Раннее выявление фенилкетонурии. Скрининг населения на фенилкетонурию.
Добавил пользователь Morpheus Обновлено: 31.01.2026
1. Клинический протокол диагностики (далее - биохимического неонатального скрининга), лечения и диспансерного наблюдения пациентов с заболеванием фенилкетонурия (далее-ФКУ), предназначен для оказания медицинской помощи новорожденным в амбулаторных и стационарных условиях районных, областных организаций здравоохранения, детям и беременным в областных и республиканских организациях здравоохранения Республики Беларусь, имеющих в своем составе медико-генетические центры (отделения, консультации).
2. Возрастная категория: детское, взрослое население.
3. Наименование нозологических форм заболеваний (шифр по МКБ-10).
Классическая фенилкетонурия – Е70.0;
другие виды гиперфенилаланинемии – Е70.1.
4. Фенилкетонурия – наследственная болезнь обмена веществ, связанная с дефицитом фермента фенилаланингидроксилазы, что приводит к повышенному содержанию фенилаланина (далее-ФА) в крови и других тканях. При отсутствии лечения заболевание проявляется задержкой моторного и психоречевого развития, микроцефалией, судорогами, нарушениями поведения и другими проявлениями. Одним из осложнений у пациентов с ФКУ, не соблюдающих диету, являются упорные экземоподобные высыпания, не поддающиеся обычным методам лечения и обусловленные метаболическим дефицитом тирозина.
5. Частота заболевания в разных популяциях различается. В Беларуси фенилкетонурия выявляется с частотой 1:6000 новорожденных, то есть ежегодно рождается 15-20 детей с этим заболеванием.
Раннее выявление пациентов и своевременное начало лечения позволяют избежать клинических проявлений заболевания.
Диагностика
6. Обследованию на ФКУ подлежат все новорожденные (материалом для исследования является капиллярная кровь, взятая из пальца традиционным способом):
6.1. взятие крови для биохимического неонатального скрининга:
взятие крови у новорожденных осуществляется в организации здравоохранения: у доношенных на 3 день жизни, у недоношенных на 7-14 день жизни;
6.2. проведение тестирования:
тестирование проводится в клинико-диагностической генетической лаборатории Республиканского научно-практического центра (далее-РНПЦ) «Мать и дитя» в сухих пробах крови,
скринирующим тестом на фенилкетонурию является определение ФА флюорометрическим методом. Границей нормальных значений ФА является концентрация 180 мкмоль/л. При превышении этих значений проводят повторное тестирование;
6.3. повторное тестирование
при превышении пограничного значения концентрации ФА (более 180 мкмоль/л) осуществляется повторное тестирование (должно быть проведено до двухнедельного возраста ребёнка).
При сохранении повышенного уровня ФА при повторном тестировании рекомендуется в трёхдневный срок госпитализировать ребенка в РНПЦ «Мать и дитя» для верификации диагноза, введения терапии и консультации врача-генетика.
7. Верификация диагноза включает:
7.1. определение концентрации ФА в сыворотке крови;
7.2. определение концентрации фенилуксусной и фенилпировиноградной кислоты в моче;
7.3. исключение прочих наследственных дефектов метаболизма по селективной скринирующей программе: аминокислоты, простые углеводы, гликозаминогликаны;
7.4. общий анализ крови;
7.5. общий анализ мочи;
7.6. молекулярно-генетический анализ (далее – анализ ДНК) пациенту и родителям: определение основных мутаций гена фенилаланигидроксилазы, приводящих к ее недостаточности.
8. Дополнительная диагностика (по показаниям):
8.1. биохимическое исследование крови: концентрация общего билирубина, общего белка, электролиты, определение активности аспарагиновой аминотрансферазы (АсАТ), аланиновой аминотрансферазы (АлАТ);
8.2. Ультразвуковое исследование (далее-УЗИ) головного мозга;
8.3. УЗИ органов брюшной полости.
9. Информация и учет пациентов с ФКУ, выявленных биохимическим неонатальным скринингом:
9.1. РНПЦ «Мать и дитя» ежеквартально (суммарную информацию) передает в областные организации здравоохранения, имеющие в своем составе медико-генетические центры (отделения, консультации), информацию обо всех выявленных детях с ФКУ.
10. Контроль полноты обследования новорожденных, родившихся в родовспомогательных больничных организациях, осуществляет РНПЦ «Мать и дитя».
Лечение
11. Основным методом лечения ФКУ является диетотерапия с ограничением естественного белка и заменой его аминокислотной смесью, не содержащей ФА. Оптимальным является введение диеты в течение первых двух недель жизни.
12. Для лечения ФКУ используются смеси аминокислот, адаптированные к возрасту пациента.
13. Введение лечения:
13.1. Дети с фенилкетонурией для введения диетотерапии госпитализируются в инфекционное отделение новорожденных РНПЦ «Мать и дитя», где наблюдаются врачом-генетиком, врачом-педиатром и (по показаниям) врачом-неврологом.
13.2. введение диетотерапии смесью аминокислот осуществляется в течение 6-7 дней: ежедневно одно кормление заменяется смесью аминокислот, под контролем уровня ФА.
13.3. смесь аминокислот для введения диетотерапии на период госпитализации обеспечивает управление здравоохранения области, комитет по здравоохранению г. Минска в соответствии с местом жительства ребенка.
13.4. симптоматическая терапия (по показаниям) согласно ранее утвержденным протоколам.
14. Диспансерное наблюдение пациентов с ФКУ, выявленных системой скрининга новорожденных, контроль лечения.
14.1 Диспансерное наблюдение пациентов с ФКУ осуществляется медико-генетическими центром (отделением, консультацией) в областных организациях здравоохранения.
14.2 Контроль лечения ФКУ проводится определением уровня ФА в сыворотке или сухом пятне крови. Исследование проводится в клинико-диагностической генетической лаборатории РНПЦ «Мать и дитя».
14.3 Частота контрольного обследования:
в течение первого года жизни – 2 раза в месяц;
с 1 года до 12 лет – 1 раз в месяц;
в дальнейшем - 1 раз в квартал.
Уровень ФА не должен превышать 600мкмоль/л.
14.4 Частота контрольного осмотра врачом-генетиком (по месту жительства):
до 1 года – 1 раз в 6 месяцев;
в дальнейшем – ежегодно.
14.5 Консультации: врача-невролога, врача-гастроэнтеролога и других специалистов (по показаниям).
15. Диетотерапия с ограничением естественного белка и заменой его смесью аминокислот должна проводиться до 18 лет (в дальнейшем - с учетом индивидуальных особенностей пациента).
16. Генетической службой проводится медико-генетическое консультирование, молекулярно-генетические исследования семей с ФКУ и осуществляется пренатальная диагностика при дальнейших беременностях (по желанию семьи).
17. При планировании беременности женщиной, страдающей ФКУ:
17.1 анализ ДНК женщины и ее супруга;
17.2 при гетерозиготном носительстве мутации обоими супругами проведение пренатальной диагностики (по желанию семьи).
17.3 рекомендуется соблюдать диету в течение не менее трех месяцев до зачатия – до нормализации уровня ФА в сыворотке крови. Рекомендуемый уровень ФА в крови не более 360 мкмоль/л.
17.4 строгое соблюдение диеты во время всей беременности с ограничение естественного белка и заменой его смесью аминокислот.
17.5 частота контрольного исследования ФА у беременных – 2 раза в неделю. Рекомендуемый уровень ФА в течение беременности - 120-360 мкмоль/л.
Раннее выявление фенилкетонурии. Скрининг населения на фенилкетонурию.
Рекомендации: Скрининг фенилкетонурии (ФКУ) рекомендуется выполнять на всех новорожденных до их выписывания из клиники. Новорожденным, которым выполнили тестирование в течение первых 24 часов жизни, следует повторить тест до третьей недели жизни. Не рекомендуется выполнять рутинный скрининг матерей на ФКУ.
Когда-то фенилкетонурия отмечалась у одного из 15 тысяч новорожденных. Из-за отсутствия лечения после родов, у большинства пациентов, пораженных фенилкетонурией, развивались тяжелые необратимые поражения умственного развития, у многих отмечали такие невралгические симптомы поведения, как приступы, тремор, нарушенная походка, атетоз, и такие нарушения психики, как аутизм. Эти клинические проявления ФКУ редко наблюдаются у детей, рожденных после середины 60-х годов, когда рутинный скрининг стал обязательным и раннее лечение ФКУ — привычным. Однако приходится считаться с тем, что сейчас вступила в активный детородный возраст большая группа женщин, которые в детстве страдали фенилкетонурией, что увеличивает риск материнской ФКУ (по оценке 1 случай на 30—40 тысяч беременностей).
Для этих женщин повышается риск рождения ребенка с умственной отсталостью, микроцефалией, врожденным пороком сердца и низким природовым весом. Считается, что если не организовать лечение для защиты новорожденного от тератогенных воздействий материнского фенилаланина, то через одно поколение частота возникновения умственной отсталости, связанной с ФКУ, может вернуться на прежний уровень7.

Эффективность скрининговых тестов на фенилкетонурию.
В течение более двух десятилетий в качестве основных скрининговых тестов на фенилкетонурию выступали автоматизированные определения фенилаланина в крови, например, тест Гутри. Хотя не существует каких-либо работ, в которых методически точно определялась бы чувствительность и специфичность теста Гутри, международный опыт использования теста на миллионах новорожденных показал что он выявляет практически все случаи, а имевшие место ошибки связаны с административными или лабораторными недоработками. В качестве альтернативного метода проверки можно использовать флюорометрические наборы, обнаруживающие превосходную чувствительность. При определении ФКУ встречаются ложно-положительные результаты, в определенных ситуациях и при тестировании некоторых групп населения соотношение ложно-положительных к истинно-положительным результатам может составить 32:1.
