Профилирование экспрессии генов и кластеризация сигнатур опухоли
Добавил пользователь Morpheus Обновлено: 29.01.2026
Анализ обогащения набора генов (GSEA) (также функциональный анализ обогащения) - это метод определения классов гены или белки которые чрезмерно представлены в большом наборе генов или белков и могут быть связаны с заболеванием фенотипы. В методе используются статистические подходы для выявления значительно обогащенных или истощенных групп генов. Технологии транскриптомики и протеомика результаты часто идентифицируют тысячи генов, которые используются для анализа. [1]
Исследователи, выполняющие высокопроизводительные эксперименты которые дают наборы генов (например, гены, которые дифференциально выразил в разных условиях) часто хотят получить функциональный профиль этого набора генов, чтобы лучше понять лежащие в основе биологические процессы. Это можно сделать, сравнивая входной набор генов с каждым из бункеров (терминов) в генная онтология - а статистический тест можно выполнить для каждого бина, чтобы увидеть, обогащен ли он входными генами.
Содержание
Задний план
Пока завершение Проект генома человека даровал исследователям огромное количество новых данных, но также оставил перед ними проблему, как их интерпретировать и анализировать. Чтобы найти гены, связанные с болезнями, исследователи использовали ДНК-микрочипы, которые измеряют уровень экспрессии генов в разных клетках. Исследователи будут выполнять эти микроматрицы на тысячах разных генов и сравнивать результаты двух разных категорий клеток, например нормальные клетки против раковых клеток. Однако этот метод сравнения недостаточно чувствителен, чтобы обнаружить тонкие различия между экспрессией отдельных генов, поскольку болезни обычно затрагивают целые группы генов. [2] Множественные гены связаны с одним биологическим путем, и поэтому именно аддитивное изменение экспрессии в наборах генов приводит к различию в фенотипической экспрессии. Был разработан анализ обогащения генетической группы [2] сосредоточиться на изменениях экспрессии в группах априори определенных наборов генов. Таким образом, этот метод решает проблему необнаруживаемых небольших изменений в экспрессии отдельных генов.
Методы GSEA
Анализ обогащения набора генов использует априори наборы генов, сгруппированные по их участию в одном и том же биологическом пути или по проксимальному расположению на хромосоме. [1] Базу данных этих предопределенных наборов можно найти на База данных молекулярных сигнатур (MSigDB). [3] [4] В GSEA, ДНК-микрочипах или сейчас РНК-Seq, по-прежнему выполняются и сравниваются между двумя категориями клеток, но вместо того, чтобы сосредоточиться на отдельных генах в длинном списке, основное внимание уделяется набору генов. [1] Исследователи анализируют, попадают ли большинство генов из набора в крайние точки этого списка: верхняя и нижняя части списка соответствуют наибольшим различиям в экспрессии между двумя типами клеток. Если набор генов попадает либо в верхнюю (сверхэкспрессию), либо в нижнюю (в недостаточно выраженную), считается, что это связано с фенотипическими различиями.
В методе, который обычно называют стандартным GSEA, аналитический процесс включает три этапа. [1] [2] Общие шаги кратко изложены ниже:
- Рассчитайте оценка обогащения (ES), который представляет количество, в котором гены в наборе чрезмерно представлены в верхней или нижней части списка. Эта оценка Колмогоров – Смирнов -подобная статистика. [1][2]
- Оцените статистическую значимость ES. Этот расчет выполняется с помощью теста на перестановку на основе фенотипа, чтобы получить нулевое распределение для ES. Значение P определяется путем сравнения с нулевым распределением. [1][2]
- Вычисление значимости этим способом проверяет зависимость набора генов от диагностических / фенотипических меток. [1][2]
- Сделайте поправку на проверку нескольких гипотез, когда одновременно анализируется большое количество наборов генов. Оценки обогащения для каждого набора нормализуются, и рассчитывается коэффициент ложного обнаружения. [1][2]
Ограничения и предлагаемые альтернативы стандартному GSEA
Когда в 2003 году было впервые предложено GSEA, возникли некоторые непосредственные опасения по поводу его методологии. Эта критика привела к использованию взвешенного по корреляции критерия Колмогорова – Смирнова, нормализованного ES и расчета коэффициента ложного обнаружения, которые в настоящее время являются факторами, определяющими стандартное GSEA. [5] Однако теперь GSEA также подвергается критике за то, что его нулевое распределение является излишним и слишком сложным, чтобы его стоило вычислить, а также за то, что его статистика, подобная Колмогорову – Смирнову, не так чувствительна, как исходная. [5] В качестве альтернативы был предложен метод, известный как анализ простого обогащения (SEA). Этот метод предполагает независимость генов и использует более простой подход для расчета t-критерия. Однако считается, что эти предположения на самом деле слишком упрощают, и корреляцию генов нельзя игнорировать. [5]
Еще одно ограничение анализа обогащения набора генов заключается в том, что результаты очень зависят от алгоритма, который объединяет гены в кластеры, и количества тестируемых кластеров. [6] Spectral Gene Set Enrichment (SGSE) - это предлагаемый неконтролируемый тест. Создатели метода утверждают, что это лучший способ найти ассоциации между наборами генов MSigDB и данными микрочипов. Общие шаги включают:
1. Расчет связи между основными компонентами и наборами генов. [6]
2. Использование взвешенного Z-метода для расчета связи между наборами генов и спектральной структурой данных. [6]
Инструменты для выполнения GSEA
GSEA использует сложную статистику, поэтому для выполнения расчетов требуется компьютерная программа. GSEA стало стандартной практикой, и существует множество веб-сайтов и загружаемых программ, которые предоставляют наборы данных и проводят анализ.
NASQAR
NASQAR (Nucleic Acid SeQuence Analysis Resource) - это веб-платформа с открытым исходным кодом для высокопроизводительного анализа и визуализации данных секвенирования. [7] [8] Пользователи могут выполнять GSEA с помощью популярного пакета clusterProfiler на основе R. [9] в простом и удобном веб-приложении. NASQAR в настоящее время поддерживает GO Term и KEGG Pathway обогащение всеми организмами, поддерживаемыми базой данных Org.Db. [10]
PlantRegMap
В генная онтология Доступна аннотация (GO) для 165 видов растений и анализ обогащения GO. [11]
MSigDB
База данных молекулярных сигнатур содержит обширную коллекцию аннотированных наборов генов, которые можно использовать с большинством программного обеспечения GSEA.
Broad Institute
Веб-сайт Broad Institute находится в сотрудничестве с MSigDB и имеет загружаемое программное обеспечение GSEA, а также общее руководство для тех, кто не знаком с этой аналитической техникой. [12]
WebGestalt
WebGestalt [13] представляет собой набор инструментов для анализа набора генов в Интернете. Он поддерживает три хорошо зарекомендовавших себя дополнительных метода анализа обогащения, включая анализ избыточного представления (ORA), анализ обогащения генетического набора (GSEA) и анализ на основе топологии сети (NTA). Анализ может быть выполнен в отношении 12 организмов и 321 251 функциональной категории с использованием 354 идентификаторов генов из различных баз данных и технологических платформ.
Enrichr
Enrichr - это инструмент анализа обогащения набора генов для наборов генов млекопитающих. Он содержит фоновые библиотеки для регуляции транскрипции, путей и взаимодействий с белками, онтологии, включая ГО, онтологии фенотипов человека и мыши, сигнатуры клеток, обработанных лекарствами, и экспрессию генов в различных клетках и тканях. Enrichr был разработан лабораторией Ma'ayan на горе Синай. [14] Фоновые библиотеки собраны из более чем 70 ресурсов и содержат более 200 000 аннотированных наборов генов. Доступ к инструменту можно получить через API, и он предоставляет различные способы визуализации результатов. [15]
GeneSCF
GeneSCF - это инструмент функционального обогащения в режиме реального времени с поддержкой нескольких организмов. [16] и предназначен для решения проблем, связанных с использованием устаревших ресурсов и баз данных. [17] Преимущества использования GeneSCF: анализ в реальном времени, пользователям не нужно полагаться на инструменты обогащения, чтобы получать обновления, для вычислительных биологов легко интегрировать GeneSCF с их конвейером NGS, он поддерживает несколько организмов, анализ обогащения для нескольких списков генов с использованием базы данных из нескольких источников за один прогон извлеките или загрузите полные термины / пути / функции GO со связанными генами в виде простой таблицы в текстовом файле. [18] [19]
ДЭВИД
ДЭВИД это база данных для аннотаций, визуализации и интегрированного обнаружения, биоинформатика инструмент, который объединяет информацию из большинства основных источников биоинформатики с целью анализа больших списков генов в высокая пропускная способность манера. [20] DAVID выходит за рамки стандартного GSEA с дополнительными функциями, такими как переключение между идентификаторами гена и белка в масштабе всего генома, [20] однако аннотации, используемые DAVID, не обновлялись с октября 2016 года, [21] что может существенно повлиять на практическую интерпретацию результатов. [22]
Метаскейп
Метаскейп портал для анализа списков генов, ориентированный на биологов. [23] Метаскейп объединяет анализ обогащения путей, комплексный анализ белков и метаанализ с несколькими списками в один цельный рабочий процесс, доступный через значительно упрощенный пользовательский интерфейс. Метаскейп поддерживает точность анализа, ежемесячно обновляя свои 40 базовых баз знаний. Метаскейп представляет результаты с использованием удобной для интерпретации графики, электронных таблиц и презентаций высокого качества и находится в свободном доступе. [24]
AmiGO 2
В Генная онтология (GO) консорциум также разработал собственный онлайн-инструмент для обогащения терминов GO, [25] позволяет проводить анализ обогащения по конкретным видам в сравнении с полной базой данных, более крупнозернистыми GO slims или настраиваемыми ссылками. [26]
ОТЛИЧНЫЙ
В 2010 году Джилл Беджерано из Стэндфордский Университет выпустил Инструмент для обогащения аннотаций геномной области (GREAT), программное обеспечение, использующее регуляторные домены чтобы лучше связать термины генной онтологии с генами. [27] Его основная цель - выявить пути и процессы, которые в значительной степени связаны с активностью, регулирующей факторы. Этот метод сопоставляет гены с регуляторными областями посредством гипергеометрического теста по генам, выявляя проксимальные регуляторные домены генов. Это достигается за счет использования общей доли генома, связанной с данным термином онтологии, в качестве ожидаемой доли областей ввода, случайно связанных с термином. Обогащение рассчитывается по всем регуляторным регионам, и для проверки GREAT было проведено несколько экспериментов, один из которых - анализ обогащения, выполненный на 8 наборах данных ChIP-seq. [28]
FunRich
Инструмент функционального анализа обогащения (FunRich) [29] в основном используется для функционального обогащения и сетевого анализа ОМИКС данные. [30]
FuncAssociate
FuncAssociate Инструмент позволяет использовать онтологию генов и настраивать анализ обогащения. Это позволяет вводить упорядоченные наборы, а также файлы взвешенного пространства генов для фона. [31]
InterMine
Экземпляры InterMine автоматически предоставлять анализ обогащения [32] для загруженных наборов генов и других биологических объектов.
Люкс ToppGene
ToppGene представляет собой универсальный портал для анализа пополнения списка генов и определения приоритетов генов-кандидатов на основе функциональных аннотаций и сети взаимодействия белков. [33] Разработано и поддерживается отделом биомедицинской информатики в Медицинский центр детской больницы Цинциннати.
QuSAGE
Количественный анализ набора для экспрессии генов (QuSAGE) представляет собой вычислительный метод анализа обогащения набора генов. [34] QuSAGE повышает мощность за счет учета межгенных корреляций и количественной оценки активности набора генов с полным функция плотности вероятности (PDF). Из этого PDF-файла P значения и доверительные интервалы легко извлекается. Сохранение PDF также позволяет проводить апостериорный анализ (например, попарные сравнения активности набора генов) при сохранении статистической прослеживаемости. Тернер и др. расширила сферу применения QuSAGE на лонгитюдные исследования путем добавления функциональности для общих линейных смешанных моделей. [35] QuSAGE использовался NIH / NIAID Консорциум проекта по иммунологии человека для определения исходных транскрипционных сигнатур, связанных с человеческими вакцинация против гриппа ответы. [36] QuSAGE доступен как Пакет R / Bioconductor, и поддерживается Лаборатория Кляйнштейна в Йельская школа медицины.
Blast2GO
Blast2GO это биоинформатическая платформа для функциональной аннотации и анализа наборов геномных данных. [37] Этот инструмент позволяет выполнять анализ обогащения набора генов (GSEA ), [38] среди других функций.
g: Профайлер
Приложения и результаты GSEA
GSEA и полногеномные ассоциации исследований
Однонуклеотидные полиморфизмы, или SNP, представляют собой одноосновные мутации, которые могут быть связаны с заболеваниями. Одно изменение основания может повлиять на белок, являющийся результатом экспрессии этого гена; однако он также может не иметь никакого эффекта. Полногеномные исследования ассоциации представляют собой сравнения между здоровым генотипом и генотипом болезни, чтобы попытаться найти SNP, которые чрезмерно представлены в геномах болезни и могут быть связаны с этим состоянием. До GSEA точность полногеномных исследований ассоциации SNP была сильно ограничена большим количеством ложноположительных результатов. [39] Теория о том, что SNP, способствующие возникновению заболевания, как правило, сгруппированы в набор генов, которые все участвуют в одном и том же биологическом пути, - это то, на чем основан метод GSEA-SNP. Это приложение GSEA не только помогает в обнаружении связанных с заболеванием SNP, но и помогает осветить соответствующие пути и механизмы заболеваний. [39]
GSEA и спонтанные преждевременные роды
Методы обогащения набора генов привели к открытию новых подозрительных генов и биологических путей, связанных с самопроизвольные преждевременные роды. [40] Exome Последовательности женщин, перенесших SPTB, сравнивали с последовательностями женщин из проекта 1000 Genome Project, используя инструмент, который оценивал возможные варианты, вызывающие заболевание. Затем гены с более высокими баллами прогонялись через разные программы, чтобы сгруппировать их в наборы генов на основе путей и групп онтологий. Это исследование показало, что варианты были значительно сгруппированы в наборы, связанные с несколькими путями, все из которых подозревались в SPTB. [40]
GSEA и профилирование раковых клеток
Анализ обогащения набора генов можно использовать для понимания изменений, которые клетки претерпевают во время канцерогенез и метастаз. В ходе исследования микроматрицы были выполнены на карцинома почек метастазы, первичные опухоли почек и нормальную ткань почек, и данные были проанализированы с помощью GSEA. [41] Этот анализ показал значительные изменения экспрессии генов, участвующих в путях, которые ранее не были связаны с прогрессированием рака почек. На основании этого исследования GSEA предоставила новые потенциальные мишени для терапии почечно-клеточного рака.
GSEA и шизофрения
GSEA можно использовать для понимания молекулярных механизмов сложных расстройств. Шизофрения - это в значительной степени наследственное заболевание, но оно также очень сложное, и начало болезни связано с множеством генов, взаимодействующих по нескольким путям, а также взаимодействием этих генов с факторами окружающей среды. Например, эпигенетические изменения, такие как Метилирование ДНК, подвержены влиянию окружающей среды, но также по своей сути зависят от самой ДНК. Метилирование ДНК является наиболее хорошо изученным эпигенетическим изменением и недавно было проанализировано с помощью GSEA в отношении промежуточных фенотипов, связанных с шизофренией. [42] Исследователи ранжировали гены по их корреляции между паттернами метилирования и каждым из фенотипов. Затем они использовали GSEA для поиска обогащения генов, которые, по прогнозам, будут нацелены на микроРНК при прогрессировании заболевания. [42]
GSEA и депрессия
GSEA может помочь предоставить молекулярные доказательства ассоциации биологических путей с заболеваниями. Предыдущие исследования показали, что долгосрочные симптомы депрессии коррелируют с изменениями иммунного ответа и воспалительных путей. [43] Это подтверждали генетические и молекулярные доказательства. Исследователи взяли образцы крови у людей, страдающих депрессией, и использовали данные об экспрессии по всему геному, а также GSEA, чтобы найти различия в экспрессии в наборах генов, связанных с воспалительными путями. Это исследование показало, что у людей с наиболее тяжелыми симптомами депрессии также наблюдались значительные различия в экспрессии этих наборов генов, и этот результат подтверждает гипотезу ассоциации. [43]
Профилирование экспрессии генов и кластеризация сигнатур опухоли
Профилирование экспрессии генов и кластеризация сигнатур опухоли
Геномика уже оказывает большие влияние на диагностическую точность и оптимизацию терапии при раке. В этой секции мы опишем, как для диагностики и лечения опухолей используют один из видов геномной технологии — профилирование экспрессии.
Предположим, у нас есть множество образцов тканей из разных опухолей и мы хотим разработать чувствительный метод, чтобы различать эти типы опухолей в будущих образцах. Для одновременного измерения уровней экспрессии мРНК некоторых или всех из приблизительно 25 000 генов человека в любом образце ткани относительно стандартного образца может использоваться метод CGH.
Измерение экспрессии мРНК в образце создает профиль экспрессии генов, специфичный для этого образца. На рисунке изображена гипотетическая идеализированная ситуация для восьми образцов, по четыре на каждый из двух типов рака, А и В, профилированных по 100 разным генам. Профиль экспрессии, полученный из массивов экспрессии в этом простом примере, достаточно достоверен, так как содержит 800 показателей экспрессии.
В реальном эксперименте по профилированию экспрессии могут анализироваться сотни образцов по экспрессии всех генов человека, что быстро создает огромные массивы данных, содержащих миллионы значений величины экспрессии. Необходимость организации и анализа данных для получения ключевой информации вызвало проблемы, потребовавшие развития сложных статистических и биоинформационных инструментальных средств. Используя эти средства, можно так организовать данные, чтобы найти группы генов с согласованной экспрессией. Группировку генов по их экспрессии в образцах называют кластеризацией.
Кластеры экспрессии генов затем могут тестироваться для нахождения корреляции с конкретными характеристиками интересующих образцов. Например, профилирование может указать, что группа генов с коррелирующими профилями экспрессии чаще выявляется в образцах из опухоли А, но не из опухоли В, тогда как другая группа генов с коррелирующими профилями, наоборот, чаще присутствует в образцах опухоли В, но не А.
Гены, экспрессия которых согласуется друг с другом и с конкретным набором образцов, составляют сигнатуру экспрессии этих образцов.
Профилирование экспрессии генов для описания опухолей может оказаться полезным по нескольким причинам.
• Во-первых, это резко увеличивает возможности различать опухоли, дополняя стандартные критерии, применяемые патологами, например гистологическую картину, цитогенетические маркеры и экспрессию специфических маркерных белков.
Если различные сигнатуры для разных типов опухолей (например, опухоль А по сравнению с опухолью В) определяются известным образом, образец экспрессии неизвестной опухоли затем можно сравнить с сигнатурами экспрессии опухолей А и В и классифицировать его как А-подобный, В-подобный или никакой, в зависимости от того, насколько хорошо его профиль экспрессии соответствует сигнатурам опухолей А и В.
• Во-вторых, разные сигнатуры могут коррелировать с разными клиническими результатами, например прогнозом, ответом на терапию или любыми другими интересующими результатами. Если это подтвердится, такие сигнатуры могли бы быть использованы для помощи в выборе лечения у вновь диагностированных пациентов.
• В-третьих, кластеризация может выявить прежде не предполагаемые функциональные взаимосвязи генов, включенных в процесс болезни, что окажется немаловажным для теоретических исследований.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Комплексное геномное профилирование
В этом разделе вы узнаете о комплексном геномном профилировании (КГП) и перспективах использования этого подхода для преобразования данных геномных исследований в клинически полезную информацию.
Этот раздел может помочь в поиске других методов специфической терапии для пациентов
с CUP-синдромом.
Что собой представляет подход комплексного геномного профилирования?
Комплексное геномное профилирование (КГП) может быть выполнено при разных злокачественных опухолях с целью преобразования данных геномных исследований
в клинически значимую информацию. Он может помочь в поиске методов терапии CUP-синдрома на основании молекулярных исследований1. 1-4
Что обнаруживает КГП?
КГП применяет технологию секвенирования следующего поколения (ССП) для обнаружения известных и новых вариантов в четырех основных классах геномных альтераций большого подтипа связанных с раком генов и выявления геномных сигнатур, т. е. мутационной нагрузки опухоли (МНО) и микросателлитной нестабильности (МСН). 4–9
Какие типы альтераций может обнаружить КГП?
Например, все службы Foundation Medicine обнаруживают четыре основных класса геномных альтераций в одном тесте. FoundationOne®CDx дополнительно определяет МНО и МСН, и FoundationOne®Liquid определяет МСН. 3-5,10
Комплексное геномное профилирование по сравнению с профилированием экспрессии генов
Как соотносятся КГП и профилирование экспрессии генов (ПЭГ)?
Как указано выше, КГП выявляет существующие альтерации опухолевой ДНК, тогда как ПЭГ анализирует паттерны экспрессии генов на основании мРНК. Паттерны экспрессии генов могут быть выявлены в большинстве метастазов, где эти паттерны могут соответствовать ткани,
из которой они происходят. 1-4
1. Chalmers Z R et al. Genome Med 2017; 9: 34.
2. Frampton GM et al. Nat biotechnol 2013; 31: 1023–31.
3. He J et al. Blood 2016; 127: 3004–14.
4. Greco FA et al. Ann Oncol 2012; 23: 298–304.
Пример пользы КГП при раке легкого
Пример КГП при немелкоклеточном раке легкого (НМКРЛ)
КГП с валидированными тестами панелей из нескольких генов выявляет геномные альтерации
в опухолевой ткани, которые могут иметь клиническое значение. Варианты терапии
могут быть спланированы в соответствии
с обнаруженным у пациента паттерном альтераций1. 1
Эти разработки главным образом основаны
на НМКРЛ, для которого существуют клинически валидированные методы таргетной терапии, продемонстрировавшие улучшенные исходы. 2–8
Ранее НМКРЛ считался одним заболеванием. Теперь мы знаем, что к раку легкого приводят многие различные альтерации. При рассмотрении разнообразия типов НМКРЛ мы понимаем, что множество геномных профилей выявляет различные типы. С помощью КГП можно различить в НМКРЛ несколько малых молекулярных подтипов.
Возможная польза КГП при CUP-синдроме
Какова возможная польза КГП при
CUP-синдроме?
В большинстве случаев (80–85 %) пациенты
с CUP-синдромом, имеют неблагоприятный профиль рисков и в момент обследования имеют неблагоприятный прогноз, несмотря на применение различных сочетаний химиотерапевтических средств в клинических исследованиях. В этой конкретной группе частота ответа на лечение низка и медиана общей выживаемости обычно менее одного года. КГП показало, что почти все образцы, полученные у пациентов с CUP-синдромом, имеют пригодные для таргетного воздействия альтерации, возможно объясняющие ограниченные возможности и неблагоприятный прогноз для пациентов с
CUP-синдромом. 1,2
1. Ross JS et al. JAMA Oncol 2015; 1: 40–9.
2. Subbiah IM et al. Oncoscience 2017; 4: 47–56.
Варианты терапии, основанные на молекулярных исследованиях
Пациенты с CUP-синдромом и плохим прогнозом получают стандартное лечение: химиотерапию двумя препаратами на основе платины или паклитаксел.
В некоторых исследованиях фазы II с применением сочетания препаратов платины и таксанов или паклитаксела общая частота ответа составляла примерно
30 % и медиана выживаемости составляла от 7 до 11 месяцев. 1-3
Возможная польза КГП при
CUP-синдроме
Пациенты с CUP-синдромом являются неоднородной популяцией, в которой присутствуют различные альтерации, возможно пригодные для таргетного воздействия.
В одной из публикаций показано, что у 30 % пациентов молекулярное профилирование опухоли обнаружило геномные альтерации, пригодные для таргетного воздействия. 4
В другой публикации выполнено КГП (исследование биопсийных тканей методом Foundation Medicine) образцов, полученных
у 200 пациентов с CUP-синдромом, с целью определения возможности выявления геномных альтераций, способных послужить основой для таргетной терапии CUP-синдрома. 5 Из этих
200 случаев CUP-синдрома 96 % имели не менее одной геномной альтерации, а одна или большее число значимых геномных альтераций обнаружены в 85 % из этих случаев (169 из 200). 5
Анализ базы данных FoundationCore TM для пациентов с CUP-синдромом
Анализ базы данных FoundationCore TM для пациентов с CUP-синдромом (состоящей из данных Foundation Medicine) также показал клиническую возможность проспективного клинического исследования терапии, основанной на молекулярных исследованиях. 6
1. Huebner G et al. British Journal of Cancer 2009; 100: 44-9.
2. Hainsworth JD et al. J Clin Oncol 1997; 15: 2385-93.
3. Greco FA et al. J Clin Oncol 2002; 15 (20): 1651-6.
4. Varghese AM et al. Ann Oncol 2017; 1 (28): 3015-21.
5. Ross JS et al. JAMA Oncol 2015; 1: 40-9.
6. Krämer A et al. J Clin Oncol 2018; 36 (15): (suppl e24162).
Технология прямой цифровой детекции NanoString
Система мультиплексного цифрового анализа nCounter SPRINT™ Profiler использует уникальную технологию NanoString и является единственным прибором, который способен в одной пробе количественно проанализировать до 800 мишеней как РНК, так и белков одновременно.
Технология NanoString позволяет проводить прямое мультиплексное измерение транскрипционной активности генов и уровня трансляции соответствующих им белков, профилирование экспрессии микроРНК и оценку копийности генов (в том числе в одной клетке) и др.. Что позволит лаборатории проводить широкий ряд исследований в области онкологии, иммунологии, нейропатологий, профилирования РНК и многих других. В основу метода положено мечение мишеней уникальными цветовыми штрих-кодами, прикрепленными к мишень-специфичным зондам и их последующая детекция. За счет исключения из технологического процесса этапа амплификации и, как следствие, связанных с ним ошибок, демонстрируется высокий уровень точности и чувствительности.
Основные преимущества метода:
- Единовременное измерение экспрессии сотен генов-мишеней/микроРНК, мишеней ДНК и десятков белков в одной реакции
- Высокая чувствительность (
- Полностью автоматизированная система
- Отсутствие этапов энзиматической обработки образцов
- Малые концентрации и объемы исходного материала
- Низкие требования к качеству исходного материала
- Широкий динамический диапазон
- Минимальная вариабельность результатов между лотами и сайтами тестирования
Технология заняла четвертое место в списке лучших инноваций 2013 года и была опубликована на обложке журнала Nature. На данный момент существует более 2300 публикаций с использованием технологии
1. Принцип технологии
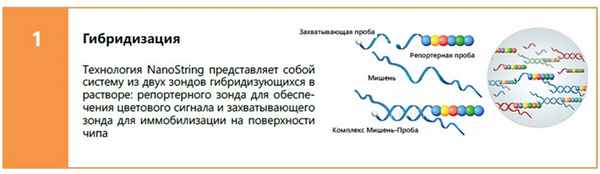
Технология nCounter основана на прямой цифровой детекции мишеней с помощью флуоресцентных штрих-кодов. На выбранные мишени синтезируются два зонда: захватывающая проба - олигонуклеотид меченный биотином и репортерная проба – олигонуклеотид на одном своем конце содержащий уникальный цветовой штрих-код из 4 разных флуорофоров в 6 позициях. Каждая такая цветовая последовательность указывает на конкретную мишень.
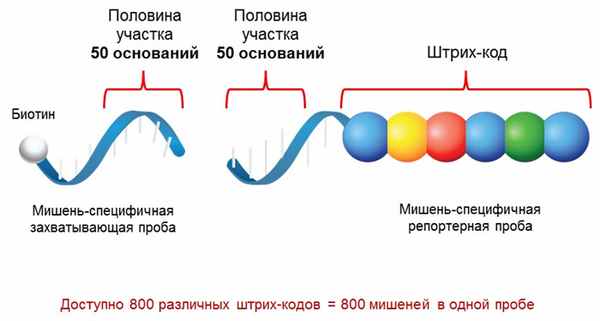
В зависимости от задач структура зонда может варьироваться
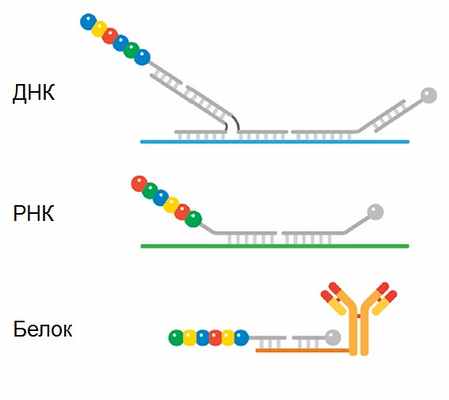


Для анализа микроРНК потребуется дополнительный этап лигирования адаптеров для увеличения длинны мишени
Для анализа экспрессии белков используются антитела, к которым пришита единичная молекула олигонуклеотида фоточувствительной связью. После того, как антитела связались с белками-мишенями и все несвязавшиеся антитела отмыты, на пробу оказывается действие UV и олигонуклеотиды отделяются от антител в жидкость. Затем олигонуклеотиды анализируются по стандартной схеме.
Сам процесс работы прост и автоматизирован, что позволяет экономить рабочее время сотрудников лаборатории, не требует длительного обучения работе на приборе а также демонстрирует высокую воспроизводимость экспериментов. Требуется ручная постановка только для этапа гибридизации образцов с зондами (5 минут рабочего времени) и загрузка реагентов в прибор (5 минут)
2. Технические преимущества метода
- Анализ без предварительной обратной транскрипции и/или амплификаци., что позволяет избежать возможных ошибок на этих этапах
- Высокая чувствительность ( Чувствительность технологии 0,1-0,5 фемтоМоль РНК мишени (7)
- Высокая воспроизводимось, не требуются технические повторы
Полученные данные показывают R2 = 0.99 (7), R2 > 0.98 (8) Также, в исследованиях (1) была показана высокая воспроизводимось при технических репликах, на основании чего были сделаны выводы о том, что технические повторы для данной методики не требуются - Минимальное влияние технических изменений (стабильность зондов, картриджа) (8)
- Не требует большого количества исходного материала.
3. Сравнение с другими методами
NanoString и ПЦР
Rémi Pescarmona с соавторами проводили сравнение NanoString и ПЦР в реальном времени для анализа экспресии генов. Полученные данные показали высокую сходимость результатов R2= 0.87
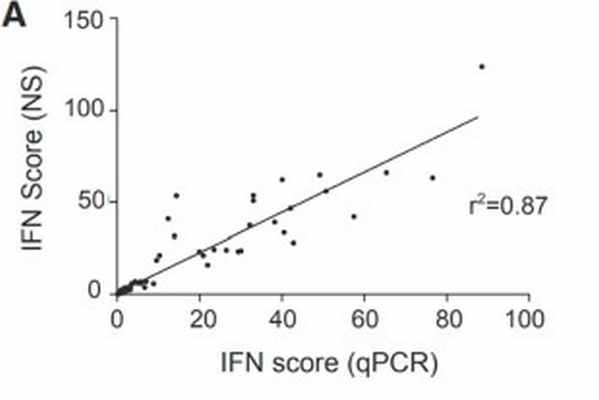
Однако, при этом технология NanoString зарекомендовала себя как более быстрая, с меньшим временем ручной работы и с большой возможностью к мультиплексированию (3)
NanoString и цифровая ПЦР
David A. Armstrong и соавторы исследовали уровень микроРНК при раке мочевого пузыря различных биологических образцах в парафиновых блоках, экзосомах выделенных из мочи, плазме крови, лейкоцитарной пленке методом NanoString. Валидацию результатов производили методом цифровой ПЦР. Результаты показали высокую корреляцию между данными полученными этими двумя методами R=0.83; R=0.88 R=0.91 (5)
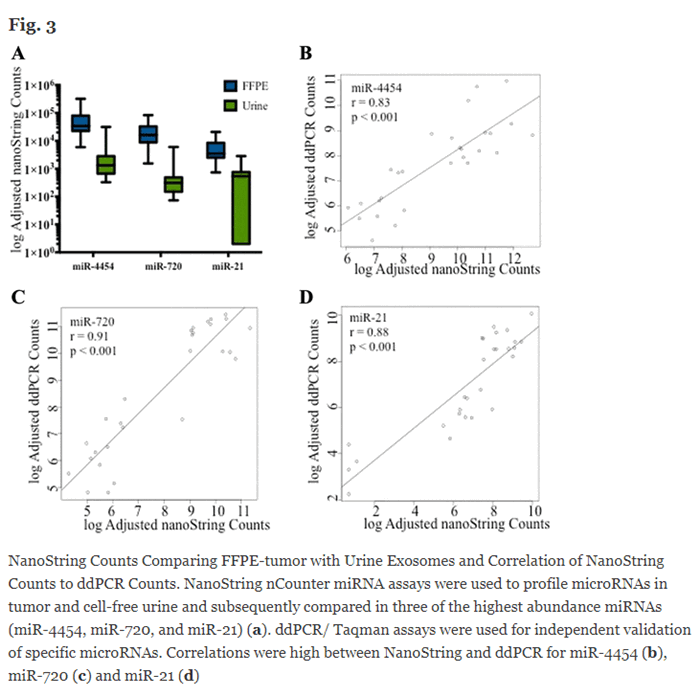
NanoString, FISH и ПЦР для детекции химерных транскриптов и генов
В исследовании Kenneth T.E. Chang и сооавторов была разработана уникальная панель для детекции 174 транслокаций с помощью технологии NanoString. В рамках исследования производилось сравнение с ПЦР-РВ и FISH, и было выявлено, что NanoString имеет ряд преимуществ:
- Подход к детекции химерных транскриптов не требует постановки нескольких реакций как FISH или ПЦР-РВ
- Анализ значительно улучшает сроки выполнения работ и затраты на рабочую силу, позволяя при этом дать полную характеристику генов партнеров и экзон-экзонному соединению, которые потенциально могут иметь важное значение для клинической практики. Например, был обнаружен один случай широко распространенной внутрибрюшной и тазовой мелкоклеточной саркомы малого таза, который при клиническом анализе FISH был положительным по перестройке EWSR1, но негативным по перестройке WT1 и FLI1. Анализ NanoString выявил слияние EWSR1-ERG в этом случае, позволяя классифицировать его как саркому Юинга, а не как десмопластическую круглоклеточную опухоль (опухоль, которая не будет реагировать на химиотерапию Юинга)
- Данные показали, что исследование одного образца на 174 фюжн вариантов технологией NanoString сопоставима по стоимости с одним FISH исследованием (6)
4. Реагенты для использования с системой
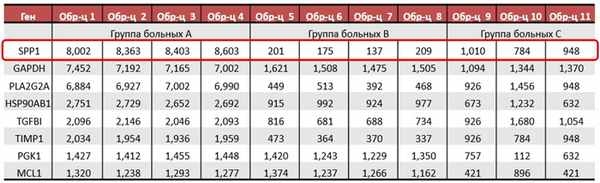
Производитель предлагает широкий перечень уже готовых панелей для различных областей исследования: онкология, онкогематология, нейропатологии, нейровоспаление, иммунология, аутоиммуные заболевания, стволовые клетки, микроРНК и многие другие. Большинство готовых панелей на экспрессию генов в своем составе, кроме генов различных сигнальных путей и сигнатур, отвечающих за определенные процессы, имеют сигнатуры для определения клеточного состава образца. Например, панель nCouner Human Neuroinflamation демонстрирует в процентном соотношении клетки ЦНС: нейроны, астроциты, микроглия, олигодендроциты, эндотелий и периферические иммунные клетки: B-клетки, дендритные клетки, истощённые цитотоксические лимфоциты, макрофаги, T-клетки, CD8+ T клетки, нейтрофилы, тучные клетки, цитотоксические клетки, регуляторные T-клетки, натуральные киллеры CD56dim, натуральные киллеры, CD45+ и T-хелперы 1. Также, эта панель исследует 23 сигнальных пути и процессов, затрагивающих три основные темы нейровоспаления: иммунитет и воспаление, нейробиология и нейропатология, метаболизм и стресс. Кроме сигнатур связанных с процессами, сигнальными путями и типами клеток, некоторые наборы включают специфические сигнатуры такие как PAM50 для определения молекулярного подтипа опухоли молочной железы или TIS (Tumor Inflammation Signature) которая показывает на вовлеченных и активацию иммунных клеток в микроокружение опухоли. Другой пример - панель nCounter IO 360 анализирует более 40 различных сигнатур для исследования вовлеченности иммунного ответа при онкологических заболеваниях
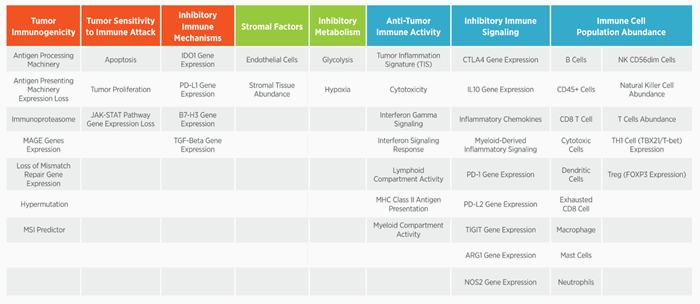
Готовые панели серии Vantage можно смешивать между собой и создавать панель для одновременного количественного анализа РНК и белков:
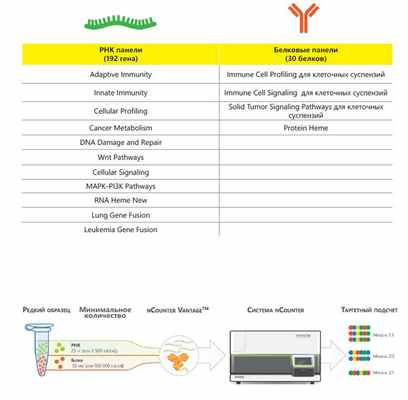
Кроме уже готовых панелей технология позволяет создавать и свои собственные панели в трех различных вариантах:
- Готовая панель по списку генов/мишеней от пользователя
- Химия Elements – заказываются универсальные зонды и захватывающие пробы, к которым пришиваются олигонуклеотиды в лаборатории пользователя
- Химия PlexSet – также заказываются универсальные зонды и захватывающие пробы, но в смеси, что позволяет мультиплексировать образцы и в одной лунке анализировать не один образец на 800 мишеней, а 8 образцов по 96 мишеней (пока только для анализа экспрессии генов)
Сейчас в разработке находится набор для пришивания олигонуклеотидов к антителам фоточувствительной связью для создания кастомизированных белковых панелей на базе пользовательских лабораторий.
5. Программное обеспечение nSolver
Программное обеспечение nSolver позволяет производить анализ полученных данных и визуализировать результаты в различных форматах без использования дополнительных биоинформатических ресурсов. Система в режиме Advanced Analysis визуализирует генные сети и отмечает цветом гиперэкспрессированные гены и гены, экспрессия которых подавлена.
Базовый анализ включает в себя: диаграммы размаха (Box Plots), тепловые карты (Heat Maps), диаграммы рассеивания (Scatter Plots), скрипичные диаграммы (Violin Plots), гистограммы
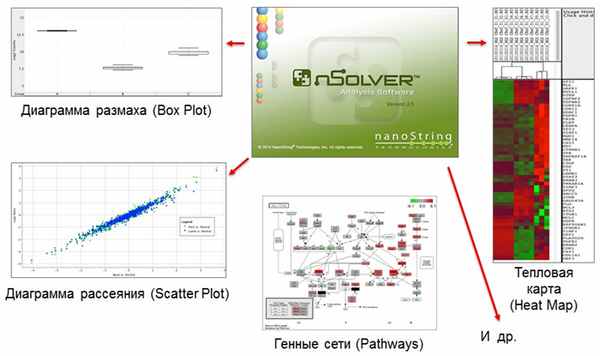
Продвинутый анализ
Дифференциальная экспрессия – позволяет следить за тем, как экспериментальные факторы влияют на изменения экспрессии на уровне сигнальных путей
Профилирование клеток – позволяет отследить, как соотношения известных маркеров различных типов клеток изменяются в зависимости от лечения, времени или других переменных.
Pathway Score – позволяет выявить участки вариабельности путем кластеризации по сигнальному пути
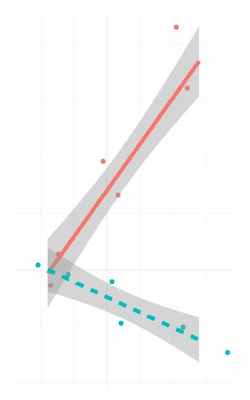
Корреляция экспрессии РНК:белок – позволяет сравнить уровень экспрессии мРНК и белков для каждой индивидуальной мишени, что позволяет выявлять новые взаимосвязи между транскрипционными и трансляционными ответами
Вести науки
В данной статье представлены современные подходы к классификации этого состояния, описаны диагностические алгоритмы, разобраны общие методы терапии острой СН.
Измерение концентрации ИЛ-6 и СРБ в 1-е сутки развития ишемического инсульта позволяет определить пациентов с наибольшим риском развития пневмонии на госпитальном этапе.
Автоматизация исследований на ВИЧ с помощью ИФА-анализатора приводит в целом к отрицательному экономическому эффекту за счет существенного превышения затрат на дополнительные расходные материалы и амортизацию оборудования над возможной экономией фонда оплаты труда.
В острой ситуации биомаркеры указывают на большую дисфункцию миокарда и воспаление у STEMI и демонстрируют более разнообразную патофизиологическую картину у пациентов с NSTEMI. Эти же биомаркеры оказались одинаково прогностическими у пациентов STEMI и NSTEMI.
Повышенные сывороточные уровни PM20D1 и его каталитических продуктов NAAA тесно связаны с дисрегуляцией глюкозы, связанной с ожирением, резистентностью к инсулину и MetS, и потенциально могут использоваться в качестве клинических биомаркеров для диагностики и мониторинга указанных расстройств.

В этом обзоре рассмотрены генерация, изменения и механизмы действия специфичных для плаценты экзосом при ГДМ, а также их перспективы в качестве прогностической и терапевтической мишени для диагностики, мониторинга и лечения гестационного диабета.
Исследование предполагает необходимость проведения тренингов и супервизий для вовлеченных сотрудников и скоординированной деятельности по предоставлению качественных услуг, отвечающих потребностям клиентов.
Основываясь на патологическом диагнозе почечной биопсии у пациентов с СД2, в настоящем исследовании были изучены связанные факторы ДН и создана модель номограммы для прогнозирования риска ДН, чтобы направлять клинический скрининг групп высокого риска ДН и формулировать более целенаправленные стратегии вмешательства.
Институт Лабораторной Медицины
Образовательная организация, реализующая мероприятия и программы в области дополнительного профессионального образования в сфере здравоохранения для специалистов с высшим медицинским и немедицинским образованием и среднего медицинского персонала.
Читайте также: