Проводящая глухота с микрогнатией и вислоухостью. Синдром Ушера
Добавил пользователь Cypher Обновлено: 30.01.2026
Морфология, наследственность и лечение синдрома Ушера
Изменения височной кости включали улитку, ее нерв и питающие ее сосуды. Отмечалась атрофия кортиева органа и эпителия внутреннего и наружного желобка в нижней части базального завитка улитки. Дегенеративные изменения уменьшались в верхнем завитке. В дополнение имелась резкая атрофия спирального узла, его периферических и центральных волокон, а также концевых ветвей сосудов лабиринта (Nager, Buch, Jorgensen). Эти изменения характерны для тина Шайбе.
Наследственность. На основании поражения сибсов, имеющих здоровых родителей, и увеличения частоты кровнородственных браков было установлено аутосомно-рецессивное наследование синдрома (Kloepfer et al., McLeod). Рассчитано, что один на 100 людей является носителем гена. У гетерозигот могут быть выявлены: отсутствие реакции на вращение (Holland et al.), повышение порога темновой адаптации (De Haas et al.) или легкая глухота (McLeod et al.).
Диагноз. В дополнение к диагнозу, установленному посредством офтальмоскопии, пигментный ретинит может быть диагностирован при помощи электроретинографии, электроокулографии, исследования полей зрения и регистрации темновой адаптации.
Пигментный ретинит в качестве изолированной патологии может наследствоваться как аутосомно-рецессивное, доминантное или Х-сцепленное состояние (Vernon). Комбинация пигментного ретинита с глухотой наблюдается при нескольких синдромах. При синдроме Альстрёма у больного имеется ожирение и может быть сахарный диабет. При синдроме Рефсума наблюдаются психические расстройства, периферические невриты и увеличение уровня фитановой кислоты в крови. При синдроме Барде — Бидля отмечаются умственная отсталость, ожирение, гипогонадизм и полидактилия. У больных с синдромом Лауренс — Муна обнрауживают умственную отсталость, гипогенитализм и спастическую параплегию.
Больные с синдромом Кокейна распознаются на основании их малого роста, глубокой умственной отсталости и птицеподобного лица.
Пигментный ретинит может наблюдаться при синдроме Кернса (Kearns), включающем прогрессирующую наружную офтальмоплегию, пигментную дегенерацию сетчатки, дефект сердечной проводимости и смешанную глухоту.
Лечение. В большинстве случаев глухота настолько глубока, что использование слуховых аппаратов не представляется возможным. Для пигментного ретинита лечения не имеется.
Прогноз. Большинство больных из-за прогрессирующей потери зрения или других расстройств, связанных с этим заболеванием, вынуждены к 30—40-летнему возрасту оставить работу.
Выводы. Синдром Ушера характеризуется: 1) аутосомно-рецессивным наследованием, 2) прогрессирующей потерей зрения в связи с пигментным ретинитом, 3) умственной отсталостью или психозами (иногда), 4) легкой атаксией, врожденной глубокой или умеренной нейросенсорной глухотой и 5) отсутствием вестибулярных реакций.
Проводящая глухота с микрогнатией и вислоухостью. Синдром Ушера
Мы имели возможность обследовать мать и 2 детей — сына и дочь с оттопыренным отвисающим ухом (исправленным за несколько лет до того, как мы их видели), длинными тонкими крыльями носа, микрогнатией и непрогрессирующей смешанной глухотой, главным образом проводящей, в пределах 30—60 дБ. В анамнезе не было указаний па инфекции, шум в ушах пли головокружения. В семье было 2 здоровых сибса.
Наружный слуховой проход был значительно сужен. При тимпанотомии у обоих обнаружена фиксация основания стремени. Задняя ножка его была укорочена, составляя около 65% нормальной длины, и не была прикреплена к основанию. Мышца стремени и ее сухожилие были рудиментарными.
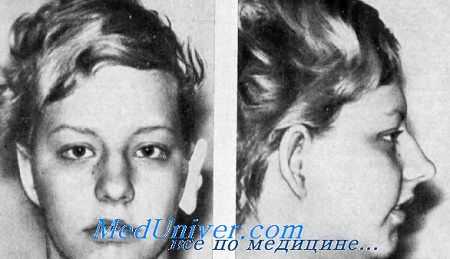
Вислоухость, микрогнатия и проводящая глухота. Мать и дочь, имевшие до хирургического вмешательства двустороннюю вислоухость и оттопыренные уши. У сына была такая же деформация ушной раковины. У всех отмечались маленькая нижняя челюсть и проводящая глухота.
Синдром Ушера - пигментный ретинит и нейросенсорная тугоухость
Наиболее частым заболеванием в группе генетических нарушений слуха в сочетании с глазными болезнями является синдром Ушера. Больные с этим синдромом страдают врожденной глухотой и прогрессирующей потерей зрения, развивающейся в связи с пигментным ретинитом. Распространенность синдрома Ушера среди детей с глубокой глухотой может достигать 10%.
При некоторых описанных в этой главе синдромах в дополнение к сочетанию глазных болезней с глухотой поражается и нервная система. При других синдромах имеется также сахарный диабет. Хотя эти синдромы можно было бы включить в другие статьи, они должны рассматриваться здесь, и это, по-видимому, правильно, ибо глазная патология является постоянным их симптомом.
Синдром Ушера характеризуется врожденной нейросенсорной потерей слуха от умеренной до резко выраженной степени, вестибулярной гипофункцией и медленно прогрессирующим пигментным ретинитом. (Hallgren). Возможно, что синдром Ушера генетически гетерогенен и что семьи, описанные Hallgren, представляют собой отдельную нозологическую форму (синдром Халлгрена).
Сочетание глухоты с пигментным ретинитом было описано очень давно, еще в 1858 г. von Gracfe, и было признано как заболевание, имеющее необычайно высокую частоту среди глухих евреев. Liebreich и Hammerschlag отметили значительное увеличение частоты кровнородственных браков между родителями больных.
Lindenov в Данин и Hallgren в Швеции установили распространенность синдрома Ушера в своих странах, составившую около 3 па 100 000 в общей популяции. Аналогичный уровень распространенности синдрома Ушера в США установили Kloepfer, Lagnaite и McLaurin. Частота больных с этим синдромом среди детей, страдающих глубокой глухотой, составляет, по данным разных исследователей, от 3 до 10% (Vernon, Fraser).
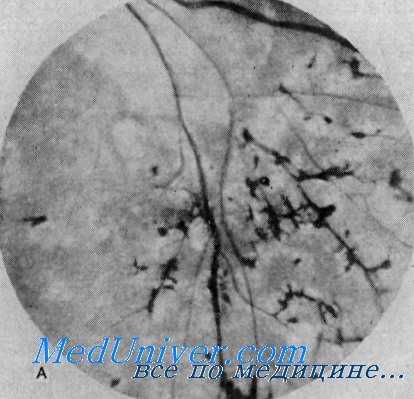
Пигментный ретинит и врожденная нейросенсорная глухота (синдром Ушера) (из А. С. McLeod et al., Arch. Otolaryngol.) Вислоухость, микрогнатия и проводящая глухота. Мать и дочь, имевшие до хирургического вмешательства двустороннюю вислоухость и оттопыренные уши. У сына была такая же деформация ушной раковины. У всех отмечались маленькая нижняя челюсть и проводящая глухота.
Клинические данные. Разделение синдрома Ушера па подтипы не производилось. Имеются факты, свидетельствующие о том, что экспрессивность синдрома может значительно варьировать (McLeod et al.).
Орган зрения. Потеря зрения выявляется обычно в возрасте около 10 лет, однако инициальные глазные симптомы в виде ночной слепоты можно обнаружить раньше, еще в дошкольном возрасте. Нарушения зрения медленно прогрессируют, достигая полной слепоты, которая в пятом десятилетии жизни наблюдается у 40%, в шестом десятилетии— у 60%, в седьмом десятилетии — у 75% больных (Hallgren). Офтальмоскопическое исследование обнаруживает типичный медленно прогрессирующий пигментный ретинит, начинающийся скоплениями гранул пигмента па глазном дне и распространяющийся по направлению к периферии. Диск зрительного нерва становится бледным, а артерии сужаются.
Другие глазные симптомы включают катаракту, потерю способности воспринимать цвет и в некоторых случаях глаукому. Примерно у 10% больных в возрасте 20 лет и у 50% больных в возрасте старше 40 лет были выявлены капсулярпые, кортикальные передние или задние катаракты (Hallgren, Cherry).
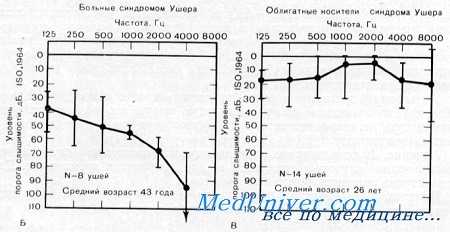
Орган слуха. Почти у 90% из 117 больных, описанных Hallgren, была обнаружена резко выраженная двусторонняя пепросен-сорная глухота, в то время как у 10% нейросенсорная потеря слуха достигала только умеренной степени (30—70 дБ) и была резче выражена на высоких частотах. Такие же изменения отметили Landau и Feinmesser, Kloepfer с сотр. и McLeod с сотр. SISI-тест был положительным на высоких частотах, а исследование по непрерывному методу Бекеши дало II тип кривой, что позволило предположить кохлеарное происхождение глухоты. McGovern и Goode с сотр. обнаружили у больных с синдромом Ушера менее глубокие нарушения слуха.
Вестибулярная система. Реакция на вестибулярную калорическую пробу обычно была патологической (Hallgren). У всех больных, обследованных Vernon и McLeod, также был обнаружен дефект вестибулярной функции. Из 6 больных, обследованных McLeod с сотр., у 4 была обнаружена нормальная реакция на колорическую пробу, у одного—слабая и еще у одного — реакция отсутствовала. У всех 6 больных имелись нарушения равновесия. Не ясно, была ли нарушена походка вследствие вестибулярной или вследствие мозжечковой патологии. Однако большинство исследователей считают, что при синдроме Ушера имеет место патология лабиринта.
Лабораторные данные. Исследования мочи и крови, а также рентгенограммы черепа были нормальными. Изменения электроэнцефалограммы не были постоянными (Krill, Stamps).
Наследственная тугоухость
Наследственная тугоухость – одна из форм врожденного нарушения слуха, обусловленная генетическими мутациями и по этой причине способная передаваться по наследству от родителей потомству. Симптомами данного состояния являются ослабление слуха различной степени тяжести, нередко возникающее в первые месяцы и годы жизни ребенка (очень редко – после 6-ти лет), а также вторичные речевые нарушения. Диагностика наследственной тугоухости осуществляется на основании данных аудиометрических исследований (аудиометр, звукореактотест), наследственного анамнеза больного и молекулярно-генетического анализа. Специфического лечения наследственной тугоухости не существует, больным нередко требуется обучение в специализированных центрах и реабилитация.
Общие сведения
Наследственная тугоухость – широкая группа различных генетических заболеваний, которые сопровождаются ослаблением слуха вплоть до полной глухоты (в зависимости от формы патологии). Этими заболеваниями обусловлено более половины случаев раннего ослабления слуха – остальные варианты являются следствием пренатального повреждения плода или приобретенной в раннем детстве тугоухости. Подобные состояния известны очень давно. Они активно изучались отоларингологами, неврологами, а в XX веке и врачами-генетиками, которые смогли определить наследственную природу у части подобных заболеваний.
Было также обнаружено, что одни разновидности наследственной тугоухости обнаруживаются в сочетании с иными симптомами генетического заболевания (синдромальные формы), а другие типы проявляются только нарушениями слуха (несиндромальные формы). Дальнейшие исследования наследственной тугоухости показали наличие огромного количества разновидностей заболевания, что потребовало создания сложной классификации данного состояния в зависимости от его типа, механизма повреждения органа слуха, типа наследования и других факторов. Наследственная тугоухость является достаточно распространенной патологией – по некоторым данным, ее встречаемость составляет 1-2 случая на 1000 новорожденных.
Причины и классификация наследственной тугоухости
Все формы наследственной тугоухости делятся по следующим признакам: сочетанию с другими нарушениями в рамках одной генетической патологии (синдромальные и несиндромальные формы), механизму передачи потомству (аутосомно-доминантные, аутосомно-рецессивные и Х-сцепленные), причине нарушения слуха (кондуктивная, сенсоневральная и смешанная тугоухость). Кроме того, клиницисты разделяют все случаи заболевания по степени ослабления слуха (легкая, умеренная, умеренно-тяжелая и глубокая) и возрасту появления нарушений относительно развития речи (прелингвальная и постлингвальная). Сложность классификации всех форм наследственной тугоухости обусловлена тем, что данное состояние может быть следствием огромного количества различных генетических мутаций, которые, к тому же, способны приводить к неодинаковым фенотипическим проявлениям патологии.
Несиндромальные формы наследственной тугоухости являются наиболее распространенными вариантами данного состояния – в различных популяциях ими обусловлено от 60 до 80% всех случаев врожденных нарушений слуха, передающихся по наследству. Обычно (порядка 75-80%) это аутосомно-рецессивные патологии, обусловленные мутацией гена GJB2, расположенного на 13-й хромосоме. Ген кодирует белок под названием коннектин-26, участвующий в формировании межклеточных связей в нейросенсорном аппарате внутреннего уха. В результате мутации у гомозигот данный протеин не образуется вообще или содержит в себе определенный дефект. Это становится причиной выраженной (чаще всего глубокой) прелингвальной наследственной тугоухости сенсоневрального характера. Другие гены, дающие схожую клиническую картину, пока досконально не изучены.
Доминантные формы несиндромальной наследственной тугоухости практически всегда представляют собой аллельные варианты других генетических заболеваний, которые, помимо прочего, характеризуются нарушениями слуха. Они составляют примерно 20% от всех случаев несиндромальных поражений слухового аппарата. Так, одна из форм доминантной наследственной тугоухости (DFNB18) обусловлена мутацией гена USH1C, расположенного на 11-й хромосоме. Продуктом экспрессии данного гена является белок PZD, принимающий активное участие в формировании волосковых клеток и других компонентов внутреннего уха. Мутация USH1C также ответственна за некоторые варианты синдрома Ушера (тип 1С).
Доминантная несиндромальная наследственная тугоухость типа DFNB12 вызывается дефектом гена CDH23, локализованного на 10-й хромосоме. Он кодирует один из основных белков-кадгеринов, которые участвуют в формировании множества нейроструктур и органов чувств, поэтому мутации CDH23 проявляются не только нарушениями слуха, но и синдромом Ушера 1D, пигментной ретинопатией и другими подобными состояниями. Аналогично аллельными заболеваниями являются доминантная наследственная тугоухость DFNB4 и синдром Пендреда, обусловленные мутацией гена SLC26A4, расположенного на 7-й хромосоме. Продуктом экспрессии данного гена является один из трансмембранных анионных каналов под названием пендрин, наиболее часто встречающийся в органе слуха, щитовидной железе и некоторых других органах. Большинство доминантных форм наследственной тугоухости развиваются уже после формирования речи у ребенка.
Намного более редкими (порядка 1-3% от всех случаев) являются формы наследственной тугоухости с Х-сцепленным механизмом наследования. Наиболее распространенным вариантом такой патологии выступают мутации гена POU3F4, который кодирует важный фактор транскрипции, необходимый для экспрессии других протеинов (досконально не изученных), участвующих в формировании органов слуха. Нарушение функционирования этого органа чувств в данном случае имеет кондуктивный характер и обусловлено аномальным циркулированием перилимфы. Приводить к Х-сцепленной наследственной тугоухости могут и мутации гена PRPS1, который кодирует последовательность одного из ферментов (фосфорибозилпирофосфат синтетазу-1) – патогенез развития нарушений слуха при этом пока остается неясным. Самой редкой описанной формой наследственной тугоухости становятся мутации гена MTRNR1, который локализован не в ядре клетки, а в митохондриальной ДНК – по этой причине заболевание передается только по женской линии или от матери детям.
Симптомы наследственной тугоухости
Главным проявлением наследственной тугоухости, как можно понять из названия патологии, является снижение слуха различной выраженности и характера. Симптомы заболевания нередко появляются в раннем детстве (прелингвальное развитие), при этом в конечном итоге может страдать развитие речи – наблюдается прямая корреляция между степенью снижения слуха и задержкой или недоразвитием речевого аппарата. Столь раннее возникновение наследственной тугоухости может указывать на аутосомно-рецессивный характер данной патологии. Постепенное постлингвальное снижение слуха (после 6-8 лет) чаще всего имеет аутосомно-доминантный характер наследования. При проведении аудиографии можно определить степень наследственной тугоухости, а также ее частотность (звуки какой именно частоты слабее всего воспринимает больной). В отдельных случаях ослабление слуха может происходить во взрослом возрасте.
Помимо собственно снижения слуха различные формы наследственной тугоухости могут проявляться и другими симптомами. Наиболее частым из них является дисфункция вестибулярного аппарата, обусловленная анатомической и функциональной близостью органов слуха и равновесия. Кроме того, могут наблюдаться признаки других генетических и врожденных пороков: нарушения со стороны щитовидной железы, кожных покровов (гиперкератозы, псориаз), мочевыделительной системы, органов зрения. Это наиболее характерно для синдромальных форм наследственной тугоухости: болезни Ваарденбурга, болезни Стиклера, болезни Ушера, болезни Пендреда и ряда других. Всего описано более 400 генетических патологий, которые могут проявляться синдромальной наследственной тугоухостью.
Диагностика и лечение наследственной тугоухости
Для определения наследственной тугоухости используют традиционные отоларингологические методики: аудиометрию, осмотр слуховых ходов, ответ слухового отдела ствола мозга (так называемая BAYER-методика), вызванную отоакустическую эмиссию. Поскольку в большинстве случаев данная патология определяется в детском возрасте, аудиометрия нередко производится в игровой форме. При наследственной тугоухости разных типов может определяться как нарушение воздушной проводимости звуковых колебаний (особенно при кондуктивных механизмах развития заболевания), так и сочетанное снижение костной и воздушной проводимости. Определенную роль в диагностике патологии могут играть аномалии развития органов слуха – например, широкий водоворот преддверия при несиндромальной наследственной тугоухости 4-го типа (DFNB4) или синдроме Пендреда.
Снижение слуха может отмечаться как в отношении определенных звуковых частот, так и в широком диапазоне колебаний. Выраженность нарушений также довольно сильно варьируется при различных формах наследственной тугоухости. При сенсоневральном механизме поражения будет отмечаться уменьшение активности импульсов в слуховом нерве, что определяется при проведении электрофизиологических исследований. Задержка речевого развития обнаруживается при прелингвальных формах наследственной тугоухости. Для ряда типов заболевания (например, обусловленных мутациями генов GJB2, SLC26A4, POU3F4) в качестве диагностики используют методы современной генетики – секвенирование последовательности генов для выявления мутаций.
Лечение наследственной тугоухости, как правило, сводится к подбору слуховых аппаратов различного типа, которые следует начать использовать как можно ранее для предупреждения патологии развития речи. В некоторых случаях при изолированных кондуктивных поражениях органов слуха может быть проведена хирургическая коррекция, однако ее результаты неоднозначны. Большинство форм наследственной тугоухости редко прогрессируют до абсолютной глухоты, поэтому пожизненное применение слуховых аппаратов обеспечивает приемлемое для больного качество жизни.
Прогноз и профилактика наследственной тугоухости
Прогноз наследственной тугоухости относительно выздоровления неблагоприятный – в большинстве случаев снижение слуха сохраняется на протяжении всей жизни. Крайне медленное прогрессирование симптомов или даже отсутствие прогрессирования позволяет использовать слуховые аппараты. В тех случаях, когда у ребенка была поздно выявлена прелингвальная тугоухость, может потребоваться коррекция дефектов речи у логопеда. Для ряда синдромальных форм наследственной тугоухости у детей прогноз во многом зависит от сопутствующих нарушений и пороков развития. Для профилактики этого состояния используют методы генетической пренатальной диагностики, а также выявление носительства дефектных генов (при рецессивных и Х-сцепленных типах заболевания) с последующим медико-генетическим консультированием родителей перед зачатием.
Синдром Ушера ( Синдром Ашера )
Синдром Ушера – это редко встречающееся генетическое заболевание, протекающее с врожденной сенсоневральной тугоухостью, прогрессирующим пигментным ретинитом и вестибулярной атаксией. В зависимости от типа синдрома у пациентов присутствуют следующие признаки: значительная потеря слуха либо глухота, снижение зрения, нарушение равновесия, когнитивные расстройства. Диагностика включает офтальмологическое (визометрия, офтальмоскопия, электроретинография), отоневрологическое (аудиометрия, вестибулярные пробы), генетическое обследование. Лечение направлено на коррекцию слуха (слухопротезирование, КИ), поддержание зрения (вит. А, Е, омега-3).
МКБ-10
Синдром Ушера (синдром Ашера) – самая частая причина наследственной слепоглухоты. Его распространенность в популяции оценивается в 3,2-6,2 случая на 100 тыс. населения (по другим данным – 1:6000). Наибольшая заболеваемость отмечается среди евреев-ашкенази, франкоканадцев, испанских аргентинцев, финнов и др. «Первооткрывателем» заболевания считается германский офтальмолог А. фон Грефе. Однако свое «имя» синдром получил в честь британского окулиста Ч. Ушера, который в 1914 г. указал на наследственный характер болезни. Молекулярно-генетические механизмы синдрома были расшифрованы в 1995 г., что открыло широкие возможности для его изучения.
Причины
В настоящее время известно более десятка генов, дефекты которых могут привести к развитию синдрома Ушера. Все эти гены, несмотря на разную локализацию и функцию, входят в состав трансмембранного белкового комплекса, участвующего в перемещении миозина, функционировании фоторецепторов сетчатки, а также волосковых клеток улитки. Наиболее часто мутации обнаруживаются в следующих генах:
- CDH23 – «ген глухоты», кодирует белок кадгерин-23;
- MYO7A – ген миозина VIIA;
- PCDH15 – ген протокадгерина-15;
- USH1С – ген гармонина;
- USH1G – ген анкириноподобного белка;
- USH2A – ген ушерина;
- ADGRV1 - ген адгезии G-белка;
- CLRN1 – ген кларина-1 и др.
Синдром Ушера наследуется аутосомно-рецессивным путем от обоих родителей, являющихся носителями дефектных генов. Чаще болеют представители закрытых этнических групп, среди которых достаточно часты близкородственные браки.
Патогенез
Белковые продукты, кодируемые названными генами, принимают непосредственное участие в развитии и функционировании рецепторного аппарата сетчатки глаза и внутреннего уха. Генетические мутации приводят к нарушению формирования воспринимающего аппарата – фоторецепторов и волосковых клеток, что сопровождается врожденными или рано дебютирующими нарушениями зрительной, слуховой и вестибулярной функции.
При синдроме Ушера на глазном дне накапливаются гранулы пигмента, которые распространяются от центра к периферии. Со временем происходит сужение полей и снижение остроты зрения. Патология также затрагивает структуры внутреннего уха: отмечается атрофия спирального узла, нервных волокон кортиева органа, сосудистой полоски улитки.
Классификация
Синдром Ушера генетически неоднороден. Выделяют 4 клинических подтипа, различающихся молекулярно-генетическими механизмами, возрастом манифестации и выраженностью симптоматики:
- Тип 1. Сопряжен с мутациями в генах MYO7A, CDH23, PCDH15, USH1G, USH1C. Протекает наиболее тяжело. Характерна врожденная глухота или глубокая тугоухость. Вестибулярные нарушения и признаки пигментного ретинита развиваются до 5 лет. Составляет около 30% всех случаев.
- Тип 2. Вызывается мутациями USH2A, DFNB31, ADGRV1. Сопровождается непрогрессирующей тугоухостью, ухудшением зрения после 10 лет. Вестибулярная функция не нарушена. Диагностируется примерно у 60% пациентов.
- Тип 3. Связан с мутациями гена CLRN1. Протекает с постепенным ухудшением слуха и зрения, часто – с вестибулярной дисфункцией. Изменения сетчатки развиваются после 20 лет. Выявляется у 3% больных.
- Тип 4. Атипичный вариант синдрома Ушера. Обусловлен дефектами генов HARS, PDZD7, CEP250, C2orf71, может наследоваться Х-сцеплено.
Симптомы
Классическая клиническая картина развивается при 1-м типе синдрома Ушера. Уже в раннем детстве у ребенка диагностируется тяжелая сенсоневральная тугоухость или полная глухота. Наблюдается задержка психомоторного развития, дети поздно начинают ходить. В дошкольном возрасте обнаруживается зрительная дисфункция: в результате усугубления пигментного ретинита быстро ухудшается периферическое зрение, развивается ночная слепота (гемералопия). Это проявляется затруднением ориентации в темноте, частыми спотыканиями, столкновениями с препятствием (другой человек, дверной проем, мебель).
Возникают вестибулярные расстройства: головокружение, нарушение равновесия, атактическая походка. В некоторых случаях может отмечаться когнитивный дефицит, психозы. При других типах болезни Ушера зрительные и слуховые нарушения развиваются позднее и выражены в меньшей степени; вестибулярный синдром не отмечается.
Осложнения
Нейросенсорная тугоухость и постепенная утрата зрения приводят к глубокой инвалидизации. Дети с синдромом Ушера нуждаются в специальном обучении, психолого-педагогическом сопровождении, создании безопасной окружающей среды. Вестибулярные нарушения могут провоцировать падения, повышать риск травматизма. Вторично страдает речь и интеллектуальная сфера. Одним из частых осложнений является катаракта. Прогрессивное снижение зрения приводит к тому, что к 40–50 годам больные синдромом Ушера могут полностью ослепнуть. В тяжелых случаях возникает слепоглухота.
Диагностика
Поскольку нарушения при синдроме Ушера затрагивают несколько анатомо-функциональных систем, диагностика должна носить мультидисциплинарный подход. Больным необходимы консультации отоларинголога, офтальмолога, генетика, проведение комплексного инструментально-лабораторного обследования:
- Офтальмологический осмотр.Визометрия и осмотр глазного дна не всегда позволяют обнаружить начальные признаки пигментной дегенерации сетчатки. В этом отношении наиболее чувствительным тестом является электроретинография, выявляющая изменения еще на доклиническом уровне. По результатам периметрии отмечается концентрическое сужение полей зрения.
- Исследование слуха. Степень снижения слуха устанавливается с помощью аудиометрии. Дополнительно регистрируется отоакустическая эмиссия, стволовые ВП, проводится электрокохлеарография. Больного осматривает врач-сурдолог.
- Исследование вестибулярного анализатора. Для выявления вестибулярных нарушений выполняется видеонистагмография, вестибулометрические пробы.
- Генодиагностика. Проводится секвенирование образцов генетического материала пациента. На основе обнаруженных изменений определяется генетический вариант синдрома Ушера. Также необходимо медико-генетическое консультирование членов семьи.
Дифференциальная диагностика
Синдром Ушера необходимо отличать от других синдромальных и несиндромальных форм глухоты и слепоты, развивающихся при:
- врожденной краснухе – врожденная тугоухость, катаракта, ВПС;
- синдроме Альстрема – дегенерация сетчатки, сенсоневральная тугоухость, ожирение, СД, кардиомиопатия, нефропатия;
- синдроме Халлгрена (акустико-ретино-церебеллярной дегенерации) – пигментный ретинит, катаракта, тугоухость, мозжечковый синдром.
Лечение синдрома Ушера
Консервативная терапия
На сегодняшний день методов полного излечения заболевания не разработано. Все предпринимаемые меры направлены на компенсацию нарушенных функций и замедление течения патологии. Больным синдромом Ушера рекомендовано проведение ежегодных поддерживающих курсов медикаментозной терапии, включающей ноотропы, сосудорасширяющие препараты, антиоксиданты. С целью торможения прогрессирования пигментного ретинита рекомендуется прием больших доз ретинола пальмитата, соблюдение диеты, богатой содержанием витаминов А, Е, ПНЖК.
Способ коррекции слуховой функции подбирается индивидуально. Выбор может быть сделан в пользу слухового протезирования или установки кохлеарного импланта. Для адаптации детей с синдром Ушера к жизни в социуме важна помощь психологов, сурдопедагогов.
Экспериментальное лечение
Сообщается о разработке генной терапии для больных с синдромом Ушера 1 типа. Клинические испытания проходит препарат UshStat, который представляет собой лентивирусный вектор для доставки неповрежденного гена MYO7A. Предполагается субретинальное введение UshStat с целью восстановления функции зрения.
Прогноз и профилактика
Качество жизни пациентов с синдромом Ушера значительно страдает из-за проблем со зрением и слухом. Около 50% лиц с 1-м и 70% со 2-м типом заболевания сохраняют остроту зрения от 20 до 40% на обоих или одном глазу. Больным необходимо пожизненное соблюдение диеты, защита глаз от ультрафиолета. Профилактика заключается в проведении генетических консультаций и лабораторного тестирования в семьях, где имеются случаи данного заболевания. Возможна пренатальная диагностика синдрома Ушера.
1. Изучение интерактома при синдроме Ашера в российской популяции для выбора приоритетных патогенетически ориентированных терапевтических подходов/ Иванова М.Е , Атарщиков Д.С. , Демчинский А.М. , Стрельников В.В. , Бар Д. , Порядин Г.В. , Балашова Л.М. , Салмаси Ж.М.// РМЖ «Клиническая Офтальмология». – 2019. - №4.
3. Этиологические аспекты врожденной тугоухости/ Коноплев О.И. и др./ Медицинский вестник Северного Кавказа. – 2019.
Нейросенсорная тугоухость
Нейросенсорная тугоухость – нарушение слуха, обусловленное поражением слухового анализатора и проявляющееся односторонним или двусторонним снижением слуха, шумом в ушах, а также возникающими в связи с этим нарушениями социальной адаптации. Диагностика заболевания основана на изучении анамнеза, данных физикального и инструментального обследования (камертональных методов, аудиометрии, МРТ, УЗИ БЦА и др.). Лечение предусматривает восстановление сниженной слуховой функции при помощи слухопротезирования, использование глюкокортикоидов, медикаментозных средств с ангиопротекторным и нейропротекторным действием.
Нейросенсорная тугоухость (НСТ, сенсоневральная глухота) – снижение функции слухового анализатора, проявляющееся частичной или полной потерей слуха. При этом патологический процесс может поражать структуры, участвующие в восприятии звука, на различных участках: в клетках внутреннего уха, в нервных проводниках, в стволе или коре головного мозга. По данным статистики, примерно 6% населения нашей планеты имеют нарушения слуха различной степени выраженности. Из них около 80-90% жалуются на шум в ушах. С возрастом нарушения слуха прогрессируют, от 30 до 60% людей старше 65-70 лет страдают тугоухостью.
Причины нейросенсорной тугоухости
Нейросенсорная потеря слуха может возникать в результате врожденных или приобретенных нарушений функции слуха.
1. Врожденная патология.
- пороки развития среднего или внутреннего уха, в том числе – обусловленные генетическими нарушениями (синдромы Ваарденбурга, Стиклера, Ушера, Пендреда, Ланге-Нильсена, Альпорта, нейрофиброматоз II типа, болезнь Рефсума);
- патология в родах (гипоксия плода).
2. Внешние факторы.
- инфекции (грипп, ОРЗ, паротит, корь, краснуха, скарлатина, менингит и др.);
- сосудистые расстройства при артериальной гипертензии, церебральном атеросклерозе;
- интоксикации (промышленные и бытовые токсины, медикаментозные средства с ототоксическим действием: аминогликозиды, антималярийные препараты, анальгетики, цитостатики и т. д.),
- травмы костей черепа;
- акустические повреждающие агенты и баротравма;
- эндокринные расстройства;
- болезни крови;
- неблагоприятные метеорологические факторы;
- физиологическое старение.
Вышеперечисленные внешние воздействия приводят к возникновению патологического процесса в слуховом анализаторе с развитием преходящей ишемии, стойкого нарушения кровообращения, а затем и гибели чувствительных клеток внутреннего уха, проводящего аппарата или корковых центров органа слуха.
Нейросенсорная тугоухость классифицируется по длительности и тяжести течения, уровню поражения, времени появления основной симптоматики и степени снижения слуха.
- Продолжительность. Симптомы НСТ могут появиться внезапно (за 3-6 часов, например, во время ночного сна) или постепенно (на протяжении 3-5 суток). Заболевание может приобретать хроническое течение со стабильным или прогрессирующим снижением слуха.
- Время появления. Ухудшение слуха может возникнуть в первые годы жизни ребенка, еще до развития полноценной речи (в долингвальный период), или уже после формирования речевой функции (постречевая тугоухость).
- Выраженность нарушений. Выделяют четыре степени тугоухости, которые определяют при сравнении с нормальными показателями. В норме слуховой порог находится в промежутке между 0 и 25 дБ, при первой степени НСТ он равен 26-30 дБ, при второй (умеренные нарушения) – 41-55, при третьей – 56-70, при четвертой (тяжелая степень) – 71-90 дБ. При полной глухоте этот показатель превышает 90 дБ.
Симптомы нейросенсорной тугоухости
Основными проявлениями заболевания являются снижение слуха и шум в ушах, реже наблюдается головокружение и сопутствующие соматоформные расстройства. Изменяется восприятие обычной разговорной и шепотной речи. При легкой степени НСТ обычный разговор слышен с расстояния 5-7 метров, а шепот – с 2-3 метров. При умеренных нарушениях эти показатели снижаются соответственно до 3-4 и 1 метра, при тяжелых разговорная речь слышна в лучшем случае с расстояния 1 метра, а шепот неразличим вообще. При IV степени нейросенсорной тугоухости человек неспособен воспринимать даже громкие звуки с самого близкого расстояния без специальных приборов.
Снижение слуха нередко сопровождается появлением шума в ушах периодического или постоянного характера. Шум может восприниматься в виде высокочастотных звуков по типу писка, звона, шипения, а также представлять собой постоянный надоедливый низкочастотный гул. При наличии сопутствующего кохлеовестибулярного синдрома больных беспокоят приступы головокружения, нередко сочетающиеся с тошнотой (иногда рвотой), признаки нарушения равновесия: ухудшается координация движений при выполнении простых бытовых манипуляций, появляется пошатывание при ходьбе, неустойчивость и высокая вероятность падения при резких поворотах.
Длительное хроническое течение нейросенсорной тугоухости со значительным нарушением слуховой функции становится причиной развития психоэмоциональных расстройств (снижение настроения, раздражительность, беспокойство, тревога), потере социальных контактов, снижению и утрате работоспособности (трудоспособности). В пожилом возрасте частичная или полная утрата слуха при отсутствии своевременной коррекции и наличии сопутствующих сосудистых заболеваний головного мозга нередко приводит к прогрессирующим нарушениям памяти, мышления, появлению бредовых и галлюцинаторных синдромов.
При остром развитии болезни клиническая симптоматика появляется внезапно (в течение 3-12 часов, часто во время ночного сна) на фоне полного благополучия. Иногда снижение слуха может быть более продолжительным (на протяжении 3-5 суток). При подостром и хроническом течении нейросенсорной тугоухости патологический процесс развивается в течение 1-3 месяцев и более.
Выявление этиологических факторов, определение выраженности нарушений слуха и наличия сопутствующих заболеваний, влияющих на течение НСТ, требует участия в диагностике врачей различных специальностей: отоларинголога, отоневролога, офтальмолога, кардиолога, эндокринолога, травматолога-ортопеда и других специалистов. Стандартное физикальное обследование, в частности, отоскопия, не дает какой-либо значимой информации, так как признаки поражения наружного уха и барабанной перепонки обычно отсутствуют. При этом простая оценка слышимости разговорной и шепотной речи на определенном расстоянии в кабинете ЛОР-врача позволяет ориентировочно оценить степень снижения слуха.
Более информативным является использование специальных инструментальных исследований: камертональных проб (Вебера, Ринне, Федеричи), тональной аудиометрии, регистрации слуховых потенциалов (электрокохлеографии), проведения вестибулометрических тестов. Для выявления сопутствующих заболеваний нервной системы и патологии позвоночника, исключения травматических повреждений может назначаться МРТ или КТ костей лицевого черепа и головного мозга, шейного отдела позвоночника, УЗИ брахиоцефальных артерий и т. д. Дифференциальная диагностика нейросенсорной тугоухости проводится с другими заболеваниями уха, горла и носа (хроническим средним отитом и связанными с ним кондуктивными нарушениями, болезнью Меньера, лабиринтитом, невриномой слухового нерва и др.), рассеянным склерозом, сосудистыми заболеваниями головного мозга (дисциркуляторной энцефалопатией, последствиями перенесенного инсульта, сосудистой деменцией).
Лечение нейросенсорной тугоухости
Основная цель лечебных мероприятий – восстановление или стабилизация функции слуха, устранение сопутствующей симптоматики (головокружения, шума в ушах, нарушений равновесия, нервно-психических расстройств), возвращение к активной жизни, социальным контактам.
- Физиотерапия, рефлексотерапия. На начальных стадиях заболевания применяется фоноэлектрофорез, электростимуляция тканей внутреннего уха, акупунктура и электропунктура, что позволяет в ряде случаев снизить интенсивность шума в ушах, избавиться от головокружения, улучшить сон и настроение.
- Медикаментозное лечение. Эффективность лекарственного воздействия наиболее высока при раннем начале лечения. При внезапно наступившей тугоухости полностью восстановить слух иногда позволяет применение ударных доз глюкокортикоидных гормонов в течение 5-8 суток. Широкое применение находят препараты, улучшающие кровообращение, проведение нервных импульсов и микроциркуляцию: пентоксифиллин, пирацетам. При сопутствующем НСТ головокружении назначают средства с гистаминоподобным действием, например, бетагистин. Используются медикаменты, оказывающие гипотензивное действие при наличии артериальной гипертензии, а также психотропные препараты при наличии нервно-психических расстройств.
- Слухопротезирование. Показано при умеренной и тяжелой степени утраты слуха. Применяются заушные, внутриушные и карманные аналоговые и цифровые аппараты для моноаурального или бинаурального слухопротезирования.
- Хирургическое лечение, кохлеарная имплантация. Практикуется транстимпанальное введение глюкокортикоидных гормонов в барабанную полость. Оперативные вмешательства проводятся при опухолях задней черепной ямки для уменьшения выраженности некоторых симптомов, сопровождающих вестибулярные расстройства. Кохлеарная имплантация выполняется при полном отсутствии слуха при условии сохранности функции слухового нерва.
Прогноз у больных с острой нейросенсорной тугоухостью при своевременном лечении в 50% случаев относительно благоприятный. Применение слуховых аппаратов и имплантации при хронической НСТ обычно позволяет стабилизировать слух. Профилактические мероприятия по предотвращению утраты слуховой функции предусматривают исключение вредных факторов внешней среды (шума и вибрации на производстве и в быту), отказ от алкоголя и приема токсичных медикаментозных средств, предупреждение травматизма, в том числе акустических и баротравм, своевременное лечение инфекционных и соматических заболеваний.
Читайте также:
- Лечение септического шока. Принципы лечения септического шока.
- Лучевая диагностика протокового рака поджелудочной железы
- Неврогенный кашель. Диагностика происхождения кашля
- Отравление аллопуринолом и его побочные эффекты
- Связи базальных ганглиев: двигательная петля, когнитивная петля, лимбическая петля, глазодвигательная петля