Хотя в течение многих лет ложно-положительные результаты рассматривались, как менее важные, чем ложно-отрицательные, поскольку их нетрудно скорректировать, повторив тест, все же следует иметь в виду, что повторный анализ вызывает значительное беспокойство родителей. На чувствительность теста Гутри влияет возраст новорожденного, когда берется проба. Имеющаяся в настоящее время тенденция выписывать детей из родильного дома как можно раньше (в результате чего скрининг на ФКУ выполняют в возрасте 1—2 дней от роду) заставляет ряд медиков высказать соображение, что выполненный в таком возрасте тест может оказаться неточным.
К тому же, по данным ученых, флюорометрические наборы имеют большую чувствительность, чем тест Гутри. Было предложено еще два решения, чтобы дополнительно повысить чувствительность — повторять тест на всех новорожденных после выписки из клиники и понизить пороговое значение уровня фенилаланина, однако такой подход не разделяется многими учеными. Повторная проверка дала бы очень незначительное число новых обнаружений при высокой стоимости такого скрининга, подсчитано, что для того, чтобы обнаружить еще один дополнительный случай ФКУ надо провести от 600 тысяч до 6 миллионов новых тестов. Если же снизить порог, то это повысит чувствительность за счет специфичности и увеличит отношение ложно-положительных к истинно-положительным случаям. Тем не менее во многих скрининговых программах критический порог снижен до 2 мг/дл. В качестве средства предотвращения осложнений плода рекомендует выполнять скрининг на беременных женщинах на материнскую ФКУ. Обычно такое нарушение наблюдается редко, а большинство женщин, больных фенилкетонурией, знают об этом. Таким образом, полезный выход от такого скрининга очень мал. В одной программе, в которой для такого пренатального скрининга было выполнено 260 тысяч тестов, было найдено 9 ранее неизвестных случаев гиперфенилаланинемии.
Из 11 последующих беременностей у 5 женщин была доброкачественная гиперфенилаланинемия, 4 новорожденных из этого числа были рождены нормальными (пятая беременность закончилась абортом). В Массачусетсе в течение 10 лет делался рутинный анализ крови, взятой из пуповины, за это время было обнаружено 22 женщины с ранее недиагносцированной гиперфенилаланинемией.
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Эффективность раннего обнаружения фенилкетонурии.
До распространения лечения пищей с ограниченным количеством фенилаланина в начале 60-х годов, часть ФКУ приводила к тяжелым расстройствам умственного развития детей. Обзор, выполненный в 1953 году, показал, что у 85% коэффициент интеллекта был ниже 40, а у 37% ниже 10, только менее 1% обнаруживали коэффициент выше 70. Большинство детей с обнаруженными случаями фенилкетонурии оказывалось в приютах для умственно отсталых.
Эффективность диетических ограничений фенилаланина никогда не проверялась в тщательно построенных методически исследованиях, но и в будущем вряд ли такое исследование будет выполнено по этическим соображениям. Тем не менее разительный контраст между детьми, пораженными фенилкетонурией, и теми, которых удалось вылечить с помощью диеты, побудили большинство правительств западных стран ввести тест на ФКУ, как обязательную скрининговую проверку с конца 60-х годов. К сожалению, имеются ограничения и касательно эффективности диеты.
Чтобы предотвратить необратимые последствия фенилкетонурии, необходимо ввести ограничения на фенилаланин с самого раннего возраста, и такую диету иногда приходится выдерживать 4—8 лет, а в настоящее время появились данные, свидетельствующие о целесообразности придерживаться такой диеты также и в подростковом возрасте, а то и взрослым. И даже если все эти меры приняты, диета не всегда обеспечивает полную защиту от некоторых более тонких проявлений ФКУ. Проверка интеллектуальных способностей лиц, перенесших ФКУ, хотя и лежит в пределах нормы относительно больших масс населения, все же значительно ниже, чем у сверстников и у родителей; сообщают также о некоторых психологических изъянах таких, как перцептуальная моторная дисфункция, трудности обучения. Здесь помощь может оказать ранее обнаружение материнской фенилкетонурии.

Частота материнской фенилкетонурии увеличивается с увеличением числа вылеченных в детстве от фенилкетонурии женщин, которые вступают в детородный возраст. Материнская гиперфенилаланинемия может вызывать тератогенные эффекты даже относительно нормального плода, не отягощенного генетической ФКУ. Если мать во время беременности не придерживается диеты с низким содержанием фенилаланина то существует очень большой риск рождения ненормального ребенка: более 90% таких детей будут умственно отсталыми, у 75% будет наличествовать микроцефалия у 40—50% замедление роста в матке, у 10—25% другие природовые дефекты. Нет уверенности, что можно предотвратить эти случаи за счет правильной диеты с ограничением фенилаланина. Хотя некоторые беременные женщины, которые питались надлежащим образом, рожали нормальных детей, в ряде исследований было показано, что диетические ограничения не позволяют полностью устранить поражение плода.
Многие полагают, что для достижения истинного эффекта женщины должны придерживаться диеты до зачатия. Высказывают также соображения, что диета со сниженным содержанием фенилаланина означает также дефицит калорий, протеинов и других веществ, необходимых для нормального роста плода. В настоящее время выполняется ряд работ, в которых изучается эффект такой диеты во время беременности.
Рутинный скрининг всех новорожденных на фенилкетонурию обязателен во всех штатах Америки. Американская академия педиатрии рекомендует брать пробу из пятки до того, как ребенок покидает клинику перед самым моментом выписки. Недоношенные дети и такие, которых лечили по поводу ФКУ, должны проверяться на 7-й день или накануне. Академия рекомендует делать повторный тест до третьей недели жизни для детей, у которых первый тест был выполнен в течение первых 24 часов жизни. Некоторые авторы выступают за рутинный пренатальный скрининг материнской ФКУ, но большинство групп медиков не поддерживают такой подход, учитывая высокую стоимость подобного скрининга.
Для всех новорожденных рекомендуется делать анализ крови на уровень фенилаланина до выписки из роддома, предпочтительно в возрасте старше 3 дней. Для новорожденных, которых проверяли в течение первых 24 часов, скрининговый тест следует повторить в течение первых 14 дней жизни. Всех родителей нужно надлежащим образом информировать относительно правильной интерпретации результатов теста на ФКУ, упоминая также о возможности ложно-положительных результатов. Проводить рутинный пренатальный скрининг на материнскую ФКУ не рекомендуется.
- Вернуться в оглавление раздела "Профилактика заболеваний"
Генетика фенилкетонурии. Наследование
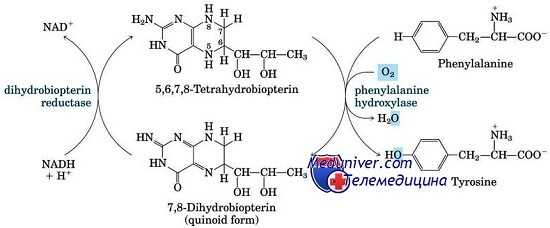
Аномалии, приводящие к увеличению уровня фенилаланина крови, чаще всего недостаточность фенилаланингидроксилаза (ФАГ) или фенилкетонурия (ФКУ), иллюстрируют почти все принципы биохимической генетики, относящиеся к дефектам ферментов. Все генетические аномалии метаболизма фенилаланина — следствие мутаций со снижением функции в гене, кодирующем ФАГ, или в генах, необходимых для синтеза или восстановления ее кофактора, ВН4.
Классическую фенилкетонурию (ФКУ) по праву считают образцовым представителем врожденных ошибок метаболизма. Это аутосомно-рецессивное заболевание распада фенилаланина, вызванное мутациями в гене, кодирующем ФАГ, фермент, преобразующий фенилаланин в тирозин. Открытие фенилкетонурии (ФКУ) Фелингом в 1934 г. впервые продемонстрировало генетический дефект как причину умственной отсталости.
Из-за неспособности к утилизации фенилаланина пациенты с фенилкетонурией (ФКУ) накапливают эту аминокислоту в жидкостях тела. Гиперфенилаланинемия повреждает формирующуюся в раннем детстве ЦНС и создает помехи функционированию зрелого мозга. Небольшая часть фенилаланина метаболизируется по альтернативным путям, производя повышенные количества фенилпировиноградной кислоты (кетокислота, по которой названа болезнь) и других метаболитов, выделяющихся с мочой.
Любопытно, что хотя ферментный дефект известен уже десятилетия, точный патогенетический механизм, каким образом увеличение фенилаланина повреждает мозг, все еще неизвестен. Важно, что развитие неврологического ущерба, вызванного метаболическим блоком при классической ФКУ, может в основном предупреждаться изменениями диеты, предохраняющими от накопления фенилаланина. Лечение фенилкетонурии (ФКУ) стало образцом для лечения многих метаболических болезней, исходы которых могут улучшаться за счет предотвращения накопления субстрата фермента и его производных.
Скрининг новорожденных на фенилкетонурию (ФКУ)
Широко используется популяционный скрининг новорожденных на фенилкетонурию (ФКУ). Фенилкетонурия (ФКУ) — образец генетических болезней, для которых оправдан массовый неонатальный скрининг; заболевание сравнительно часто встречается в ряде популяций (до 1 на 2900 живых новорожденных). Лечение, начатое в начале жизни, весьма эффективно; без лечения неизбежно развивается тяжелая умственная отсталость. Скрининг-тесты выполняют через несколько дней после рождения.
Капельку крови, полученную при проколе пятки, наносят на бумажный фильтр, высушивают и отправляют в централизованную лабораторию для оценки уровня фенилаланина в крови и соотношения фенилаланин/ тирозин. В прошлом образцы собирали перед выпиской ребенка из роддома. Тенденция к ранней выписке матери и новорожденного после родов изменила эту практику. Тест предпочтительно не делать до возраста 24 ч, поскольку уровень фенилаланина при фенилкетонурии (ФКУ) повышается только после рождения. Положительные результаты теста должны быть быстро подтверждены, поскольку задержка начала лечения более 4 нед после родов не позволяет избежать влияния на интеллектуальное состояние пациентов с фенилкетонурией (ФКУ).
Различные формы фенилкетонурии и гиперфенилаланинемия
Поскольку фенилкетонурия (ФКУ) связана с выраженной недостаточностью активности фенилаланингидроксилазы (ФАГ) (менее 1% по сравнению с контролем), мутантная ФАГ, имеющая остаточную активность, вызывает менее тяжелые фенотипические проявления, так называемую гиперфенилаланинемию и атипичную фенилкетонурию (ФКУ).
Гиперфенилаланинемию, отличную от фенилкетонурии (ФКУ), диагностируют, если концентрация фенилаланина в плазме ниже 1 ммоль/л на фоне нормальной диеты. Эта степень гиперфенилаланинемии только в 10 раз выше нормы и значительно ниже, чем концентрации, обнаруживаемые при классической фенилкетонурии (ФКУ) (>1 ммоль/л). Умеренное увеличение фенилаланина при гиперфенилаланинемии не способно повреждать функции мозга и может даже быть благоприятным, если увеличение небольшое (
Атипичная фенилкетонурия (ФКУ) — категория, включающая пациентов с промежуточным уровнем фенилаланина между классической ФКУ и гиперфенилаланинемией; такие пациенты требуют некоторого ограничения фенилаланина в диете, но меньшего, чем для пациентов с классической фенилкетонурии (ФКУ). Комплекс из этих трех клинических фенотипов с мутациями в гене ФАГ — пример клинической гетерогенности.
Гиперфенилаланинемии: аллельная и локусная гетерогенность при фенилкетонурии (ФКУ)
Молекулярные дефекты в гене фенилаланингидроксилазы. У пациентов с гиперфенилаланинемией, включая классическую фенилкетонурию (ФКУ), атипичную фенилкетонурию (ФКУ) и доброкачественные гиперфенилаланинемии, обнаружена поразительная степень аллельной гетерогенности в локусе фенилаланингидроксилазы (ФАГ) (более 400 различных мутаций по всему миру).
Подавляющее большинство аллелей фенилаланингидроксилазы (ФАГ) — достаточно редкие мутации, нарушающие ферментные свойства фенилаланингидроксилазы (ФАГ) и приводящие к гиперфенилаланинемии, хотя также обнаружены и доброкачественные полиморфизмы или менее частые доброкачественные варианты.
В популяциях европейского происхождения около двух третей известных мутантных хромосом представлены шестью мутациями. Шесть других мутаций ответственны за чуть более 80% мутаций фенилаланингидроксилазы (ФАГ) в азиатских популяциях. Остальные патогенные мутации встречаются реже. Чтобы сделать эту информацию широкодоступной, международным консорциумом разработана база данных мутаций в гене фенилаланингидроксилазы (ФАГ).
Во всех популяциях существует выраженная генетическая гетерогенность фенилаланингидроксилазы (ФАГ). Благодаря высокой степени аллельной гетерогенности в локусе, большинство пациентов с фенилкетонурией (ФКУ) во многих популяциях — компаундные гетерозиготы (т.е. у них присутствуют два разных патогенных аллеля), что полностью соответствует наблюдаемой ферментативной и фенотипической гетерогенности при нарушениях фенилаланингидроксилазы (ФАГ).
Сначала казалось, что знание генотипа фенилаланингидроксилазы (ФАГ) надежно предсказывает детали фенотипа; это ожидание оправдалось не полностью, хотя обнаружена определенная корреляция между генотипом ФАГ и биохимическим фенотипом.
В общих чертах мутации, которые полностью подавляют или резко снижают активность фенилаланингидроксилазы (ФАГ), вызывают классическую фенилкетонурию (ФКУ), тогда как мутации, приводящие к достаточно большой остаточной активности фермента, связаны с легкими фенотипами.
Тем не менее некоторые мутации фенилаланингидроксилазы (ФАГ) у гомозиготных пациентов определяют весь спектр фенотипов, от классической фенилкетонурии (ФКУ) до доброкачественной гиперфенилаланинемии.
Таким образом, стало очевидно, что в формировании фенотипа, наблюдаемого при специфическом генотипе, участвуют другие неопознанные биологические факторы, несомненно, включая гены-модификаторы. Это наблюдение, признанное в настоящее время общей характеристикой множества моногенных болезней, указывает на то, что даже моногенные болезни, подобные фенилкетонурии (ФКУ), — генетически не простые заболевания.
Дефекты в метаболизме тетрагидробиоптерина при фенилкетонурии (ФКУ)
Первоначально считали, что все дети с наследственной гиперфенилаланинемией имеют первичную недостаточность фенилаланингидроксилазы (ФАГ). Сейчас ясно, что примерно у 1-3% пациентов ген ФАГ нормален, а их гиперфенилаланинемия — результат генетического дефекта в одном из нескольких других генов, задействованных в синтезе или регенерации кофактора ФАГ, ВН4. Ассоциация одного фенотипа, например гиперфенилаланинемии, с мутациями в разных генах — пример локусной гетерогенности.
Как показывают мутации в генах, кодирующих белок фенилаланингидроксилазы (ФАГ) и метаболизм его кофактора биоптерина, белки, закодированные генами, демонстрирующими локусную гетерогенность, обычно входят в одну цепочку биохимических реакций. Пациенты с недостаточностью ВН4 сначала были выявлены из-за того, что, несмотря на успешное поддержание в диете низкой концентрации фенилаланина, у них рано развивались глубокие неврологические проблемы.
Плохие результаты частично объясняются необходимостью кофактора ВН4 для активности двух других ферментов, тирозингидроксилазы и триптофангидроксилазы. Обе этих гидроксилазы критичны для синтеза моноаминовых нейротрансмиттеров, таких как дегидроксифенилаланин, норэпинефрин, эпинефрин и серотонин. Пациенты с недостаточностью ВН4 имеют нарушение или в его биосинтезе из ГТФ, или в регенерации ВН4. Подобно классической фенилкетонурии (ФКУ), нарушение наследуется по аутосомно-рецессивному типу.
Очень важно отличать пациентов с дефектами в метаболизме ВН4 от больных с мутациями в фенилаланингидроксилазы (ФАГ), поскольку их лечение заметно разнится. Во-первых, так как белковая структура фенилаланингидроксилазы (ФАГ) у больных с нарушениями ВН4 нормальная, ее активность может восстанавливаться, если этим пациентам давать большие дозы ВН4, что приводит к снижению уровня фенилаланина плазмы. Следовательно, степень ограничения фенилаланина в диете пациентов с дефектами в метаболизме ВН4 может быть значительно уменьшена, а некоторые пациенты могут перейти на нормальную диету (т.е. без ограничения фенилаланина).
Во-вторых, необходимо также постараться нормализовать уровень нейротрансмиттеров в мозге этих пациентов, назначая продукты тирозингидроксилазы и триптофангидроксилазы: L-dopa и 5-гидрокситриптофан соответственно. По этим соображениям всем новорожденным с гиперфенилаланинемией показано обследование для определения аномалий в метаболизме ВН4.
Реакция на тетрагидробиоптерин при мутациях в гене ФАГ при фенилкетонурии (ФКУ)
У большинства пациентов с мутациями в гене фенилаланингидроксилазы (ФАГ), а не в метаболизме ВН4, отмечено отчетливое уменьшение уровня фенилаланина крови на фоне перорального приема больших доз кофактора фенилаланингидроксилазы (ФАГ) ВН4. Лучше отвечают на такое лечение пациенты со значимой остаточной активностью фенилаланингидроксилазы (ФАГ) (т.е. пациенты с атипичной фенилкетонурией (ФКУ) и гиперфенилаланинемией), но также поддается лечению небольшое число пациентов даже с классической фенилкетонурией (ФКУ). В то же время наличие остаточной активности ФАГ не дает гарантии влияния на уровень фенилаланина плазмы при назначении ВН4.
Наиболее вероятно, что степень ответной реакции на ВН4 зависит от специфических свойств каждого мутантного белка фенилаланингидроксилазы (ФАГ), отражающих лежащую в основе мутаций ФАГ аллельную гетерогенность. Показано, что введение в диету ВН4 оказывает лечебный эффект через несколько механизмов, вызванных повышением количества нормального кофактора, входящего в контакт с мутантным.
Эти механизмы включают стабилизацию мутантного фермента, защиту фермента от разложения клеткой, увеличение поступления кофактора к ферменту, имеющего низкое сродство с ВН4, и другие полезные эффекты в кинетических и каталитических свойствах фермента. Обеспечение повышенного количества кофактора — общая стратегия, применяемая в лечении многих врожденных ошибок метаболизма.
Материнская фенилкетонурия
Обычно успешное лечение фенилкетонурии (ФКУ) позволяет больным гомозиготам вести полноценную жизнь и иметь практически нормальные перспективы деторождения. В прошлом диету с низким содержанием фенилаланина прекращали у большинства пациентов с ФКУ в среднем детстве, основываясь на предположении (ошибочном, как установлено в настоящее время), что функционирование зрелой нервной системы не нарушается при возврате гиперфенилаланине-мии. Впоследствии было обнаружено, что почти все потомство женщин с фенилкетонурией (ФКУ), не получавших лечения, аномально; большинство этих детей с задержкой умственного развития, многие имеют микроцефалию, задержку роста и пороки развития, особенно сердца.
Как предсказывают принципы менделирующего наследования, все эти дети — гетерозиготы. Таким образом, задержка их развития вызвана не собственной генетической конституцией, а высокотератогенным эффектом высоких уровней фенилаланина в материнской крови. Соответственно необходимо, чтобы женщины с фенилкетонурией (ФКУ), планирующие беременность, начинали соблюдать диету с низким содержанием фенилаланина еще до зачатия.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Фенилкетонурия ( Болезнь Феллинга , Фенилпировиноградная олигофрения )
Фенилкетонурия – это наследственное нарушение аминокислотного обмена, обусловленное недостаточностью печеночных ферментов, участвующих в метаболизме фенилаланина до тирозина. Ранними признаками фенилкетонурии служат рвота, вялость или гиперактивность, запах плесени от мочи и кожи, задержка психомоторного развития; типичные поздние признаки включают олигофрению, отставание в физическом развитии, судороги, экзематозные изменения кожи и др. Скрининг новорожденных на фенилкетонурию проводится еще в родильном доме; последующая диагностика включает молекулярно-генетическое тестирование, определение концентрации фенилаланина в крови, биохимический анализ мочи, ЭЭГ, МРТ головного мозга. Лечение фенилкетонурии заключается в соблюдении специальной диеты.
МКБ-10
Общие сведения
Фенилкетонурия (болезнь Феллинга, фенилпировиноградная олигофрения) – врожденная, генетически обусловленная патология, характеризующаяся нарушением гидроксилирования фенилаланина, накоплением аминокислоты и ее метаболитов в физиологических жидкостях и тканях с последующим тяжелым поражением ЦНС. Фенилкетонурия впервые описана А. Феллингом в 1934 г.; встречается с частотой 1 случай на 10 000 новорожденных.
В неонатальном периоде фенилкетонурия не имеет клинических проявлений, однако поступление фенилаланина с пищей вызывает манифестацию заболевания уже в первом полугодии жизни, а в дальнейшем приводит к тяжелым нарушениям развития ребенка. Именно поэтому пресимптоматическое выявление фенилкетонурии у новорожденных является важнейшей задачей неонатологии, педиатрии и генетики.
Причины фенилкетонурии
Фенилкетонурия является заболеванием с аутосомно-рецессивным характером наследования. Это означает, что для развития клинических признаков фенилкетонурии ребенок должен унаследовать по одной дефектной копии гена от обоих родителей, являющихся гетерозиготными носителями мутантного гена.
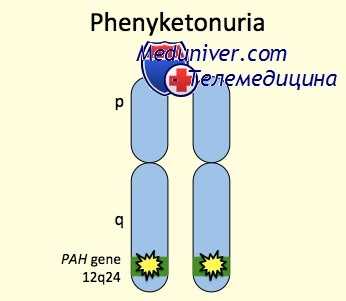
Чаще всего к развитию фенилкетонурии приводит мутация гена, кодирующего фермент фенилаланин-4-гидроксилазу и расположенного на длинном плече 12 хромосомы (локус12q22-q24.1). Это, так называемая, классическая фенилкетонурия I типа, составляющая 98% всех случаев заболевания. Гиперфенилаланинемия может достигать 30 мг% и выше. При отсутствии лечения данный вариант фенилкетонурии сопровождается глубокой умственной отсталостью.
Кроме классической формы, различают атипичные варианты фенилкетонурии, протекающие с той же клинической симптоматикой, но не поддающиеся коррекции диетотерапией. К ним относятся фенилкетонурия II типа (недостаточность дегидроптеринредуктазы), фенилкетонурия III типа (дефицит тетрагидробиоптерина) и другие, более редкие варианты. Вероятность рождения ребенка, больного фенилкетонурией, повышается при заключении близкородственных браков.
Патогенез
В основе классической формы фенилкетонурии лежит недостаточность фермента фенилаланин-4-гидроксилазы, участвующего в конверсии фенилаланина в тирозин в митохондриях гепатоцитов. В свою очередь, производный тирозина – тирамин является исходным продуктом для синтеза катехоламинов (адреналина и норадреналина), а дийодтирозин – для образования тироксина. Кроме этого, результатом метаболизма фенилаланина служит образование пигмента меланина.
Наследственная недостаточность фермента фенилалаиин-4-гидроксилазы при фенилкетонурии приводит к нарушению окисления фенилаланина, поступающего с пищей, в результате чего его концентрация в крови (фенилаланинемия) и спинномозговой жидкости значительно возрастает, а уровень тирозина соответственно падает. Избыточное содержание фенилаланина устраняется путем повышенной экскреции с мочой его метаболитов - фенилпировиноградной, фенилмолочной и фенилуксусной кислот.
Нарушение обмена аминокислот сопровождается нарушением миелинизации нервных волокон, снижением образования нейромедиаторов (дофамина, серотонина и др.), запускающими патогенетические механизмы задержки умственного развития и прогредиентное слабоумие.
Симптомы фенилкетонурии
Новорожденные с фенилкетонурией не имеют клинических признаков заболевания. Обычно манифестация фенилкетонурии у детей происходит в возрасте 2-6 месяцев. С началом кормления в организм ребенка начинает поступать белок грудного молока либо его заменителей, что приводит к развитию первых, неспецифических симптомов – вялости, иногда – беспокойства и гипервозбудимости, срыгивания, мышечной дистонии, судорожного синдрома. Одним из ранних патогномоничных признаков фенилкетонурии служит упорная рвота, которая нередко ошибочно расценивается как проявление пилоростеноза.
Ко второму полугодию становится заметным отставание ребенка в психомоторном развитии. Ребенок становится менее активным, безучастным, перестает узнавать близких, не пытается садиться и вставать на ножки. Аномальный состав мочи и пота обусловливают характерный «мышиный» запах (запах плесени), исходящий от тела. Часто наблюдается шелушение кожи, дерматиты, экзема, склеродермия.
У детей с фенилкетонурией, не получающих лечения, выявляется микроцефалия, прогнатия, позднее (после 1,5 лет) прорезывание зубов, гипоплазия эмали. Отмечается задержка речевого развития, а к 3-4 годам выявляется глубокая олигофрения (идиотия) и практически полное отсутствие речи.
Дети с фенилкетонурией имеют диспластическое телосложение, нередко - врожденные пороки сердца, вегетативные дисфункции (потливость, акроцианоз, артериальную гипотонию), страдают запорами. К фенотипическим особенностям детей, страдающих фенилкетонурией, следует отнести светлую кожу, глаза и волосы. Для ребенка с фенилкетонурией характерны специфическая поза «портного» (согнутые в суставах верхние и нижние конечности), тремор рук, шаткая, семенящая походка, гиперкинезы.
Клинические проявления фенилкетонурии II типа характеризуются тяжелой степенью умственной отсталости, повышенной возбудимостью, судорогами, спастическим тетрапарезом, сухожильной гиперрефлексией. Прогрессирование заболевание может приводить к гибели ребенка в возрасте 2-З лет. При фенилкетонури III типа развивается триада признаков: микроцефалия, олигофрения, спастический тетрапарез.
В настоящее время диагностика фенилкетонурии (а также галактоземии, врожденного гипотиреоза, адрено-генитального синдрома и муковисцидоза) входит в программу неонатального скрининга, осуществляемого всем новорожденным. Основные и дополнительные методы диагностики:
- Скрининг-тест. Проводится на 3-5 день жизни доношенного и 7 день жизни недоношенного ребенка путем забора образца капиллярной крови на специальный бумажный бланк. При обнаружении гиперфенилаланемии более 2,2 мг% ребенка направляют к детскому генетику для повторного обследования.
- Биохимические исследования. Для подтверждения диагноза фенилкетонурии проверяется концентрация фенилаланина и тирозина в крови, определяют активность печеночных ферментов (фенилаланингидроксилазы), выполняется биохимическое исследование мочи (определение кетоновых кислот), метаболитов катехоламинов в моче и др.
- Неврологическое обследование. Дополнительно проводится ЭЭГ и МРТ головного мозга, осмотр ребенка детским неврологом.
- Пренатальная диагностика. Генетический дефект при фенилкетонурии может быть обнаружен еще на этапе беременности в ходе инвазивной пренатальной диагностики плода (хорионбиопсии, амниоцентеза, кордоцентеза). В остальных случаях окончательный диагноз выставляется по результатам ДНК-диагностики после рождения.
Дифференциальный диагноз фенилкетонурии проводят с внутричерепной родовой травмой новорожденных, внутриутробными инфекциями, другими нарушениями обмена аминокислот.
Лечение фенилкетонурии
Основополагающим фактором в лечении фенилкетонурии является соблюдение диеты, ограничивающей поступление белка в организм. Лечение рекомендуется начинать при концентрации фенилаланина >6 мг%. Для грудных детей разработаны специальные смеси - Афенилак, Лофенилак; для детей старше 1 года – Тетрафен, Фенил-фри; старше 8 лет - Максамум-ХР и др. Основу диеты составляют низкобелковые продукты - фрукты, овощи, соки, белковые гидролизаты и аминокислотные смеси. Расширение диеты возможно после 18 лет в связи с возрастанием толерантности к фенилаланину. В соответствии с российским законодательством обеспечение лиц, страдающих фенилкетонурией, лечебным питанием, должна осуществляться бесплатно.
Больным назначается прием минеральных соединений, витаминов группы В и др.; по показаниям - ноотропные средства, антиконвульсанты. В комплексной терапии фенилкетонурии широко используется общий массаж, ЛФК, иглорефлексотерапия. Атипичные формы фенилкетонурии, не поддающиеся лечению диетой, требуют назначения гепатопротекторов, противосудорожных средств, заместительной терапии леводопой, 5-гидрокситриптофаном.
Дети, страдающие фенилкетонурией, находятся под наблюдением участкового педиатра и психоневролога; нередко нуждаются в помощи логопеда и дефектолога. Необходим тщательный мониторинг нервно-психического статуса детей, контроль уровня фенилаланина в крови и показателей электроэнцефалограммы.
Прогноз и профилактика
Проведения массового скрининга на фенилкетонурию в неонатальном периоде позволяет организовать раннюю диетотерапию и предотвратить тяжелые церебральные повреждения, нарушения функции печени. При раннем назначении элиминационной диеты при классической фенилкетонурии прогноз развития детей хороший. При поздно начатом лечении прогноз в отношении умственного развития неблагоприятный.
Профилактика осложнений фенилкетонурии заключается в проведении массового скрининга новорожденных, раннего назначения и длительного соблюдения диетического питания.
С целью оценки риска рождения ребенка с фенилкетонурией предварительное генетическое консультирование должны пройти супружеские пары, уже имеющие больного ребенка, состоящие в кровнородственном браке, имеющие родственников с данным заболеванием. Женщины с фенилкетонурией, планирующие беременность, должны соблюдать строгую диету до зачатия и во время беременности для исключения повышения уровня фенилаланина и его метаболитов и нарушения развития генетически здорового плода. Риск рождения ребенка с фенилкетонурией у родителей-носителей дефектного гена, составляет 1:4.
Читайте также: