Синдром Герстмана-Штраусслера-Шайнкера. Фатальная семейная бессонница.
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Ученым удалось приблизиться к разгадке механизма действия прионных болезней. Это целая группа нейродегенеративных заболеваний, которые уничтожают мозг человека, не оставляя шанса на спасение. Ученые надеются, что открытие приблизит их к разработке лечения.
Эти странные прионы
Прионы — это особые белки с аномальной структурой, которые подобно вирусам способны инфицировать млекопитающих, включая человека. Они размножаются в живых клетках, заставляют собственные белки организма превращаться в прионы, особенно часто они поражают мозг. Это единственная известная инфекция, которая размножается без РНК и ДНК.
Прионы очень стабильны, они накапливаются с пораженных тканях, повреждая и разрушая их. При этом их очень сложно химически или физически разрушить. Из-за этого до сих пор нет лечения ни от одной прионной болезни, все они смертельны в 100% случаев. Все вместе прионные болезни называют трансмиссивные губчатые энцефалопатии.
Куру — это нейродегенеративное прионное заболевание, распространенное среди аборигенов Новой Гвинеи. В основном она расправилась среди каннибалов, которые съедали мозг недруга в ритуальных целях. После проявления болезни больной живет не больше года, однако инкубационный период может длиться десятилетиями.
Синдром Герстмана — Штраусслера — Шейнкера встречается редко. Это наследственное заболевание, которое проявляется у людей от 20 до 60 лет. Начинается с проблем с координацией, а заканчивается глубокой деменцией. Инкубационный период составляет от 5 до 30 лет.
Фатальная семейная бессонница — пожалуй, одна из самых жутких прионных болезней. Пациенты буквально теряют способность спать и умирают от бессонницы за несколько месяцев. В это время их мучают панические атаки и галлюцинации.
Болезнь Крейтцфельдта — Якоба также известна как коровье бешенство. Имеет несколько форм. Первая — классическая — возникает спонтанно у людей старше 50 лет, постепенно разрушает мозг, убивая за 8 месяцев. Вторая получила название «новый вариант», она появилась в конце 80-х в великобритании. Предположительно первые пациенты заразились от плохо обработанного мяса бешеных коров. Существует наследуемая форма, а еще этой болезнью можно заразиться, например, во время медицинского вмешательства.
Помешать заражению
Поражая мозг, прионы накапливаются в аксонах— длинных отростках нейронов. В итоге они увеличиваются в размере и теряют способность правильно проводить сигналы. Подобным образом идет развитие и других нейродегенеративных заболеваний белковой природы, например, болезни Альцгеймера. Если ученым удастся понять, как и из-за чего это происходит, то получится если не избавить людей полностью от этих недугов, то хотя бы научиться их сдерживать.
Исследователи внимательно наблюдали за поведением прионов, белки поражают только аксоны, но не трогают сами тела нейронов. Оказалось, что именно в этой особенности и лежит разгадка. Обычно нейроны способны очищать себя от лишнего, в том числе и от сгустков прионов. Однако с аксонами дело обстоит труднее, клетки просто не справляются с очисткой.
Судя по всему, обычно нейроны избавляются от прионов с помощью везикул— маленьких капсул, которые клетки используют для транспорта веществ. Нейроны отправляют свои везикулы по системам микротрубочек в аксоны, как по рельсам. Если в капсуле оказывается прион, то, попав в аксон, он сливается с другими, образует агрегаты, от которых уже не получается избавиться. В итоге эти агрегаты набухают и перекрывают пути для импульсов, вызывая скорую гибель нейрона.
Поняв механизм, ученые смогли ему противостоять. Они нашли четыре белка, которые ответственны за отправку везикул с прионами в аксоны. Когда они подавляли выработку хотя бы одного из них, то в аксоны начинало поступать гораздо меньше прионов. При этом нейроны функционировали нормально или почти нормально, а выживали так же хорошо, как и здоровые клетки мозга.
Пока результат получен на мышах. Однако он вызывает оптимизм у исследователей, они надеятся, что метод позволит если не излечить болезни, связанные с накоплением белков, то хотя бы замедлить их развитие.
«Мы с большим энтузиазмом относимся к открытию молекул, которые могут ингибировать этот путь формирования агрегатов, и изучению эффектов таких ингибиторов на животных моделях прионов и других нейродегенеративных заболеваний» , — говорит один из авторов исследования, Сандра Энкалада, доктор философии.
Синдром Герстмана-Штраусслера-Шейнкера
Синдром (болезнь) Герстмана-Штраусслера-Шейнкера, или синдром ГШШ – нейродегенеративное прионное заболевание. За этим исчерпывающим и сугубо специальным определением скрывается тяжелая, редкая, в некоторых отношениях уникальная патология, и хотя бы поэтому каждое слово заслуживает более подробного рассмотрения.
Дегенерацией, – применительно к единичному организму, – называют патологический процесс перерождения (вырождения) на клеточном уровне, в ходе которого та или иная ткань постепенно утрачивает нормальную, естественную для нее структуру, становясь все более примитивной по строению и все хуже справляясь с возложенными на нее функциями. Как правило, такие процессы обусловлены нарушениями клеточного метаболизма (дистрофия) и/или дефицитом кровоснабжения (ишемия), конечным результатом чего является полная деградация, сокращение в объеме и отмирание (атрофия, некроз, инфаркт) паренхиматозной, т.е. функциональной, узкоспециализированной ткани какого-либо органа. Соответственно, термином «нейродегенеративные заболевания» обозначают большую группу болезней, обычно наследственных и на сегодняшний день неизлечимых, при которых дегенерирует сложнейшая паренхима центральной нервной системы, т.е. нейронное вещество спинного и/или головного мозга, а также нервных узлов и сплетений периферической нервной системы. Начинаясь, как правило, с незначительных нарушений памяти, речи, моторных функций, большинство таких заболеваний в терминальной стадии обусловливают тотальную деменцию (органическое слабоумие) и сугубо вегетативное существование пациента. К наиболее известным нейродегенеративным процессам относятся болезни Альцгеймера, Пика, Паркинсона, амиотрофический боковой и рассеянный склероз, и пр.
Прионные же заболевания, или спонгиоформные (губчатые) энцефалопатии – совершенно особый вид нейродегенерации, причиной которого является недавно открытые (1982) белки-прионы, до сих пор остающиеся предметом интенсивных научных дискуссий: некоторые исследователи склонны считать прионы особой формой жизни, другие рассматривают их исключительно как биохимический феномен. Так или иначе, но прионные молекулы способны in vivo модифицировать другие белки таким образом, что те превращаются в идентичные прионы; в результате клетка погибает, а ткань в целом вырождается в пористую биомассу, не имеющую никакого отношения к первоначальной специализации. Поскольку этот процесс прогрессирует в веществе головного мозга (причем значительно быстрее, чем другие виды нейродегенерации), его проявления и последствия трудно назвать иначе, как катастрофическими. Широкую огласку получили прионные эпизоотии сельскохозяйственного скота (овечья почесуха, коровье бешенство), однако прионными оказались и некоторые заболевания человека, ранее приписываемые неизвестным вирусам, – болезнь Крейтцфельдта-Якоба, куру, фатальная семейная инсомния.
Что касается синдрома Герстмана-Штраусслера-Шейнкера, то как самостоятельная нозологическая единица он выделен и описан еще в 1936 году, однако этиопатогенез этой специфической нейродегенерации стал ясен лишь с открытием прионов и осуществлением хромосомных исследований. К счастью, эта патология не просто редка, а встречается исключительно редко: примерно в одном случае на 10 млн населения.
2. Причины
Наряду с фатальной семейной бессонницей и генетическим вариантом болезни Крейтцфельдта-Якоба, синдром ГШШ является не трансмиссивным (заразным), а наследственным заболеванием. Дефектный ген PRPN запускает «цепную реакцию» воспроизводства белков-прионов в мозговой ткани. Как правило, это происходит в возрастном интервале от 20 до 60 лет, однако о реальной распространенности, о возрастных закономерностях, о влиянии фактора пола и других характеристиках процесса трудно судить с уверенностью, поскольку накопленный объем наблюдений, по статистическим меркам, совершенно недостаточен.
3. Симптоматика, диагностика
Некоторые клинические и патоморфологические особенности синдрома ГШШ роднят его с болезнью Альцгеймера, другие – с различными нейродегенеративными формами атаксии (неупорядоченности, двигательной дискоординации). Это дополнительно осложняет диагностику и, видимо, нередко приводит к диагностическим ошибкам, которые могут быть однозначно устранены только при посмертном патоморфологическом исследовании.
В симптоматике выявляются феномены, характерные как для органического поражения мозжечка (нарушения координации движения, способности к пространственной ориентации), так и т.н. бульбарная симптоматика, специфическая для поражений продолговатого мозга: ослабление и постепенное отмирание глотательного рефлекса, прогрессирующие нарушения артикуляторных составляющих речи и т.д. В терминальной стадии клиническая картина носит столь же тотальный и катастрофический характер, как и при других видах нейродегенерации. Однако от сходных заболеваний именно прионного генеза синдром ГШШ отличается более медленным течением: усредненный период от манифестации до летального исхода составляет примерно 5-6 лет (в разбросе от 3 месяцев до 13 лет).
4. Лечение
Как раскрыть тайны прионов
Две недели назад в журнале PLOS Pathogens был описан метод, позволяющий изучать прионные заболевания на клеточных культурах.
Прионные болезни сильно выделяются на фоне других инфекций. С фундаментальной научной точки зрения они необычны тем, что вызывается патогеном, не имеющим генома и не содержащим вообще никаких нуклеиновых кислот. Обычных людей гораздо больше беспокоит то, что все эти болезни, хотя и развиваются обычно довольно медленно и встречаются редко, абсолютно смертельны и эффективного способа их лечения пока не найдено. Более того, хотя некоторые сопровождающие прионную инфекцию процессы изучены, до сих пор непонятно, из-за чего развиваются конкретные симптомы и наступает смерть. Именно в этом должен помочь новый метод.
Раковые клетки, бактерии и вирусы размножаются благодаря тому, что у них есть свой собственный геном, в котором записана информация о необходимых им белках. Вирусы не вполне самостоятельны в этом смысле, им нужна помощь со стороны клетки хозяина, но, если нет вирусной ДНК (РНК), нет и инфекции. Одно из направлений в лечении ВИЧ/СПИДа как раз занимается разработкой ферментов, которые уничтожали бы копии вирусного генома в клетках.
У приона нет никакого генома, это просто один белок, только в неправильной конформации. Он может взаимодействовать с аналогичным белком правильной конформации, меняя его конформацию на прионную, неправильную. Предположительно, при этом α-спираль превращается в β-слой. Обычно белки в клетке, немного поработав, разбираются специальными ферментами обратно на аминокислоты, из которых строятся новые белки. Белки с испорченной конформацией, однако, утилизировать не удается, и они накапливаются. Более того, они накапливаются экспоненциально, поскольку новообращенные прионы приобретают все свойства патогена и тоже могут менять конформацию нормальных белков.
Хотя мы до сих пор употребляли слово «прионы» во множественном числе, для животных известен ровно один такой белок. Это белок, который кодирует ген PRNP. Его нормальную форму принято обозначать как PrP C (от слова cellular – клеточный), а прионную – PrP Sc (от слова scrapie – почесуха; эта овечья болезнь была первым описанным прионным заболеванием). Примечательно, что нет полной ясности в вопросе о том, зачем этот белок (его нормальная форма) вообще нужен. Известно, что его особенно много в нервных клетках (поэтому прионные заболевания поражают нервную систему), обычно он находится на их поверхности. Предпринимались попытки вывести мышей, у которых этого гена вообще не было бы. Такие мыши вполне жизнеспособны. В самых ранних экспериментах вообще не удалось обнаружить достоверные отличия между ними и обычными здоровыми мышами. Дальнейшие исследования, однако, показали, что белок играет роль в формировании воспоминаний, ориентации в пространстве и поддержании режима сон/бодрствование. Кроме того, вероятно, у него есть функции за пределами нервной системы: в мышцах и клетках крови.
У человека и животных есть несколько болезней, вызываемых PrP Sc , и все они, хотя и поражают нервную систему, характеризуются немного разными симптомами. Первая описанная еще в начале XVIII века болезнь – почесуха овец. Из-за поражений центральной нервной системы у овец появляется сильный кожный зуд, и они начинали чесаться о заборы деревья и камни. Животное умирало через несколько недель после появления первых симптомов. Похожие болезни есть у крупного рогатого скота (коровье бешенство), у оленей и лосей, у норок, и более экзотических копытных, например, некоторых видов антилоп, и даже у кошачьих. По-видимому, все случаи прионных болезней у животных носят инфекционную природу, то есть здоровое животное заражается от больного. Обычно речь идет об орально-фекальном пути заражения. Кроме того, определенную роль могут играть жидкости, попадающие на землю, когда инфицированное животное рожает.
К сожалению, из-за длительности инкубационного периода и сложности лабораторных методов диагностики выявить зараженных животных на ранних стадиях болезни не удается. У возбудителя болезни нет никакой ДНК, чтобы можно было провести полимеразную цепную реакцию. Вызывающий болезнь белок не вполне чужеродный, кроме того, большинство событий происходит по ту сторону гематоэнцефалического барьера, поэтому никаких антител в крови не образуется, и иммунохимическая диагностика тоже невозможна.
У людей прионные болезни бывают не только инфекционными. Хотя, возможно, людей просто лучше изучают. У людей найдены мутации в гене PRNP, которые приводят к тому, что собственный белок без воздействия внешних агентов принимает неправильную конформацию. Таковы, например, фатальная семейная бессонница и синдром Герстмана — Штраусслера — Шейнкера. При фатальной семейной бессоннице люди действительно теряют способность уснуть и вскоре погибают. Такой набор симптомов хорошо согласуется с упомянутой выше ролью PrP в поддержании циркадных ритмов. Синдром Герстмана — Штраусслера — Шейнкера больше похож на классическое нейродегенеративное заболевание. Его симптомы – деменция и нарушения моторики – нарастают постепенно.
Самое распространенное прионное заболевание человека – болезнь Крейцфельда-Якоба может быть и инфекцией, и генетической болезнью. Заразиться можно, например, съев мяса больного животного или в ходе медицинской манипуляции. Мутация, заставляющая собственный белок принимать неправильную конформацию, может наследоваться, а может возникать спонтанно. Иногда прионная конформация вообще возникает сама собой. После ее возникновения помочь человеку уже невозможно. Появившийся белок PrP Sc начинает превращать нормальный вариант PrP C в себе подобный PrP Sc и процесс нарастает лавинообразно. Неправильный белок накапливается в клетках мозга в виде, напоминающем амилоидные бляшки, образующиеся при болезни Альцгеймера и состоящие из белка β-амилоида.
Недавно в ходе расследования трагического случая обнаружилось, что у этих двух болезней может быть даже больше общего, чем казалось. Около полувека назад, когда механизм прионной инфекции только начинали изучать, отставание в росте больных пытались лечить гормоном роста человека. Гормон роста выделяли из гопофизов трупов. Уже в XXI веке обнаружилось, что несколько порций гормона оказались контаминированы PrP Sc и несколько человек погибли. На вскрытии обнаружилось, что, кроме агрегатов из PrP Sc в головном мозге этих людей присутствуют амилоидные бляшки, характерные для болезни Альцгеймера. Заболевшие люди не принадлежали к группе риска и вообще были довольно молоды для Альцгеймера, так что на простое совпадение ситуация не тянет. Она заставляет задуматься о том, что у болезни Альцгеймера может быть схожий инфекционный механизм. «Дурное семя», попав в головной мозг, запускает процесс сборки бляшек, которые клеточные ферменты не в состоянии растворить.
Куру – экзотическое прионное заболевание, распространившееся среди аборигенов Папуа – Новой Гвинеи из-за каннибализма. Ученые обнаружили болезнь в середине XX века, предания народа форе свидетельствуют о том, что первые случаи пришлись на начало века. Болезнь практически исчезла, когда каннибализм прекратился. Последний заболевший умер в 2005 году. Примечательно, что за неполных 100 лет да еще и при длинном инкубационном периоде среди форе успела появиться и даже немного распространиться мутация в гене PRNP, дающая иммунитет к болезни. Видимо, белок PrP Sc не может изменить конфомацию мутантного белка. Это означает, между прочим, что для развития болезни совершенно необходим нормальный клеточный вариант PrP c .
У людей и животных, как мы видим, на всех один прион. То ли дело у грибов. У них описано 12 (11 из них достоверно) разных белков (семь из них – у дрожжей), обладающих прионными свойствами. Это белки с разными функциями и локализацией. Более того, как таковых заболеваний у грибов они не вызывают, и уж точно не вызывают смертельных заболеваний, хотя в некоторых исследованиях показано, что переход к прионной форме снижает жизнеспособность организмов. Однако другие исследования приводят к обратному выводу: переход к прионной форме может помогать адаптироваться к новым условиям. Например, такая трансформация дрожжевого белка Ure2p позволяет дрожжам лучше усваивать азот.
На сегодняшний день у людей лучше всего изучены морфологические проявления финальных стадий прионных инфекций. После вскрытия легко увидеть патологическую губчатую структуру мозга и увидеть в клетках скопления аномального белка. Про ранние стадии патогенеза известно гораздо меньше. Эксперименты на клеточных культурах затруднены из-за того, что нейроны вообще трудно культивировать, а в других клетках патологический процесс вроде как и не наблюдается.
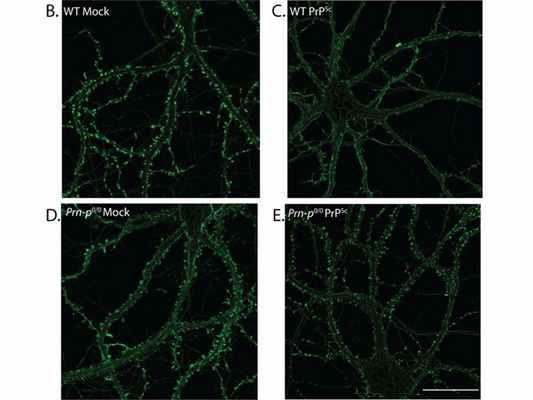
Авторы исследования, с которого мы начали свой рассказ, как раз смогли поддерживать в культуре клеток нейроны гиппокампа и проследить, какие события происходят в клетках в первые часы после инфекции. Белок PrP c находится на поверхности нейронов, поэтому для имитации заражения достаточно добавить прионную форму к среде для культивирования клеток. Оказалось, что уже через 24 часа после инфекции резко сокращается количество дендритных шипиков – выростов на поверхности дендритов, ответственных за формирование синапсов и играющих важную роль в механизмах памяти и обучения. Сокращения числа шипиков не происходило, когда эксперимент проводился на мышах, полностью лишенных белка PrP. Это, с одной стороны, согласуется с тем, что нормальный белок нужен для развития инфекции, а с другой – что оценка числа шипиков – неплохой критерий оценки инфекции.
Таким образом, предложенная модель может оказаться полезной для исследования клеточных и молекулярных механизмов прионной инфекции.
Уточнен патогенез прионных заболеваний
Прионы — отдельный класс инфекционных агентов, имеющих протеиновую основу при отсутствии индивидуального генома, представленного нуклеиновыми кислотами. В исследовании, проведенном научными сотрудниками отделения нейрохирургии Клинического нейробиологического центра Университетской клиники Цюриха (Clinical Neuroscience Centre, University Hospital Zürich), Швейцария, сообщается о том, что диссеминация прионов не связана с преодолением гематоэнцефалического барьера. Новое понимание механизмов распространения прионных агентов может способствовать разработке инновационных эффективных стратегий предотвращения нейродегенеративных изменений даже после инфицирования нервных структур.
Концепция прионных заболеваний предполагает идею новой протеиновой наследственности. Преобладающее большинство прионных частиц являются патогенами. Однако их уникальность заключается в распространении без репликации индивидуальных ДНК или РНК в организме хозяина. Трансмиссивные губчатые энцефалопатии — прогрессирующие, неизменно летальные нейродегенеративные заболевания, вызванные модификациями прионного белка. К данному классу относят ряд состояний, среди которых болезнь Крейтцфельдта — Якоба, фатальная семейная бессонница, синдром Герстмана — Штраусслера — Шейнкера и другие. До настоящего времени терапия указанных заболеваний все еще не разработана, что во многом связано с необходимостью точного понимания механизма распространения прионов от места инфицирования непосредственно в структуры головного мозга.
Новый взгляд на пути трансмиссии прионов
Ранее было известно о том, что прионы, присутствуя в крови, могут проникать через гематоэнцефалический барьер напрямую в ткани мозга. Однако исходным тезисом нового исследовательского проекта послужили данные о возможности лимфоретикулярной прионной инвазии с последующим ретроградным переносом патогенных частиц через периферические нервы. В новом проекте ученые Цюрихского университета (Zürich University), Швейцария, сосредоточили внимание на исследовании возможностей диссеминации прионных частиц в организме и проникновения их в ткани головного мозга непосредственно через кровеносные сосуды.
Исследование проводили в группе лабораторных мышей с генетически модифицированными структурами гематоэнцефалического барьера. Однако, оценив профиль выживаемости животных основной и контрольной групп, не выявили достоверной разницы между изучаемыми показателями. В частности, продемонстрировано, что проницаемость структур гематоэнцефалического барьера может не иметь взаимосвязей с началом активного патологического процесса даже в случаях парентерального внедрения. Это позволило авторам выдвинуть предположение о том, что скорость преодоление прионами гематоэнцефалического барьера может не влиять на особенности патогенеза. Исходя из полученной информации, прионы, вероятно, проникают в структуры головного мозга, подобно рабдовирусам и герпесвирусам.
Выводы
Таким образом, результаты исследований на животных с моделированным проницаемым гематоэнцефалическим барьером расширяют понимание вопроса о диссеминации прионов с периферии в ткани головного мозга, указывая на отсутствие прямого перехода через гематоэнцефалический барьер. Комментируя важность работы, авторы пришли к заключению, что, помимо значимости базового понимания прионной нейроинвазии, новая информация может послужить основой для будущей разработки стратегий эффективной постконтактной профилактики прионных заболеваний, предотвращающей развитие нейродегенеративных изменений даже после экстраневрального инфицирования.
История редких нейродегенеративных заболеваний
Нейродегенеративные заболевания – группа медленно прогрессирующих болезней, затрагивающих работу нервной системы. Одни встречаются чаще, другие – реже. Они могут передаваться по наследству или быть приобретенными. Некоторые из них поддаются лечению, а для других лекарства еще не изобрели.
Сегодня мы поговорим о двух редких нейродегенеративных заболеваниях – болезни куру и фатальной семейной бессоннице (FFI). От первой в свое время пострадало целое племя в Папуа-Новой Гвинее, вторая до сих пор не дает покоя одному итальянскому семейству. На первый взгляд между этими болезнями ничего общего, но как обычно бывает в жизни, на самом деле все не так прямолинейно.
Болезнь куру

Почти никто в мире до 1930-х годов не знал, живет ли кто-нибудь в нагорьях Папуа-Новой Гвинеи. Общество оставалось в неведении до тех пор, пока австралийские золотоискатели не изучили местность и не обнаружили около миллиона человек.
Первые исследователи отправились в эту местность в 1950-е годы, и сразу же обнаружили нечто тревожное. В племени форе численностью около 11 тысяч человек ежегодно умирало 2% населения от неизвестной науке болезни. Племенные жители называли ее «куру», что означает «дрожь» или «порча».
Местные знали: если появились первые симптомы, то смерть неминуема. Сначала больные испытывали затруднения при ходьбе, что в дальнейшем приводило к полной потере контроля над конечностями. Они также утрачивали способность управлять своими эмоциями. По этой причине некоторые издания, которые впоследствии писали об этой болезни, называли ее «хохочущей смертью». Через год больные уже не могли вставать с пола, самостоятельно есть и контролировать свое тело. Эту болезнь подробно описали в 1957 году два врача: Даниел Карлтон Гайдузек и Винсент Зигас.
Форе были убеждены, что куру вызывают злобные шаманы. В первую очередь болезнь затрагивала взрослых женщин и детей младше 8 лет. Некоторые деревни полностью потеряли молодых женщин. Жители были одержимы в попытках спасти себя, потому что чувствовали, что их племя находится на грани вымирания.
Что же вызвало эту болезнь? Ответ на этот вопрос не давался ученым в течение многих лет. После того, как ученые проверили местность и исключили влияние загрязняющих веществ, они решили, что скорее всего болезнь генетическая. Это было первое главное заблуждение среди ученых относительно природы заболевания. Впоследствии ученые выяснили, что куру – не генетическая болезнь, поскольку она затрагивает женщин и детей в одних и тех же социальных группах, но не в генетических. Впервые куру появилась в северных деревушках на рубеже веков, а затем в течение долгих десятилетий двигалась на юг.
После того, как ученые исключили возможность передачи куру по наследству, они решили, будто болезнь – проявление медленного вируса. Чтобы подтвердить эту гипотезу, группа вплотную занялась исследованиями.
Позже Гайдузек и Зигас догадались, что происходит: болезнь связана с похоронными обрядами, принятыми в племени форе, а именно поеданием тел умерших. Во многих деревнях, когда человек умирал, женщины удаляли из тела мозг, смешивали его с папоротником и варили его в бамбуковой посуде. Оставшиеся части тела готовили на огне и съедали все, кроме желчного пузыря, в знак любви и печали. В ритуале участвовали в основном взрослые женщины, поскольку считалось, что их тела укрощали опасный дух, который будет сопровождать труп в мир иной. Иногда в честь праздника женщины давали небольшие порции такой пищи своим детям. Этим объясняется смертность среди женщин и детей.

Тем не менее, у ученых не было прямых доказательств теории. Тогда они провели эксперимент на шимпанзе: им ввели материалы мозга инфицированного человека. У животных проявились симптомы куру, что заставило ученых думать, будто причиной действительно является медленный вирус с аномально длительным инкубационным периодом: у людей он составлял от 2 до 23 лет. За открытие инфекционного характера болезни куру Карлтон Гайдузек удостоился в 1976 году Нобелевской премии по физиологии и медицине.
Позже его теорию признают ошибочной: агентом куру был не вирус, бактерия, грибок или паразит – это был совершенно новый по тем временам инфекционный агент, который не имел генетического материала, мог выжить при кипячении и при этом не являлся живым. Как позже выяснит группа ученых под руководством Стенли Прузинера, возбудителем куру окажется белок с аномальной третичной структурой и не содержащий нуклеиновые кислоты – прион.
Структура белка приона и его репликация имеет основополагающее значение в изучении куру. Хотя точные детали, касающиеся структуры прионов, изначально были неясны, Прузинер выдвинул три гипотезы. Он предполагал, что это либо вирусы, либо белки, связанные с небольшим полинуклеотидом, либо белки, лишенные нуклеиновой кислоты. В ходе многочисленных исследований удалось подтвердить последнее предположение ученого. В 1997 году Призонер получил Нобелевскую премию за открытие прионов – нового биологического источника инфекций.
В нормальных условиях эти клеточные белки безобидны, но они обладают способностью превращаться в устойчивые структуры, вызывающие ряд нейродегенеративных заболеваний, в том числе и куру. Прионы «подчиняют» своей воле здоровые белки и превращают их в себе подобных. В конечном счете такая своеобразная цепная реакция приводит к образованию количества прионов, достаточного для убийства пучков нервных клеток мозга.
Эти белки буквально превращают мозжечок в решето, пронизывая его насквозь, потому больной и теряет координацию движений. Они также образуют клубки, затрудняющие протекание естественных процессов в мозге. Как правило, больной куру проходит через три стадии. Предшествуют заболеванию головная боль и боли в суставах – общие симптомы, на которые больные зачастую не обращают должного внимания. На первом этапе человек с куру теряет контроль над телом, испытывают трудности в балансировке и поддержании осанки. На втором этапе, или «сидячей» стадии, человек лишается возможности ходить. Появляется тремор в конечностях и непроизвольные подергивания. На третьем этапе больной, как правило, прикован к постели и не способен контролировать большинство функций своего организма. Может проявляться слабоумие или изменение поведения. На этом же этапе у больного возникают трудности с глотанием и он теряет способность принимать пищу традиционным образом. В конечном итоге большинство больных куру умирают от пневмонии.

Заболеть куру можно только при поедании зараженного мозга или вступая в контакт с открытыми ранами или язвами больного, поэтому нельзя говорить о широкой распространенности этого заболевания. Тем не менее, именно изучение болезни куру привело к открытию прионов, которые вызывают ряд других нейродегенеративных заболеваний: фатальной семейной бессонницы, болезни Крейтцфельда-Якоба, синдрома Герстмана — Штраусслера — Шейнкера и других.
К сожалению, вылечить куру пока нельзя. Прионы, вызывающие эту болезнь, трудно поддаются разрушению. Мозг, в котором поработали эти клеточные белки, остается заразным даже если хранить его в формальдегиде в течение многих лет. Поэтому в данном случае лучшее лекарство – это профилактика. Так правительства и общества в середине XX века стремились предотвратить заболевание, препятствуя социальной практике каннибализма. С 1950-х годов племя форе отказалось от своих похоронных обрядов, и сейчас болезнь почти полностью исчезла. Сегодня куру диагностируется редко: симптомы, похожие на куру, с большей вероятностью указывают на другое серьезное неврологическое расстройство или губчатое заболевание.
Тем не менее, исследования болезни ведутся до сих пор. В 2009 году группа ученых Совета по медицинским исследованиям Великобритании обнаружила, что некоторые люди, пережившие эпидемию куру, несут в своем организме генетическую мутацию V127, которая придает сильное сопротивление болезни. Возможно, когда-нибудь ученым удастся найти лекарство, способное противостоять разрушительной деятельности прионов.
Фатальная семейная бессонница
В 1797 году в небольшом городке недалеко от Венеции родился человек по имени Джакомо. Члены его семьи, как правило, были все как на подбор: высокие, плечистые и мускулистые (впрочем, и нынешнее поколение сохранило за собой эти привлекательные черты). Однажды осенью 1836 года Джакомо свалился с необъяснимой болезнью, начал страдать от слабоумия. В конце концов болезнь окончательно приковала его к постели, где он лежал в мучениях без сна. Вскоре после этого он скончался.
У Джакомо осталось трое детей, один из них оставил после себя еще шестерых наследников. Следующие полтора века его потомки процветали: члены семьи становились видными итальянскими врачами и бизнесменами. Их состояние позволило бы им владеть 130 квартирами в Венеции, в том числе палаццо на Большом канале. Но параллельно с высоким положением в обществе в приходских книгах напротив каждой фамилии стояла запись о преждевременной смерти. На протяжении десятилетий в них фиксировались странности вроде эпилепсии, жара, лихорадки, сопровождающейся желудочным расстройством. Позже в свидетельствах о смерти членов семьи будут указывать менингит, энцефалит Экономо, болезнь Альцгеймера, лейкоэнцефалит, алкогольную энцефалопатию и другие заболевания.
На самом деле причина смерти во всех случаях была одна – фатальная семейная бессонница. Это генетическое заболевание не было официально определено до 1986 года. Оно настолько редкое, что долгое время только потомки Джакомо были единственными людьми на планете, пострадавшими от этой болезни. С тех пор было найдено еще 30 семей, подвергнувшихся фатальной семейной бессоннице.
Общая картина симптомов выглядит достаточно мрачно. Первые признаки фатальной семейной бессонницы можно обнаружить в возрасте 32-62 лет, средний возраст – 51 год. Но бывали случаи, когда болезнь наступала и в 18 лет, и в 72 года. Самым первым и главным признаком заболевания является прогрессирующая со временем бессонница.
На первом этапе больной будет пытаться компенсировать недостаток сна послеполуденной дремотой, но как правило, ему это не удается. Зрачки становятся крошечными, давление – повышенным. Наблюдается сильная потливость, у мужчин наступает импотенция. Около четырех месяцев он страдает от приступов паники и необъяснимых фобий.
В течение следующих месяцев борьбы с болезнью больной будет пытаться заснуть, но каждый раз, закрывая глаза, он достигнет максимум легкого оцепенения, транса. Мозг перестанет отдыхать. Панические атаки становятся серьезнее, и будут длиться еще около пяти месяцев.
На третьем этапе общая бессонница вызывает быструю потерю веса и ограничения в умственном функционировании. Эта стадия длится около трех месяцев. На последнем этапе пациент полностью ослаблен и страдает от слабоумия и невосприимчивости к окружающему миру около полугода. Затем его ждет кома и смерть. Один из самых трагических аспектов болезни заключается в том, что несмотря на то, что больной демонстрирует все признаки деменции, он четко понимает то, что с ним происходит.
По меньшей мере 30 из потомков Джакомо умерли таким образом еще в прошлом веке – 13 с 1973 года и еще 7 за последнее десятилетие. Среди живых около 25 людей являются носителями гена, который вызывает это заболевание. В области Венеция в Италии, где до сих пор живет большая часть семьи, долгое время широко была распространена версия, что потомки Джакомо прокляты. Местные жители не перестают обсуждать трагическую судьбу семьи, что не может не отразиться на ее дальнейшем существовании. Молодым девушкам из этой семьи трудно найти себе спутника жизни, несмотря на их внешнюю привлекательность и приличное состояние. Доходит даже до того, что члены семьи не могут получить страховку.
Почти в каждом случае фатальная семейная бессонница вызвана мутацией в гене PRNP. Эта мутация означает, что в белке, который содержит этот ген, аспарагиновая кислота заменяет аспарагин в позиции 178, превращая белок в прион. Но для возникновения болезни этого мало. Чтобы симптомы болезни проявились, в прионе должна присутствовать аминокислота метионин в позиции 129 протеина. Вместе с аминокислотами, аспарагином и метионином в этих конкретных позициях обычные прионы обретают патологическую форму.
Бывают редкие случаи, когда болезнь возникает не из-за изменений в генах. По состоянию на 2016 год зафиксировано только 24 таких случая. Здесь фатальная семейная бессонница возникает, когда некоторые из нормальных прионов человека спонтанно переходят в ненормальную форму, которая вызывает болезнь, а затем изменяет прионы в других клетках, как и в случае с болезнью куру.
Прионы, обретающие аномальную форму, вызывают изменения в таламусе – области головного мозга, отвечающей за перераспределение информации от органов чувств (за исключением обоняния) к коре головного мозга. Эта же область управляет циклом сна и бодрствования, чувством равновесия, ощущением боли, аспектами обучения, памяти, речи и понимания языка. Даже эмоциональные переживания и характер зависят от таламуса.
Когда фатальная семейная бессонница вызывает мутацию в гене PRNP, она передается по наследству аутосомно-доминантным способом. Это означает, что для возникновения болезни достаточно наличия одного мутанного аллеля в неполовой хромосоме. В некоторых случаях человек наследует мутацию от пораженного болезнью родителя. В других случаях болезнь может возникнуть в результате новых мутаций в генах.
Человек с фатальной семейной бессонницей в 50% случаев передает ген своим детям. Иногда случается, что болезнь не передается по наследству при условии, что болезнь вызвана спонтанным изменением в прионах, а не обуславливается генетикой.
Как и куру, фатальную семейную бессонницу нельзя вылечить или замедлить ее прогрессирование. Можно только облегчить симптомы и создать максимально возможную комфортную обстановку вокруг больного.
Читайте также: