-4).
Непрекращающаяся икота, тошнота и неукротимая рвота, а также дыхательные нарушения обнаруживаются у 17% больных ремиттирующим ОМ вследствие распространения цервикального очага на продолговатый мозг. Другими симптомами поражения ствола мозга являются головокружение, потеря слуха, слабость лицевой мускулатуры, тригеминальные боли, диплопия, птоз и нистагм.
В отдельных случаях ОМ развиваются задняя возвратная энцефалопатия, эндокринопатии (чаще всего гипоталамические нарушения). Наиболее часто встречаются аменорея, галакторея и гиперпролактинемия.
терно вторичное прогрессирование (при ОМ встречается лишь в 2% случаев).
Определяющим методом исследования при ОМ является магнитно-резонансная томография (МРТ). У большей части пациентов МРТ, выполненная в острую фазу миелита, позволяет выявить обширный непрерывный очаг поражения спинного мозга, который распространяется по длиннику более чем на три позвоночных сегмента, однако отсутствие очага или короткие очаги (менее двух сегментов) могут определяться в периоды ремиссий или в отдаленные периоды заболевания, когда формируется атрофия спинного мозга. В острый период спинной мозг набухший и отечный, очаг может накапливать контрастное вещество, иногда в течение нескольких месяцев. В отличие от ОМ при РС очаги демиелинизации в спинном мозге по длине обычно не превышают 1-2 позвоночных сегментов.
При МРТ головного мозга на начальных стадиях заболевания нормальная картина наблюдается у 55-84% пациентов с ОМ, однако возможно появление очагов в белом веществе в течение развития заболевания. Церебральные очаги по своей локализации имеют предрасположенность к тем участкам головного мозга, где отмечается высокий уровень иммунореактивности к аквапорину-4 (гипоталамус, ствол головного мозга, III и IV желудочки).
Для диагностики оптического неврита используются следующие методы:
• оптическая когерентная томография (показывает истончение ретинальных волокон);
• вызванные потенциалы (замедление проведения по данным зрительных вызванных потенциалов (пик Р100) вплоть до полного отсутствия ответа);
• МРТ (в острую фазу может наблюдаться отечность зрительного нерва и накопление им контрастного вещества).
В цереброспинальной жидкости (ЦСЖ) при ОМ как правило не выявляются изменения, может отмечаться небольшой плейоцитоз (>50 лейкоцитов в 1 мм3), преимущественно лимфоцитарный с наличием нейтрофилов и иногда эозинофилов (в 30% случаев); повышение уровня белка (в 45-75% случаев). Олигоклональные IgG при анализе ЦСЖ, встречаемые при РС в 90% случаев, обнаруживаются при ОМ лишь в 20-40% случаев. Также в ликворе может быть выявлен такой неспецифический маркер аксональной нейродегенерации, как тяжелые цепи нейрофиламентов.
Антитела к аквапорину-4 (NMO-IgG) - специфический биологический маркер ОМ; поскольку чувствительность и специфичность его весьма высоки (75 и 85-99% соответственно), их выявление у пациентов со зрительно-спинальным паттерном демиелинизирующего заболевания позволяет проводить дифференциальный диагноз между ОМ и РС, идиопатическим поперечным миелитом, рекуррентным или билатеральным невритом зрительного нерва.
NMO-IgG могут выявляться у пациентов задолго до формирования целостной картины ОМ. Не установлено связи между количеством обострений, длительностью заболевания, наличием патологических очагов в головном мозге и изменениями в ЦСЖ с выявлением антител в крови. У пациентов, имеющих положительный NMO-IgG статус, обострения более частые, больше выражен неврологический дефицит. Также было показано, что вероятность ассоциации с NMO-IgG повышалась при наличии >3 пери-вентрикулярных очагов в головном мозге, очагов в белом веществе головного мозга, а также при более протяженных очагах демиелинизации в спинном мозге в течение ремиссии. Уровень антител к аквапорину-4 коррелирует с активностью заболевания и снижается при лечении ритук-симабом, азатиоприном, циклофосфамидом. Титры антител также снижались при лечении метилпреднизолоном и оставались низкими в течение ремиссии. Наиболее высокие титры антител определялись у пациентов с полной потерей зрения и выраженными поражениями головного мозга. Также было выявлено, что титры NMO-IgG коррелировали с длиной очага в спинном мозге в период ремиссии при минимуме неврологической симптоматики. Показана корреляция титра антител с тяжестью зрительных нарушений.
Диагностические критерии ОМ
Первые диагностические критерии (1999 г.) основывались исключительно на клинической картине заболевания. В 2006 г. были разработаны, а в 2008 г. уточнены современные диагностические критерии для постановки диагноза ОМ, включающие в себя три основных критерия, которые должны присутствовать обязательно, хотя могут быть разнесены во времени, и подтверждающие критерии, из которых должен быть выявлен хотя бы один.
1) оптический неврит с поражением одного или обоих глаз;
2) поперечный миелит с клинической картиной полного или частичного поражения поперечника спинного мозга и гиперинтенсивным очагом в Т2-режиме МРТ, распространенным на три и более сегмента спинного мозга;
3) отсутствие данных за саркоидоз, васкулиты, клиническую манифестацию системной красной волчанки (СКВ) и другие возможные причины синдрома.
1) МРТ головного мозга (в норме или с изменениями), не удовлетворяющие полностью критериям Barkhof и МсDonald: отсутствие изменений при МРТ головного мозга при дебюте заболевания (характерное для РС);
2) положительный тест на обнаружение антител к аква-порину-4 в сыворотке крови или цереброспинальной жидкости.
Данные по дифференциальной диагностике в случае типичных вариантов демиелинизирующих заболеваний приведены в таблице.
Дифференциальный диагноз ОМ (наиболее типичные ситуации)
Признаки ОМ РС ОРЭМ
Предшествующая инфекция или иммунизация Вариабельно Вариабельно Типично
Пол (женщины : мужчины) 9 : 1 2 : 1 Нет разницы
Средний возраст начала заболевания, годы 39 29
Миелит Подострый, симметричный Подострый, асимметричный Подострый, мультифокальный
Симптомы Оптический неврит, миелит Разнообразие клинических симптомов, диссеминация в пространстве и времени Разнообразие клинических симптомов, диссеминация в пространстве
Начало и течение Острое начало в 100% случаев, 80-90% - рецидивирующее, 10-20% - монофазное. Отсутствие вторичного прогрессирования 10-15% - прогрессирующее течение, 85% - ремиттирующее, 50% - переход во вторичнопрогрессирующее Монофазное
Находки при МРТ
головного мозга В дебюте нет очагов, 10% - гипоталамус, ствол, III и IV желудочки Перивентрикулярные и субкортикальные очаги Субкортикальные очаги, могут поражать серое вещество
спинного мозга Протяженность очагового поражения более 3 сегментов, центральная локализация Протяженность очагового поражения более 1-2 сегментов, периферическая локализация Вариабельные
ЦСЖ Плейоцитоз нейтрофильный >50 кл/мл3. Олигоклональные комплексы 15-30% Плейоцитоз лимфоцитарный 50 кл/мл3. Олигоклональные комплексы отсутствуют
Антитела к аквапорину-4 встречаются не только при ОМ, но также и при ОМ-ассоциированных заболеваниях, имеющих клиническую картину, не совсем типичную для ОМ: наличие очагов поражения головного мозга, сопутствующих заболеваний нервной системы или системных заболеваний. В настоящее время к ОМ-ассоциированным расстройствам относят: 1) ограниченные формы ОМ: “идиопатический” продольно-распространенный миелит (очаг в спинном мозге >3 позвоночных сегментов) и билатеральный единичный или повторный оптический неврит; 2) азиатский оптикоспинальный РС; 3) оптический неврит или продольно-распространенный миелит, ассоциированный с системными аутоиммунными заболеваниями;
4) оптический неврит или миелит, ассоциированный со “специфическими” для ОМ повреждениями головного мозга (гипоталамус, перивентрикулярное пространство, ствол головного мозга).
Миелит и оптический неврит - редкие состояния, которые могут возникнуть у пациентов с аутоиммунными заболеваниями, такими как системная красная волчанка (СКВ) и синдром Шегрена (СШ). Половина из этих пациентов с СКВ и треть с СШ являются серопозитивными по NMO-IgG, тогда как пациенты без проявлений миелита и оптического неврита являются серонегативными. Также у серопозитивных пациентов с СШ значимо чаще выяв-
ляются признаки поражения зрительного нерва и спинного мозга по данным МРТ У NMO-IgG-положительных пациентов чаще выявлялись антинуклеарные антитела и SSA/SSB-антитела (Sjogren syndrome autoantibody), чем у NMO-IgG-отрицательных. Также у пациентов с СКВ и СШ, серопозитивных по NMO-IgG, чаще выявляются другие системные заболевания. По всей видимости, пациенты, позитивные по NMO-IgG, имеют два параллельно текущих аутоиммунных заболевания, а неврологические симптомы служат не осложнением основного заболевания, а проявлением ОМ. Также антитела NMO-IgG и клиника оптического миелита могут выявляться при аутоиммунном тиреоиди-те и ANCA-ассоциированных васкулитах (ANCA - антитела к цитоплазме нейтрофилов) - микроскопический полиангиит, гранулематоз Вегенера и синдром Черджа-Стросс. При выявлении положительного титра антинуклеарных антител или SSA/SSB при отсутствии клинических данных системных заболеваний нельзя исключать ОМ.
Описаны случаи сочетания миастении (преимущественно генерализованной формы) с ОМ или поперечным миелитом, в подавляющем большинстве случаев клиника миастении предшествует ОМ или поперечному миелиту. Средняя длительность между началом миастении и ОМ/поперечного миелита составляет 15 лет. У 80% пациентов проводилась тимомэктомия или лучевая терапия тимуса до развития ОМ или поперечного миелита. Сочетание миастении и ОМ развивается преимущественно у женщин с ювенильной
Рис. 1. Алгоритм диагностики изолированного оптического неврита (по Miller D.H. et al., 2008, с изменениями).
Рис. 2. Алгоритм диагностики изолированного миелита (по Miller D.H. et а. 2008, с изменениями).
Рис. 3. Диагностический поиск при ОМ.
формой или ранним началом миастении, при этом обнаруживается относительно мягкое течение миастении. Таким образом,рекомендуется исследование антител к аквапори-ну-4 у пациентов с миастенией в случае наличия атипичных зрительных или двигательных нарушений.
Приведенные подходы к диагностике ОМ (рис. 1-3) позволяют своевременно установить диагноз и начать патогенетическую терапию. Это крайне важно для прогноза заболевания, так как при своевременной и адекватной терапии 5-летняя выживаемость при ОМ повышается с 68 до 91%.
Аутоиммунные заболевания в неврологии: Клиническое руководство / Под ред. И.А. Завалишина и др. Т. 1. М., 2014. Рассеянный склероз / Под ред. Е.И. Гусева и др. М., 2011. Шмидт Т.Е. // Журн. неврол. и психиатр. 2012. Т. 112. Вып. 2. № 9.С. 5.
Jacob A. et al. // J. Neurol. Neurosurg. Psychiatry. 2013. V. 84. P. 922.
Kira J. // J. Neurol. Sci. 2011. V. 311. P 69.
Lennon V. et al. // Lancet. 2004. V 364. Р. 2106.
Miller D.H. et al. // Mult. Scler. 2008. V 14. Р. 1157.
Trebst C. et al. // J. Neurol. 2014. V. 261. P. 1.
WingerchukD.M. etal. //Lancet Neurol. 2007. V. 6. P. 805.
Оптиконевромиелит на МРТ
ФГБУ «Научный центр неврологии», Москва
ФГБУ "Научный центр неврологии" РАМН, Москва
ФГБУ «Научный центр неврологии», Москва
ФГБУ «ФНИЦЭМ им. Н.Ф. Гамалеи» Минздрава России, Москва, Россия
Научный центр неврологии РАМН, Москва
Оптикомиелит и аквапорин-ассоциированные синдромы
Журнал: Журнал неврологии и психиатрии им. С.С. Корсакова. Спецвыпуски. 2015;115(2‑2): 31‑37
ФГБУ «Научный центр неврологии», Москва
При оптикомиелите, а также связанных с ним расстройствах — оптическом неврите и продольном распространенном миелите имеет большое значение определение антител к аквапорину-4 (AQP4). Это важно для своевременной постановки диагноза с целью начала патогенетической терапии, а также для дифференциальной диагностики при оптикомиелит-ассоциированных расстройствах. Рассмотрен спектр заболеваний, при которых выявляются антитела к AQP4. Приведены собственные данные по выявлению антител к AQP4 при заболеваниях центральной нервной системы. Данные антитела обнаружены у 86,36% пациентов с оптикомиелитом, у 12,5% пациентов с изолированными синдромами и у 100% пациентов с СКВ с синдромом продольного распространенного миелита или оптического неврита. Выявлена корреляция между степенью поражения спинного мозга на МРТ и обнаружением антител к AQP4.
ФГБУ «Научный центр неврологии», Москва
ФГБУ "Научный центр неврологии" РАМН, Москва
ФГБУ «Научный центр неврологии», Москва
ФГБУ «ФНИЦЭМ им. Н.Ф. Гамалеи» Минздрава России, Москва, Россия
Научный центр неврологии РАМН, Москва
(оптиконевромиелит, болезнь Девика, ОМ) — идиопатическое аутоиммунное воспалительное заболевание центральной нервной системы (ЦНС), характеризующееся преимущественным демиелинизирующим поражением зрительных нервов и спинного мозга при относительной сохранности головного мозга.
ОМ — вторая по частоте форма воспалительного демиелинизирующего заболевания после рассеянного склероза (РС). Заболевание преимущественно распространено у лиц африканского и азиатского происхождения, среди которых частота РС невысока. В Японии среди всех демиелинизирующих заболеваний встречаемость ОМ составляет 20—30%, в Юго-Восточной Азии, включая Гонконг и Китай, — 36%, в Индии — 10—23%, в Латинской Америке — 15%. В европейских странах и США доля ОМ среди остальных демиелинизирующих заболеваний ЦНС составляет 1—5% [1, 2]. Распространенность О.М. у представителей европеоидной расы, по данным различных эпидемиологических исследований, составляет 0,3—4,5 на 100 000 населения [3]. Однако многие случаи ОМ нередко ошибочно трактуются как РС, рецидивирующий поперечный миелит, рецидивирующий ретробульбарный неврит.
Длительное время велись споры: следует ли считать ОМ самостоятельным заболеванием, вариантом РС, острого рассеянного энцефаломиелита (ОРЭМ) или других демиелинизирующих заболеваний ЦНС. Последние клинические, эпидемиологические, патофизиологические и иммунологические данные свидетельствуют о том, что ОМ является отдельной нозологической формой. Это учтено в МКБ-10, в которой оптиконевромиелит кодируется как G36.0.
. На данный момент ОМ является единственным демиелинизирующим заболеванием, при котором выявлены специфические биомаркеры: в 2004 г. из сыворотки пациентов были выделены антитела, названные NMO-IgG (neuromyelitis optica — immunoglobulin G). В 2005 г. был идентифицирован белок — антиген, являющийся мишенью для NMO-IgG [4]. Это аквапорин-4 (aquaporin-4, AQP4) — селективный водный транспортер. К семейству аквапоринов относятся более 10 белков, аквапорин-4 наиболее представлен в ЦНС, где является основным доминантным мембранным каналом для молекул воды, который расположен на мембране астроцитарных ножек, участвующих вместе с капиллярами в формировании гематоэнцефалического барьера (ГЭБ) [5]. Мембраны концевых отростков ножек астроцитов находятся в контакте с эндотелием церебральных сосудов, с мягкой оболочкой головного мозга и с эпендимными клетками. При этом аквапорин-4 имеет первостепенную важность в поддержании жидкостного гомеостаза в ЦНС. В очагах поражения выявляется селективная утрата белка AQP4, они возникают преимущественно в тех участках головного мозга, в которых отмечается высокая иммунореактивность к AQP4 (гипоталамус, перивентрикулярные зоны).
AQP4 состоит из мономеров, каждый из которых имеет размер около 30 кДа и содержит 6 внутримембранных доменов и 2 коротких спиральных сегмента, один из которых окружает цитоплазматические ворота водной поры, а другой — внеклеточные. Существует 2 основные изоформы AQP4, образующиеся в результате альтернативного сплайсинга: относительно длинные М1-изоформы и короткие М23-изоформы [6]. Методом замораживания-скалывания и иммунозолотого маркирования было установлено, что AQP4 формируют в мембранах клеток ортогональные массивы частиц, причем М23-изоформы формируют большие массивы, тогда как М1 рассеяны по мембране в виде тетрамеров. Считается, что одна из задач ортогональных массивов частиц изоформы М23 — поддержание целостности ГЭБ [7]. Cамое высокое соотношение мРНК изоформ M1: M23 — в зрительном нерве и спинном мозге, т. е. в структурах, которые преимущественно поражаются при О.М. При этом у больных ОМ данное соотношение значительно снижается за счет уменьшения экспрессии изоформы М1 [8].
В основе патогенеза ОМ лежит связывание NMO-IgG с AQP4, что в конечном итоге приводит к образованию очагов повреждения ЦНС. Связывание происходит не с отдельными тетрамерами AQP4, а с ортогональными массивами частиц, которые состоят преимущественно из М23-изоформы AQP4 [9]. В результате происходят интернализация рецептора внутрь клетки и активация системы комплемента, за чем следует повреждение астроцита. Нарушается целостность ГЭБ, и в очаг поражения под действием воспалительных цитокинов, таких как интерлейкин-17 (ИЛ-17), интерлейкин-8 (ИЛ-8), гранулоцит колониестимулирующий фактор, проникают гранулоциты (нейтрофилы и эозинофилы); происходит гибель олигодендроцитов и в конечном счете — гибель нейрональных клеток. Таким образом, в отличие от РС, в патогенезе которого лежат преимущественно клеточные механизмы, ОМ является аутоиммунной астроцитопатией/каналопатией, которая имеет гуморально-опосредованный механизм [10].
Нарушение клеточных механизмов транспорта воды, повреждение ГЭБ и активная инфильтрация периваскулярного пространства клеточными элементами крови (нейтрофилами, эозинофилами) способствует развитию демиелинизации, сосудистой гиперплазии с гиалинизацией стенок капилляров, некрозу олигодендроцитов и нейронов и образованию специфических полостей как в белом, так и в сером веществе спинного мозга [11]. Кроме гуморального иммунитета, в патогенезе ОМ участвует и клеточный: отмечаются макрофагальная инфильтрация, периваскулярная гранулоцитарная и эозинофильная инфильтрация. Также в периваскулярном пространстве обнаруживают небольшое количество CD3+ и CD8+ Т-клеток. Немаловажное значение имеют В-клеточно-индуцированные реакции, что не характерно для типичного течения РС, но наблюдается при тяжелых его формах [12].
Участие гуморальной системы в образовании патологических очагов подтверждается и патоморфологически: отмечаются макрофагальная и эозинофильная инфильтрация, повышенная активность комплемента, гиалиноз и фиброз сосудов. В активных очагах иммуноглобулины и компоненты комплемента располагаются в характерных васкулоцентричных «ободках и розетках» [11]. Такая морфологическая картина напоминает аутоиммунное воспаление при васкулитах. Предполагается, что при ОМ и заболеваниях соединительной ткани имеют место сходные патогенетические механизмы [13].
Другими патогенетическими механизмами при ОМ являются нарушение транспорта и утилизации глутамата. При действии NMO-IgG происходит уменьшение связанных с AQP4 транспортеров возбуждающих аминокислот EAAT2, что нарушает транспорт глутамата. Также в пораженных астроцитах снижается синтез глутаминсинтетазы, превращающей глутамат в глутамин, и нарушается утилизация последнего. Таким образом, происходят высвобождение глутамата из астроцитов, его внеклеточное накопление с последующей гибелью олигодендроцитов по типу эксайтотоксичности [14].
На протяжении длительного времени считалось, что ОМ характеризуется монофазным течением, проявляющимся появлением одновременно или через короткий срок двустороннего поражения зрительных нервов и поперечного миелита. В настоящее время выделяют две формы ОМ — монофазную, возникающую в 10—20% случаев, и ремиттирующую — в 80—90%.
При монофазном течении симптомы оптического неврита и продольного распространенного миелита возникают одновременно или последовательно, но не более чем через 30 дней. Рецидивов при этом не наблюдается [15].
При ремиттирующей форме рецидивы отмечаются рано, кластерно и с непредсказуемыми интервалами. В течение первого года рецидив случается у 60% больных, а в течение 3 лет — у 90%. Ремиттирующая форма чаще возникает у женщин и в более позднем возрасте. Для нее характерны менее тяжелые двигательные нарушения, чем при монофазном течении. Ремиттирующее течение ОМ, высокая частота обострений в первые два года после 1-го проявления болезни, тяжесть первой атаки и сопутствующие аутоиммунные заболевания являются предикторами неблагоприятного прогноза. При О.М. 5-летняя выживаемость составляет 68% [16].
Возникновение неврита зрительного нерва одновременно с обеих сторон или последовательно с одной и другой стороны в короткий срок свидетельствует о высокой вероятности наличия ОМ. У больных с ремиттирующим ОМ слепота минимум на один глаз развивается в 60% случаев, у пациентов с монофазным течением — в 22% случаев [15]. При офтальмологическом обследовании у больных с оптическим невритом могут выявляться атрофия зрительного нерва, бледность диска зрительного нерва [17]. При исследовании полей зрения чаще всего отмечается центральная скотома, но возможно также выявление парацентральной скотомы, битемпоральной гемианопсии, нарушения цветовосприятия. Оптическая когерентная томография при ОМ показывает истончение слоя сетчатки, а также распространенное аксональное повреждение [18]. При морфологическом исследовании обнаруживаются демиелинизация и некроз, в основном в центральной части нерва; иногда — образование полостей.
Вовлечение спинного мозга обычно представляет собой полный поперечный миелит с пара- и тетрапарезами, практически симметричный уровень потери чувствительности и дисфункцию сфинктеров [13]. Для сравнения симптомы повреждения спинного мозга при РС менее выражены и асимметричны и являются следствием острого частичного поперечного миелита. В 1/3 случаев ремиттирующего ОМ наблюдаются корешковая боль, пароксизмальные тонические спазмы и синдром Лермитта. Вследствие расширения очага на продолговатый мозг возникают тошнота и непрекращающаяся икота. Другими симптомами поражения ствола мозга являются рвота, головокружение, потеря слуха, слабость лицевой мускулатуры, тригеминальные боли, диплопия, птоз и нистагм [19]. Вследствие вовлечения в процесс центров нейромышечного дыхательного контроля, могут возникнуть нейрогенная дыхательная недостаточность и последующая за этим смерть.
Определяющим методом диагностики при ОМ является магнитно-резонансная томография (МРТ). У большей части пациентов МРТ, выполненная в острую фазу миелита, выявляет обширный непрерывный очаг поражения спинного мозга, который распространяется по длиннику более чем на 3 позвоночных сегмента, однако отсутствие очага или короткие очаги (менее 2 сегментов) могут определяться в периоды ремиссий или в отдаленные периоды заболевания, когда формируется атрофия спинного мозга. В острый период спинной мозг набухший и отечный, очаг может накапливать контрастное вещество, иногда в течение нескольких месяцев. В отличие от ОМ, при РС очаги демиелинизации в спинном мозге по длине обычно не превышают одного-двух сегментов [12]. В 15% случаев у пациентов с ОМ в патологический процесс кроме зрительного нерва и спинного мозга вовлекаются другие отделы ЦНС.
В цереброспинальной жидкости (ЦСЖ) при ОМ как правило особых изменений не отмечается. Может выявляться небольшой плеоцитоз (>50 лейкоцитов в 1 мм), преимущественно лимфоцитарный с наличием нейтрофилов и иногда эозинофилов — в 30%; небольшое повышение уровня белка. Олигоклональные IgG при анализе ЦСЖ, встречаемые при РС в 90% случаев, обнаруживаются при ОМ лишь в 20—40% случаев [12].
Первые диагностические критерии (1999) основывались исключительно на клинической картине заболевания. В 2006 г. были разработаны, а в 2008 г. уточнены современные диагностические критерии для постановки диагноза ОМ [20]. Они включают в себя 3 основных, которые должны присутствовать обязательно (хотя могут быть разнесены во времени) и подтверждающие критерии, из которых должен быть выявлен хотя бы один.
Основные критерии: 1) оптический неврит с поражением одного или обоих глаз; 2) поперечный миелит с клинической картиной полного или частичного поражения поперечника спинного мозга и гиперинтенсивным очагом в Т2-режиме МРТ, распространенным на 3 и более сегмента спинного мозга; 3) отсутствие данных о саркоидозе, васкулитах, клинической манифестации системной красной волчанки (СКВ) и других возможных причинах синдрома.
Малые критерии: 1) МРТ головного мозга в норме или с изменениями, не удовлетворяющими полностью критериям Баркхоффа, отраженным в критериях МакДоналда (2005): отсутствие изменений при МРТ головного мозга при дебюте заболевания (характерное для РС); 2) положительный тест на обнаружение антител к AQP4 в сыворотке крови или цереброспинальной жидкости.
Антитела к AQP4 выявляются не только при классическом ОМ, но и при других расстройствах ЦНС, имеющих картину, не совсем типичную для ОМ: наличие очагов поражения головного мозга, наличие сопутствующих заболеваний нервной системы или системных заболеваний, оптикоспинальный или азиатский Р.С. Азиатский Р.С. имеет ряд отличий от западного, и в настоящее время стоит вопрос, является ли он РС или ОМ.
Было показано, что если у пациентов в крови обнаруживали антитела к AQP4, то у них с высокой вероятностью впоследствии развивался О.М. Это послужило причиной объединения вышеперечисленных состояний под термином [1]. В настоящее время к оптикомиелит-ассоциированным расстройствам относят ОМ, ограниченные формы ОМ: продольно-распространенный миелит и билатеральный единичный или повторный оптический неврит, оптикоспинальный РС (ОСРС), оптический неврит или продольно-распространенный миелит, ассоциированный с системными заболеваниями соединительной ткани (СЗСТ), оптический неврит или миелит, ассоциированный со «специфическими» для ОМ повреждениями головного мозга (гипоталамус, перивентрикулярное пространство, ствол головного мозга).
NMO-IgG находят в 37,9—50% случаев у пациентов с продольным распространенным миелитом [21] и в 14,3—20% случаев у пациентов с рецидивирующим изолированным оптическим невритом [22]. В проспективных исследованиях были обследованы пациенты с однократным эпизодом поперечного идиопатического миелита. Из них 50% были серопозитивными. Среди пациентов с однократным или рецидивирующим оптическим невритом 25% имели NMO-IgG. В течение последующего года у половины из них отмечался повторный эпизод миелита, или развился оптический неврит. Эти сведения подтверждают, что изолированный или рецидивирующий миелит, билатеральный оптический неврит, рецидивирующий оптический неврит являются в ряде случаев неполной, или незавершенной формой О.М. Обнаружение этих антител определяет прогноз заболевания и повышает вероятность конверсии заболевания в О.М. Выявление NMO-IgG в сыворотке крови пациентов с изолированным оптическим невритом ассоциировано с более тяжелыми первоначальными проявлениями оптического неврита [23].
Несмотря на сходные с ОМ клинические проявления азиатского ОCРС, он традиционно считается отдельным заболеванием из-за более мягкого течения и чаще обнаруживаемых очагов в головном мозге. Были предприняты попытки выявить «чистые формы» ОCРС без очагов в головном мозге и с продольным поперечным миелитом. Исследование плазмы крови этих пациентов не выявило разницы между ОСРС и ОМ. 60% больных ОСРС были серопозитивными по NMO-IgG и не отличались от ОМ, где число серопозитивных пациентов составило 73%. Некоторые из пациентов с ОСРС, вероятно, являлись больными с типичным «европейским» РС, что объясняет более низкие показатели серопозитивной реакции в нескольких японских и азиатских исследованиях.
Вероятность обнаружения NMO-IgG у пациентов с частичным поперечным миелитом, при котором длина очага менее 3 вертебральных сегментов, очень мала: только у 1 (4,5%) из 22 пациентов с миелитом
Оптиконевромиелит (синдром Девика) — редкий вариант поражения нервной системы при системной красной волчанке
Авторы: Виноградова Е.С. 1 , Новиков П.И. 2 , Моисеев С.В. 3
1 Факультет фундаментальной медицины МГУ, Москва
2 Клиника нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева, Москва
3 Факультет фундаментальной медицины МГУ, Москва; Клиника нефрологии, внутренних и профессиональных болезней им. Е.М. Тареева УКБ № 3, Первый МГМУ им. И.М. Сеченова, Минздрава России (Сеченовский Университет), Москва
Оптиконевромиелит, или синдром Девика — это воспалительное демиелинизирующее заболевание центральной нервной системы, поражающее преимущественно зрительный нерв и спинной мозг. В 50–70% случаев определяется ассоциация с системными заболеваниями соединительной ткани, такими как системная красная волчанка, болезнь Шегрена и др. В этом случае остается открытым вопрос о вторичном характере болезни. Клиническая картина характеризуется сочетанием синдромов оптического неврита и продольно-поперечного миелита. Для диагностики используются визуализирующие методы, такие как оптическая когерентная томография, метод вызванных потенциалов, магнитно-резонансная томография. В настоящее время ключевым методом постановки верного диагноза является обнаружение специфического серологического маркера — антител к аквапорину-4 (NMO-IgG), титр которых коррелирует с активностью заболевания и помогает проведению дифференциальной диагностики. Выявление синдрома Девика в сочетании с ревматическими заболеваниями определяет тактику более активного иммуносупрессивного лечения. В статье приводится описание клинического случая молодой пациентки с системной красной волчанкой и синдромом Девика. Прогноз и тяжесть заболевания определяло наличие оптиконевромиелита. В качестве индукционной терапии использовались сверхвысокие дозы глюкокортикоидов, циклофосфамид с заменой на азатиоприн в качестве поддерживающего лечения с положительным клинико-лабораторным ответом.
Ключевые слова: оптиконевромиелит, синдром Девика, аутоиммунная патология, системная красная волчанка, антитела к аквапорину-4, аквапорин-ассоциированные синдромы, демиелинизирующие заболевания центральной нервной системы.
Для цитирования: Виноградова Е.С., Новиков П.И., Моисеев С.В. Оптиконевромиелит (синдром Девика) — редкий вариант поражения нервной системы при системной красной волчанке. РМЖ. 2018;12(II):103-106.
Neuromyelitics optica (Devic’s syndrome) is a rare variant of the nervous system impairment in systemic lupus erythematosus
E.S. Vinogradova 1 , P.I. Novikov 2 , S.V. Moiseev 1,2
1 Faculty of Fundamental Medicine of the Moscow State University, Moscow
2 Tareev Clinic of Nephrology, Internal and Occupational Diseases, University Clinical Hospital No.3,
Sechenov University, Moscow
Neuromyelitics optica or Devic’s syndrome is an inflammatory demyelinating disease of the central nervous system, affecting mainly the optic nerve and spinal cord. In 50–70% of cases, association with systemic diseases of the connective tissue, such as systemic lupus erythematosus, Sjogren disease, and others, is determined. In this case, the question of the secondary nature of the disease remains open. The clinical picture is characterized by a combination of optic neuritis syndromes and/or longitudinal-transverse myelitis. Imaging methods are used for diagnostics, such as optical coherence tomography, event-related potentials method, magnetic resonance imaging. At present, the key method of making the correct diagnosis is the detection of a specific serological marker — antibodies to aquaporin-4 (NMO-IgG), the titer of which correlates with disease activity and helps the differential diagnosis. Detection of Devic’s syndrome in combination with rheumatic diseases determines the tactics of more active immunosuppressive treatment. The article describes the clinical case of a young patient with systemic lupus erythematosus and Devic’s syndrome. The prognosis and severity of the disease determined the presence of neuromyelitics optica. As induction therapy, ultrahigh doses of glucocorticosteroids, cyclophosphamide replaced with azathioprine as a supportive treatment with a positive clinical and laboratory response were used.
Key words: neuromyelitics optica, Devic’s syndrome, autoimmune pathology, systemic lupus erythematosus, antibodies to aquaporin-4, aquaporin-associated syndromes, demyelinating diseases of the central nervous system.
For citation: Vinogradova E.S., Novikov P.I., Moiseev S.V. Neuromyelitics optica (Devic’s syndrome) is a rare variant of the nervous system impairment in systemic lupus erythematosus // RMJ. 2018. № 12(II). P. 103–106.
В статье приводится описание клинического случая молодой пациентки с системной красной волчанкой и синдромом Девика (оптиконевромиелит)
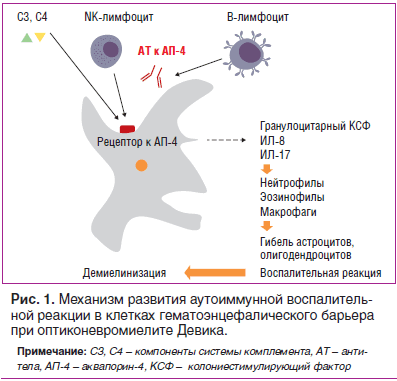
Оптиконевромиелит (ОМ) относится к группе воспалительных демиелинизирующих заболеваний центральной нервной системы (ЦНС) и является вторым по частоте после рассеянного склероза. При ОМ очаги воспаления и повреждения миелина затрагивают почти исключительно оптический нерв и спинной мозг (поперечный миелит на уровне грудных, реже — шейных сегментов). ОМ чаще встречается у лиц африканского и азиатского происхождения. Распространенность ОМ среди европеоидов составляет 0,3–4,4 человека на 100 000 населения. Возраст дебюта варьирует со снижением заболеваемости после 50 лет. Женщины болеют гораздо чаще (85% случаев), чем мужчины. Довольно часто ОМ (50–70%) сочетается с другими аутоиммунными заболеваниями — синдромом Шегрена, системной красной волчанкой (СКВ), аутоиммунным тиреоидитом и др. [1].
В 1894 г. E. Devic и его ученик F. Gault предложили выделить ОМ в отдельную нозологическую форму, при которой очаги воспаления и повреждения миелина затрагивали главным образом оптический нерв и спинной мозг (поперечный миелит на уровне грудных, реже — шейных сегментов) [2]. В 2004 г. V. Lennon et al. была доказана аутоиммунная природа болезни путем обнаружения сывороточных аутоантител NMO-IgG (neuromyelitis optica — Immunoglobulin G). В настоящее время ОМ — единственное демиелинизирующее заболевание, при котором выявлен специфический биомаркер. В основе патогенеза заболевания лежит селективная связь NMO-IgG с аквапорином-4, одним из основных белков водных каналов ЦНС, локализующихся в ножках астроцитов, образующих гематоэнцефалический барьер (ГЭБ). Наибольшая концентрация аквапорина-4 в ЦНС отмечена в сером веществе спинного мозга, гипоталамусе, перивентрикулярных областях. Это приводит к развитию воспалительной реакции, активации системы комплемента, повышенной продукции воспалительных цитокинов (интерлейкинов ИЛ-17, ИЛ-8, гранулоцитарного колониестимулирующего фактора). Нарушение клеточных механизмов транспорта воды, повреждение ГЭБ и активная инфильтрация периваскулярного пространства нейтрофилами и эозинофилами способствуют развитию демиелинизации, сосудистой гиперплазии с гиалинизацией стенок капилляров, некрозу олигодендроцитов и нейронов и образованию специфических полостей как в белом, так и в сером веществе спинного мозга (рис. 1). Описанные особенности морфологических изменений в спинном мозге могут напоминать аутоиммунное воспаление, протекающее по типу васкулита [3].
Клиническая картина характеризуется сочетанием синдромов оптического неврита и продольно-поперечного миелита (табл. 1). Типичными симптомами миелита выступают мышечная слабость, спастичность, дискоординация, атаксия, симптом Лермитта (ощущение удара током при сгибании шеи), задержка мочи, вегетативная дисфункция, возможны расстройства ниже уровня поражения спинного мозга. Поражения зрительных нервов и спинного мозга в некоторых случаях возникают одновременно, но чаще — с интервалом, который может составлять месяцы, годы и даже десятилетия. Симптоматика нарастает в течение нескольких дней, а регрессирует неделями и месяцами, зачастую с сохранением остаточного неврологического дефицита.
В настоящее время допускается, что ОМ может иметь как монофазный, так и ремиттирующий тип течения [4].
Для диагностики оптического неврита используются оптическая когерентная томография (показывает истончение ретинальных волокон), вызванные потенциалы (замедление проведения по данным зрительных вызванных потенциалов вплоть до полного отсутствия ответа), магнитно-резонансная томография (МРТ) (в острую фазу может наблюдаться отечность зрительного нерва и накопление им контрастного вещества).
МРТ спинного мозга, выполненная в острую фазу миелита, позволяет выявить обширный непрерывный очаг поражения спинного мозга, который распространяется по длиннику более чем на 3 позвоночных сегмента, однако отсутствие очага или короткие очаги (менее 2 сегментов) могут определяться в периоды ремиссий или в отдаленные периоды заболевания, когда формируется атрофия спинного мозга.
При МРТ головного мозга на начальных стадиях заболевания нормальная картина наблюдается у 55–84% пациентов с ОМ, однако возможно появление очагов в белом веществе при развитии заболевания (рис. 2) [5]. Церебральные очаги по своей локализации имеют предрасположенность к тем участкам головного мозга, где отмечается высокий уровень иммунореактивности к аквапорину-4 (гипоталамус, ствол головного мозга, III и IV желудочки) [6].
Ключевым методом диагностики является обнаружение серологического маркера — антител к аквапорину-4 (NMO-IgG), чувствительность 75%, специфичность 85–99%. Уровень антител к аквапорину-4 коррелирует с активностью заболевания и снижается при иммуносупрессивной терапии и остается низким в течение ремиссии. Показана корреляция титра антител с тяжестью зрительных нарушений [3].
Прогноз заболевания серьезный, вплоть до инвалидизации и летального исхода. При своевременной и адекватной терапии возможно достижение длительных ремиссий, пятилетняя выживаемость при ОМ повышается с 68 до 91%.
В настоящее время общепринятого стандарта лечения синдрома Девика нет. Одним из вариантов лечения является применение препаратов, частично блокирующих В-клетки, таких как ритуксимаб. Для лечения атаки миелита и оптического неврита применяют высокие дозы кортикостероидов. Наряду с этим эффективно использование и плазмафереза. Эффективность превентивной иммуномодулирующей терапии у пациентов с ОМ формально не изучена. Терапией выбора считают комбинацию преднизолона и азатиоприна [7].
Клиническое наблюдение
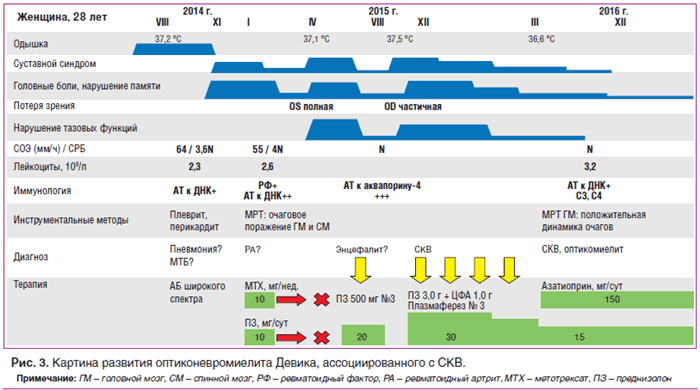
Приводим собственное клиническое наблюдение больной оптиконевромиелитом Девика, ассоциированным с СКВ (см. рис. 3).
Больная К., 28 лет, домохозяйка.
В течение 2013–2014 гг. в связи с эпизодами субфебрилитета, одышкой, персистирующими рентгенологическими признаками нижнедолевой инфильтрации слева, устойчивыми к антибактериальной терапии широкого спектра, наблюдалась и получала противотуберкулезную химиотерапию в противотуберкулезном диспансере Хабаровска. Результаты многочисленных бактериологических исследований, полимеразно-цепной реакции, бронхо-альвеолярного лаважа и кожных туберкулиновых проб (диаскин-тест) были отрицательными на протяжении всего периода наблюдения. В августе 2014 г. — левосторонний плеврит, перикардит, лейкоцитопения до 2,3×10 9 /л, повышение маркеров острофазового воспаления (скорость оседания эритроцитов (СОЭ) 52–64 мм/ч, содержание С-реактивного белка (СРБ) 3,5N), впервые выявлены антитела к нативной ДНК. В ноябре 2014 г. возникли жалобы на головные боли, боли при пальпации и движении, ограничение подвижности в шейном отделе позвоночника, боли в верхнем плечевом поясе, артриты мелких суставов кистей рук. В январе 2015 г. при обследовании: ревматоидный фактор (РФ)+, антитела к нативной ДНК 10N, лейкоцитопения 2,6×10 9 /л, СОЭ 55 мм/ч, СРБ 4N. Состояние расценено как ревматоидный артрит, проводилась пульс-терапия глюкокортикоидами, далее назначен преднизолон 10 мг/сут, метотрексат 10 мг/нед. с положительным эффектом. В феврале 2015 г. самостоятельно прекратила прием глюкокортикоидов и метотрексата, на фоне чего отметила внезапное выпадение нижней половины поля зрения, а затем полную потерю зрения на левый глаз. В марте 2015 г. при МРТ-исследовании головного мозга патологии не выявлено.
В апреле 2015 г. была госпитализирована с диагнозом «энцефалопатия неясной этиологии». Объективно: сознание нарушено до степени заторможенности, ригидность затылочных мышц, координационные пробы не выполняет, нарушение функции тазовых органов (задержка мочи). МРТ головного мозга: в белом веществе лобных, теменных, височных долей выявлены множественные очаги измененного МР-сигнала (гиперинтенсивные в режиме Т2 и FLAIR, изо- и гипоинтенсивные в режиме Т1), полигональной формы с нечеткими и неровными контурами, сливного характера, размерами от 0,3 до 3,0 см. При постконтрастном исследовании: 2 супратенториальных очага (в левой лобной доле), минимально накапливающих контраст в виде небольших очагов на периферии. МРТ шейного, поясничного отделов спинного мозга: множественные интрамедуллярные зоны гиперинтенсивного характера протяженностью 5–13 мм. Анализ ликвора: цитоз 12/мм 3 (нейтрофилы 5, лимфоциты 7), глюкоза 2,8 ммоль/л, общий белок 0,8 г/л. В лейкоцитарной формуле: лейкопения, лимфопения, значительный моноцитоз. Антитела к вирусу Эпштейна — Барр IgM, антитела к Herpes simplex IgM, антитела к кардиолипинам не обнаружены. Проведена пульс-терапия метилпреднизолоном, далее назначен преднизолон в дозе 20 мг/сут с положительным эффектом в виде постепенного восстановления памяти, функции тазовых органов. В августе 2015 г. на фоне отмены глюкокортикоидов отметила снижение остроты зрения справа, снижение чувствительности нижних конечностей, головные боли, возникли артриты мелких суставов кистей рук. Осмотр окулиста: полная атрофия зрительного нерва слева, нисходящая частичная атрофия справа, миопия средней степени. Антитела к аквапорину-4 (NMO) выявлены в высоком титре 1:320 (N <1:10). Впервые установлен диагноз системной красной волчанки, двустороннего оптикомиелита. Возобновлена активная иммуносупрессивная терапия: выполнено 3 сеанса плазмафереза, проведена комбинированная пульс-терапия глюкокортикоидами и циклофосфамидом (по 1000 мг/мес.), назначен преднизолон в дозе 30 мг/сут. В декабре 2015 г. по данным МРТ головного мозга с контрастным усилением отмечена положительная динамика очагового поражения на фоне иммуносупрессивной терапии. С марта 2016 г. переведена на поддерживающее лечение, циклофосфамид был заменен на азатиоприн в дозе 150 мг/сут, доза преднизолона постепенно снижена до 15 мг/сут. В декабре 2016 г. впервые госпитализирована в клинику им. Е.М. Тареева. Учитывая клинико-анамнестические данные о полисерозитах, артритах мелких суставов кистей рук, демиелинизирующем очаговом поражении центральной нервной системы, а также лейкопению, гипокомплементемию, выявление антител к нативной ДНК в диагностически значимых титрах, положительную динамку состояния на фоне проводимой иммуносупрессивной терапии, диагноз системной красной волчанки сомнений не вызывает (SLE SLICC 4 клинических и 2 иммунологических критерия). Особенностью данного случая является развитие двустороннего оптиконевромиелита, NMO-IgG-ассоциированного, осложнившегося полной потерей зрения слева. По результатам проведенного обследования данных за активность системного заболевания не выявлено, иммунологические тесты в норме. Ввиду наличия инвалидизирующего и потенциально жизнеугрожающего поражения ЦНС рекомендованы: продолжение иммуносупрессивной терапии азатиоприном в прежней дозе — 150 мг/сут, постепенное медленное снижение дозы преднизолона по 1,25 мг/мес. до 10 мг/сут, динамический МР-контроль очаговых изменений головного и спинного мозга, наблюдение ревматолога, невролога, окулиста. Ввиду ремиттирующего течения заболевания строго противопоказана самостоятельная отмена препаратов.
Таким образом, представленное наблюдение демонстрирует редкий вариант поражения ЦНС при системной красной волчанке. Остается открытым вопрос, следует ли рассматривать развитие ОМ в качестве самостоятельной нозологической формы или как проявление основного заболевания, в нашем случае системной красной волчанки. С одной стороны, наличие специфических антител (NMO-IgG) и возможное ухудшение течения ОМ вне контекста системных проявлений являются аргументами в пользу нозологической самостоятельности этой патологии. С другой стороны, эффективность контроля активности основного заболевания и проявлений ОМ, совпадение периодов обострения ОМ с системными проявлениями, однонаправленные изменения иммунологических маркеров ОМ и СКВ позволяют трактовать ОМ у нашей пациентки как вторичный. Важно, что определение титра NMO-IgG помогает контролировать активность болезни. Так, уровень антител к аквапорину-4 снижается и остается низким в течение ремиссии. Хороший ответ на лечение позволяет рассчитывать на длительный контроль активности при сохранении приверженности терапии.
1. Аутоиммунные заболевания в неврологии: Клиническое руководство / под ред. И.А. Завалишина и др. Т.1. М., 2014 [Autoimmunny`e zabolevaniya v nevrologii: Klinicheskoe rukovodstvo / рod red. I.A. Zavalishina i dr. T.1. M., 2014 (in Russian)].
2. Maticlo M., Weinshshenker B. Neuromyelitis optica // Multiple sclerosis. 2010. P.258–275.
3. Lennon V.A., Kryzer T.J., Pittock S.J. et al. IgG marker of optic-spinal multiple sclerosis binds to the aquaporin-4 water channel // J Exp Med. 2005. Vol. 202. P.473–477.
4. Мироненко Т.В., Хубетова И.В. Оптикомиелит (болезнь Девика). Научный обзор и собственное клиническое наблюдение // Международный неврологический журнал. 2015. T.1(71). C.141–147 [Mironenko T.V., Xubetova I.V. Optikomielit (bolezn` Devika). Nauchny`j obzor i sobstvennoe klinicheskoe nablyudenie // Mezhdunarodny`j nevrologicheskij zhurnal. 2015. T.1(71). C.141–147 (in Russian)].
5. Kitley J., Woodhall M., Waters P. et al. Myelin-oligodendrocyte glycoprotein antibodies in adults with a neuromyelitis optica phenotype // Neurology. 2012. Vol. 79(12). P.1273-1277. DOI: 10.1212/WNL.0b013e31826aac4e.
6. Симанив Т.О., Воробьёва А.А., Смирнова Н.В. и др. Оптикомиелит и аквапорин-ассоциированные синдромы // Журнал неврологии и психиатрии им. С.С. Корсакова. 2015. №2(2). С.31–37 [Simaniv T.O., Vorob`yova A.A., Smirnova N.V. i dr. Optikomielit i akvaporin-associirovanny`e sindromy` // Zhurnal nevrologii i psixiatrii im. S.S. Korsakova. 2015. №2(2). S.31–37 (in Russian)].
7. Marios C. Papadopoulos, Jeffrey L. Bennett, Alan S. Verkman. Treatment of neuromyelitis optica: state-of-the-art and emerging therapies // Nature Reviews Neurology. 2014. Vol. 10. P.493–506.
Контент доступен под лицензией Creative Commons «Attribution» («Атрибуция») 4.0 Всемирная.
Читайте также: