Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
Добавил пользователь Alex Обновлено: 07.01.2026
Фенотипическими формами дефицита лизосомной кислой липазы являются
1) младенческая и взрослая формы
2) болезнь Куфса, болезнь Бильшовского – Янского
3) болезнь Гентингтона, болезнь Баттена
4) болезнь Вольмана, болезнь накопления эфиров холестерина (+)
При диагностике болезни тея – сакса измеряют уровень активности фермента — гексозаминидазы
1) A (+)
2) B
3) C
4) D
Синдром дузе чаще всего ассоциирован с мутацией в гене
1) SCN1A (+)
2) SCN8A
3) SCN9A
4) SCN2A
Врожденная глаукома (гидрофтальм) характеризуется мутацией преимущественно в гене
1) PITX2
2) MYOC
3) CYP1B1 (+)
4) PAX6
Буллезный эпидермолиз необходимо дифференцировать с
1) пигментной ксеродермой
2) синдромом LEOPARD
3) эктодермальной дисплазией
4) энтеропатическим акродерматитом (+)
Повреждающим фактором при болезни вильсона – коновалова является
1) отложение меди в тканях (+)
2) повышение уровня метаболитов пуринового обмена
3) наличие кольца Кайзера – Флейшера
4) отложение гемосидерина в тканях
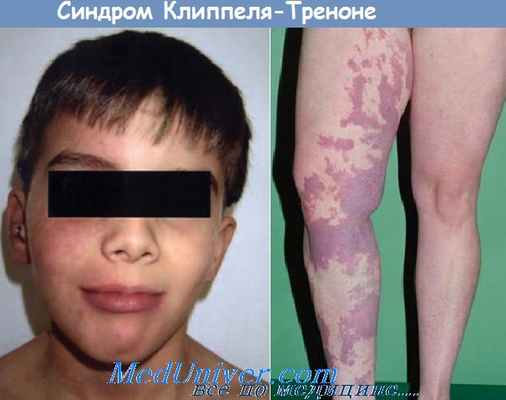
Минимальные диагностические признаки, такие как, асимметричная гипертрофия конечностей и гемангиомы, наиболее характерны для синдрома
1) Мартина-Белл
2) Клиппеля-Треноне-Вебера (+)
3) Марфана
4) Ларсена
Самый распространенный генетический вариант дистальной спинальной мышечной атрофии с преимущественным поражением ног обусловлен мутациями в гене
1) DYNC1H1 (+)
2) SCN1A
3) MPZ
4) FKRP
Симптомокомплекс врожденной эритрокератодермии, мягкого пластинчатого ихтиоза, гиперкератоза (ладоней, подошв, локтей, коленей), снижения потоотделения, нейросенсорной тугоухости, наблюдается при
1) синдроме KID (синдром кератита-ихтиоза- тугоухости) Шегрена – Ларссона (+)
2) синдроме тугоухости и атопического дерматита
3) пальмоплантарной кератодерме с тугоухостью
4) синдроме Bart-Pumphrey
К митохондриальным заболеваниям относят
1) синдром Питта — Хопкинса
2) атаксию-телеангиэктазию
3) болезнь Фабри
4) синдром Кернса — Сейра (+)
К лизосомным болезням накопления относят
1) болезнь Александера
2) болезнь Гирке
3) фукозидоз (+)
4) Х-сцепленную адренолейкодистрофию
Кардиомиопатия (дилатационная или гипертрофическая) является одним из основных симптомов синдрома
1) Вольфа – Паркинсона – Уайта
2) удлиненного интервала QT
3) Бругада
4) Барта (+)
Кондуктивное нарушение слуха характерно для синдрома
1) Джервелла – Ланге-Нильсена
2) микротии с атрезией наружного слухового прохода и проводящей глухоты (+)
3) Альстрема
4) Ашера
1) Вильямса (+)
2) Дауна
3) Ангельмана
4) Крузона
Мутации в гене bsnd характерны для синдрома
1) нейросенсорной тугоухости с легкой почечной дисфункцией (+)
2) Пендреда
3) Альпорта
4) Маршалла
У пациентов с синдромом морриса выявляют
1) повышенные уровни гонадотропинов и сниженный уровень дигидротестостерона
2) повышенный уровень гонадотропинов и сниженный уровень тестостерона
3) нормальные уровни гонадотропинов и повышенный или нормальный уровень тестостерона (+)
4) сниженные уровни гонадотропинов и тестостерона
Увеличение толщины стенки левого желудочка, которое не объясняется исключительно повышением нагрузки давлением, характерно для
1) аритмогенной кардиомиопатии правого желудочка
2) гипертрофической кардиомиопатии (+)
3) синдрома некомпактного миокарда левого желудочка
4) дилатационной кардиомиопатии
Аномалад пьера робена включает
1) гидронефроз вследствие стеноза мочеточника
2) гидроцефалию, косолапость и нарушение функций органов малого таза у ребенка со спинномозговой грыжей
3) лицевые аномалии и гипоплазию легких вследствие агенезии почек у плода и маловодия
4) расщелину нёба вследствие выраженной микрогении и глоссоптоза (+)
Дифференциальную диагностику при болезнях недостаточности ферментов дыхательной цепи необходимо проводить с
1) нервно-мышечными заболеваниями (+)
2) хромосомными синдромами
3) пероксисомными заболеваниями
4) лизосомными заболеваниями
Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
Синдром Клиппеля ‒ Треноне ‒ Вебера ‒ состояние, влияющее на развитие кровеносных сосудов, кожи, мышц и костей. Данное расстройство имеет характерные черты в виде классической триады: капиллярную гемангиому, называемую винным пятном, гипертрофию мягких тканей и костей, пороки развития вен. Это состояние было впервые описано французскими врачами Морисом Клиппелем и Паулем Треноне в 1900 году, которое они его называли «nevus vasculosus osteohypertrophicus». Немецко-британский врач Фредерик Паркес Вебер описал случаи 1907 и 1918 годов, которые были похожи, но не идентичны тем, которые описывали Клиппель и Треноне. Врожденный дефект поражает мужчин и женщин в равной степени и не ограничивается в пределах какой-либо расовой группы. Не имеется абсолютного подтверждения, что патология носит генетический характер, хотя изучение ее все еще продолжается.
4. Sfaihi L, Aissa K, Fourati H, Kamoun F, Mnif Z, Kamoun T, et al. Klippel Trenaunay syndrome in association with Sturge Weber syndrome about one case. Tunis Med. 2016 Feb. 92(2):173-4.
5. Sung HM, Chung HY, Lee SJ, Lee JM, Huh S, Lee JW, et al. Clinical Experience of the Klippel-Trenaunay Syndrome. Arch Plast Surg. 2015 Sep. 42 (5):552-8.
7. Vahidnezhad H, Youssefian L, Uitto J. Klippel-Trenaunay syndrome belongs to the PIK3CA-related overgrowth spectrum (PROS). Exp Dermatol. 2016 Jan;25(1):17-9.
В 1900 году известные французские медики Клиппель и Треноне впервые описали синдром у 2-х пациентов с винным пятном, варикозным расширением вен конечности, а также с гипертрофией костных и мягких тканей пораженных конечностей. Они назвали синдром «naevus vasculosus osteohypertrophicus». В 1907 году Паркс Вебер, не подозревая об открытии Клиппеля и Треноне, описал пациента с тремя вышеупомянутыми симптомами, а также с артериовенозной мальформацией пораженной конечности. Он назвал этот процесс гемангиэктатической гипертрофией [1].
Определение и клиника
Синдром Клиппеля - Треноне - Вебера - состояние, влияющее на развитие кровеносных сосудов, кожи, мышц и костей. Данное расстройство имеет характерные черты в виде классической триады: капиллярную гемангиому, называемую винным пятном, гипертрофию мягких тканей и костей, пороки развития вен [4].
Большинство людей с синдромом Клиппеля - Треноне - Вебера с рождения имеют винное пятно, которое появляется в результате отека мелких кровеносных сосудов вблизи поверхности кожи [2]. Винные пятна плоские, имеют четкие границы, варьируются от бледно-розового до темно-бордового цвета, обычно локализуются на латеральной части одной конечности. С возрастом пораженный участок может стать светлее или темнее. Глубина гемангиомы может быть различной: она может быть ограничена кожей или проникать в низ лежащие слои, включающие мышцы и кости [1]. Висцеральные органы, такие как селезенка, печень, мочевой пузырь и толстая кишка, а также плевра, тоже могут быть вовлечены в патологический процесс [7].
Гипертрофия костных и мягких тканей является третьим признаком синдром Клиппеля - Треноне - Вебера. Гипертрофия конечности может быть вторичной по отношению к увеличенной длине (костное участие) и / или увеличению обхвата (вовлечение мягких тканей) [3]. Гипертрофия может быть оценена при рождении, а также в первые годы жизни. Большую степень гипертрофии можно наблюдать у пациентов с имеющейся у них артериовенозной мальформацией [1].
Обычно паталогический рост ограничен одной конечностью, чаще всего ногой, однако, разрастание также может затронуть руки, реже шею, голову и поясницу [5]. Аномальный рост может вызывать боль, чувство тяжести, снижение объема активных движений в зоне поражения. Также чрезмерный рост одной ноги приводит к проблемам с ходьбой. Иногда вовлеченная в процесс конечность может быть атрофирована, а не гипертрофирована [6].
Пороки развития вен являются третьей важной особенностью клинического течения синдрома Клиппеля - Треноне - Вебера. В эту группу аномалий входят варикозные расширения вен (обычно на латеральных сторонах бедер и на икрах ног), которые иногда замечаются при рождении [7]. Варикоз может не визуализироваться до тех пор, пока ребенок не начнет ходить. Варикозы могут быть обширными, хотя чаще всего они локализуются в пределах подкожной вены ноги [2].
Глубокие вены конечностей также могут вовлекаться в патологический процесс, повышая риск возникновения тромбоза, называемого тромбозом глубоких вен. Впоследствии оторвавшийся тромб, проходя с током крови через легкие, может привести к развитию легочной эмболии. В редких случаях варикозное расширение сосудов наблюдаются в мочевом пузыре, толстой кишке и легких [4].
Варикозы могут остаться стабильными в размере или постепенно расширяться, о чем будут свидетельствовать появление болей и лимфостаз. Эти симптомы могут ухудшиться во время беременности [1].
Другими особенностями клинического течения этого синдрома являются: лимфатическая обструкция, расщепление позвоночника, гипоспадия, синдактилия, олигодактилия, полидактилия, гипергидроз, гипертрихоз, парестезии, декальцификация костей, хроническая венозная недостаточность, застойный дерматит, плохое заживление ран, изъязвление, тромбоз, ангиосаркома. Орофасциальные аномалии могут потребовать специализированного ухода за зубами и анестезию [2].
Синдромом Клиппеля - Треноне - Вебера страдает не менее 1 на 100 000 человек по всему миру. Нет подтверждения о том, что данная патология развивается у представителей определенной расы. Страдают как мужчины, так и женщины в равной степени [3].
Точная причина синдрома Клиппеля - Треноне - Вебера неизветна, хотя существуют несколько теорий. Близнак и Стэпл предложили теорию внутриутробного повреждения симпатических ганглиев или латерального промежуточного тракта, что приводит к дилатации микроскопических артериовенозных анастомозов [3]. Сервелль считал, что аномалии развития глубоких вен, с результирующей обструкцией венозного потока, приводят к венозной гипертензии, развитию варикоза и гипертрофии конечностей [6]. МакГрори и Амадио полагали, что лежащая в основе смешанная мезодермальная и эктодермальная дисплазия, вероятно, ответственны за развитие данного синдрома [7].
Синдром Клиппеля - Треноне - Вебера почти всегда является спорадическим, что означает, что он возникает у людей, не имеющих данной патологии в своей семье [2]. Исследования показывают, что это состояние является следствием генных мутаций, которые не наследуются. Эти генетические изменения, вызываемые соматическими мутациями, возникают случайным образом в одной клетке на ранних стадиях развития до рождения. По мере того как клетки продолжают делиться в период внутриутробного развития организма, одни дочерние клетки, возникшие от мутировавшей материнской, будут иметь мутации, а другие нет. Эта смесь клеток с генетической мутацией и без нее именуется понятием мозаицизм [1].
Также Кихичаком было высказано предположение, что синдром Клиппеля - Треноне - Вебера может быть вызван мутацией гена PIK3CA. Этот ген кодирует белок р110α, который является субъединицей фермента, называемого фосфатидилинозитол-3-киназа (PI3K). PI3K играет важную роль в клеточной жизнедеятельности, в том числе влияние на их пролиферацию, миграцию и выживаемость.
При возникновении мутаций в гене PIK3CA, подвергается изменению как белок p110α, так и фермент PI3K, вследствие чего у последнего неестественным образом повышается активность, позволяющая клеткам непрерывно расти и делиться. Повышенная клеточная пролиферация приводит к патологическому росту мягких тканей, костей и кровеносных сосудов [1].
Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
Синдром Клиппеля-Треноне-Вебера. Синдром Ларсена у плода
При данном синдроме отмечается сочетание больших гемангиом кожи с гипертрофией соответствующих костей и мягких тканей.
Синонимы. Синдром Клиппеля-Треноне (Klippel-Trenaunay) и синдром ангиоостео-гипертрофии.
Распространенность. Встречается редко.
Согласно предположению R. Happle парадоминантный тип наследования может наиболее удовлетворительно объяснять характер возникновения заболевания. Гетерозиготные по моногенному дефекту индивидуумы являются фенотипически нормальными. Заболевание может проявляться только тогда, когда на ранней стадии эмбриогенеза происходит дополнительная соматическая мутация нормального аллельного гена. В таких случаях эмбрион будет иметь мозаицизм с наличием клеточных линий гомозиготных или гетерозиготных по отношению к данным мутациям. Это объясняет локализованное распределение клинически обнаруживаемых дефектов.
Риск рецидива. Возможно, отсутствует, однако, учитывая аутосомно-доминантный тип наследования, в некоторых случаях риск может возрастать.
Диагностика. При ультразвуковом исследовании могут обнаруживаться водянка плода (вследствие сердечной недостаточности), с отеком конечностей и гипертрофией их мягких тканей (с превышением показателей размеров окружности над длиной), асцит, патологические гемангиоматоз-ные объемные образования внутрибрюшной локализации, а также гепатомегалия. Описан случай правильного установления диагноза с помощью трехмерной эхографии.
Патогенез. Аномалии развития капилляров, атипические формы варикоза сосудов и венозные мальформации, которые приводят к формированию ограниченных объемных образований.
Генетические нарушения. Вероятно, причиной является моногенный дефект, локализованный на длинном плече хромосомы 5q или на ее коротком плече 5р11.
Сочетанные аномалии. Заболевание может осложняться развитием синдрома Касабаха-Мерритта (Kasabach-Merritt), который сопровождается тромбоцитопенией, обусловленной потреблением тромбоцитов тканями гемангиом, а также явлениями сердечной недостаточности.
Дифференциальный диагноз. Лимфангиомы и синдром Протеуса (Proteus).
Прогноз. При обнаружении в пренатальном периоде данное заболевание обычно имеет более тяжелую форму с неблагоприятным прогнозом, особенно если оно сочетается с развитием сердечной недостаточности.
Акушерская тактика. При тяжелых формах синдрома может быть предложено прерывание беременности, в других случаях акушерская тактика ведения беременности не изменяется.
Синдром Ларсена у плода
При данном синдроме отмечаются низкий рост, множественные врожденные вывихи суставов и аномальные черты лица. Существуют две формы синдрома: летальная и нелетальная. Летальность при первой форме заболевания обусловлена гипоплазией легких.
Распространенность. Встречается редко.
Этиология. Формирование аномального коллагена, который характеризуется снижением показателя отношения е.-1/01-2 цепей для коллагена I типа.
Диагностика. Аномалии суставов с вывихами (вывих бедра, сгибание коленного сустава под углом, открытым кпереди, косорукость вследствие наличия добавочных запястных костей, косолапость и другие) могут сочетаться с ризомелическим укорочением верхних конечностей и гипоплазией малоберцовой кости и иногда с фронтальными расщелинами тел позвонков. Аномалии лица при этом синдроме включают глубокую переносицу, выступающий лоб и гипертелоризм.
Генетические нарушения. Сообщалось как об аутосомно-доминантном, так и об аутосомно-рецессивном типах наследования. Ген, в котором происходят мутации, локализуется в хромосоме Зр21.1 -р14.1.
Дифференциальный диагноз. При отсутствии семейного анамнеза у таких больных обычно устанавливают диагноз артрогипоза.
Прогноз. Своевременное выявление и обеспечение мероприятий, направленных на коррекцию трахеомаляции, легочной недостаточности и вертебральной нестабильности (в виде кифоза, обусловленного выраженной гипоплазией одного или двух тел шейных позвонков, обычно IV или V), может улучшать прогноз, хотя это бывает возможно только при наличии нелетальной формы.
Синдром ларсена у плода
Информация на сайте подлежит консультации лечащим врачом и не заменяет очной консультации с ним.
См. подробнее в пользовательском соглашении.
Синдром Эллерса-Данло тип VI, PLOD ч.м.
Синдром Элерса - Данло тип VI (EDS, СЭД, OMIM225400) - гетерогенная группа наследственных заболеваний соединительной ткани, в основе которых лежит недостаточное развитие коллагеновых структур в различных системах организма. Назван в честь двух дерматологов, Эварда Элерса и Генри Данлоса. Но ещё раньше синдром был описан Николаем Черногубовым. Проявляется патологией кожи, опорно-двигательного аппарата, сердечно-сосудистой системы, глаз. Относится к моногенным заболеваниям с различными типами наследования: аутосомно-доминантным, аутосомно-рецессивным и Х-сцепленным.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена, одного из основных белков соединительной ткани. Конечный результат каждого из механизмов формирования патологии один и тот же - уменьшение стабильности коллагенового волокна. Генерализованность клинических проявлений при синдроме Элерса - Данло обусловлена тем, что элементы поражённой соединительной ткани присутствуют практически во всех тканях и системах организма. Диагноз устанавливают на основании анамнестических сведений (задержка моторного развития, плохая заживляемость ран, склонность к кровотечениям и экхимозам, вывихи и подвывихи), характерной клинической картины (гиперэластичносгь и хрупкость кожи, гиперподвижность суставов в сочетании с патологией сердца, глаз и др.).
Согласно классификации наследственных болезней соединительной ткани, выделяют 9 форм синдрома Элерса - Данло, различающихся особенностями клинической картины и типом наследования. Характерными признаками синдрома Элерса - Данло типа VI являются аутосмно-рецессивный тип наследования, мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии. У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
У большинства больных причиной заболевания являются мутации гена PLOD1, приводящие к снижению активности фермента лизин-гидроксилазы, катализирующего образование гидроксилизина в коллагенах. Гидроксильные группы гидроксилизиновых остатков являются сайтами связывания единиц углеводов: галактозы и гликозилгалактозы, кроме того, наличие гидроксилизина необходимо для поддержания стабильности межмолекулярных коллагеновых соединений.
В основе развития патологического процесса лежит нарушение различных этапов биосинтеза коллагена — одного из основных белков соединительной ткани, приводящих к уменьшение стабильности коллагенового волокна. Элементы пораженной соединительной ткани присутствуют практически во всех тканях и системах организма, что приводит к генерализованному поражении организма при заболевании. Характерными признаками синдрома Элерса-Данло типа VI являются: мышечная гипотония, кифосколиоз, офтальмопатологии (миопия, разрывы глазного яблока, роговицы в результате минимальной травмы, спонтанная отслойка сетчатки), и другие признаки соединительно-тканной патологии.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации.
ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Интерпретация результатов исследований содержит информацию для лечащего врача и не является диагнозом. Информацию из этого раздела нельзя использовать для самодиагностики и самолечения. Точный диагноз ставит врач, используя как результаты данного обследования, так и нужную информацию из других источников: анамнеза, результатов других обследований и т.д.
- Мутация не выявлена.
- Мутация выявлена в гетерозиготном состоянии.
- Мутация выявлена в гомозиготном состоянии.
- Мутация выявлена в компаунд –гетерозиготном состоянии.
*Заполнение «анкеты молекулярно-генетического исследования» необходимо для того, чтобы врач-генетик, на основании полученных результатов, во-первых, имел бы возможность выдать пациенту максимально полное заключение и, во-вторых, сформулировать для него конкретные индивидуальные рекомендации. ИНВИТРО гарантирует конфиденциальность и неразглашение предоставляемой пациентом информации в соответствии с законодательством Российской Федерации.
Последовательность Пьера Робена - Pierre Robin sequence
Последовательность Пьера Робена [а] ( / п j ɛər р ɔː ˈ б æ̃ / ; [3] сокращенный ССН) это врожденный порок наблюдается у людей, который характеризуется лицевые аномалии. Три основные особенности: микрогнатия (аномально маленькая нижняя челюсть), что вызывает глоссоптоз (смещенный вниз или втянутый язык), что, в свою очередь, вызывает проблемы с дыханием из-за закупорки верхние дыхательные пути. Широкий U-образный волчья пасть также обычно присутствует. PRS - это не просто синдром, а скорее это последовательность - серия конкретных пороков развития, которые можно объяснить одной причиной. [4]
Содержание
Признаки и симптомы
PRS отличается необычно маленьким нижняя челюсть, смещение или втягивание языка кзади и обструкция верхних дыхательных путей. Расщелина неба (неполное закрытие неба) присутствует у большинства пациентов. Потеря слуха и трудности с речью часто связаны с PRS. [ нужна цитата ]
Причины
Механическая основа
Физические черепно-лицевые деформации PRS могут быть результатом механической проблемы, при которой ограничивается внутриутробный рост определенных структур лица или изменяется положение нижней челюсти. [4] Одна теория для этиология PRS заключается в том, что в начале первого триместра беременность, какой-то механический фактор приводит к аномальному сгибанию шеи, так что кончик нижней челюсти становится сжатым против грудинно-ключичный сустав. Это сжатие подбородка препятствует развитию тела нижней челюсти, что приводит к микрогнатии. Вогнутое пространство, образованное корпусом гипопластический нижняя челюсть слишком мала, чтобы вместить язык, который продолжает беспрепятственно расти. Поскольку больше некуда идти, основание языка смещено вниз, в результате чего кончик языка оказывается между левым и правым. небные полки. Это, в свою очередь, может привести к тому, что левая и правая небные полки не срастутся по средней линии, чтобы сформировать твердое небо. [1] Это состояние проявляется волчьей пастью. На более поздних сроках беременности (примерно от 12 до 14 недель) удлинение шеи плода снимает давление на нижнюю челюсть, позволяя ей нормально расти с этого момента. Однако при рождении нижняя челюсть все еще намного меньше (гипопластична), чем она была бы при нормальном развитии. После рождения ребенка нижняя челюсть продолжает расти, пока ребенок не достигнет зрелости. [ нужна цитата ]
Генетическая основа
Кроме того, ССБ может быть вызвано генетическое расстройство. В случае PRS, вызванного генетическим заболеванием, наследственный постулируется, но обычно это происходит из-за de-novo мутация. В частности, мутации на хромосоме 2 (возможно, на GAD1 ген), хромосома 4, хромосома 11 (возможно, на PVRL1 ген) или хромосома 17 (возможно, на SOX9 ген или KCNJ2 ген) были вовлечены в PRS. [5] Некоторые данные свидетельствуют о том, что генетическая дисрегуляция гена SOX9 (кодирующего фактор транскрипции SOX-9) и / или гена KCNJ2 (который кодирует Kir2.1 входной выпрямительный калиевый канал ) нарушает развитие определенных структур лица, что может привести к ССН. [6] [7]
PRS может возникать изолированно, но часто является частью основного заболевания или синдрома. [8] Расстройства, связанные с ССН, включают: Синдром Стиклера, Синдром ДиДжорджи, алкогольный синдром плода, Синдром Тричера Коллинза, и Синдром Патау. [9]
Диагностика
PRS обычно диагностируется клинически вскоре после рождения. У младенца обычно возникают проблемы с дыханием, особенно когда лежа на спине. Небная расщелина часто имеет U-образную форму и шире, чем у других людей с волчьей пастью. [ нужна цитата ]
Управление
Цели лечения младенцев с PRS сосредоточены на дыхании и кормлении, а также на оптимизации роста и питания, несмотря на предрасположенность к затрудненному дыханию. Если есть признаки обструкции дыхательных путей (фырканье, апноэ, затрудненное дыхание или снижение уровня кислорода), тогда младенца следует поместить в положение лежа на боку или на животе, что помогает выдвинуть основание языка вперед у многих детей. Одно исследование 60 младенцев с PRS показало, что 63% младенцев ответили на положение лежа. [10] Пятьдесят три процента младенцев в этом исследовании нуждались в некоторой форме помощи при кормлении, будь то кормление через назогастральный зонд или гастростомический зонд (кормление непосредственно в желудок). В отдельном исследовании 115 детей с клиническим диагнозом PRS, проходивших лечение в двух разных больницах Бостона, [11] В 56% случаев респираторный дистресс успешно справился без операции (либо путем положения лежа на животе, кратковременной интубации или установки носоглоточного дыхательного пути). В этом исследовании питание через гастростомический зонд применялось у 42% этих младенцев из-за трудностей с кормлением. [ нужна цитата ]
Гастроэзофагеальный рефлюкс (ГЭРБ), по-видимому, чаще встречается у детей с ССН. [12] Поскольку рефлюкс кислого содержимого в задней части глотки и верхних дыхательных путях может усилить симптомы PRS, в частности, за счет ухудшения обструкции дыхательных путей, важно максимально эффективно лечить GER у детей с PRS и симптомами рефлюкса. Лечение может включать вертикальное положение на клине (если ребенок находится в положении лежа, может потребоваться подтяжка), небольшие и частые кормления (для минимизации рвоты) и / или фармакотерапия (например, ингибиторы протонной помпы). [ нужна цитата ]
В носоглотке канюляция (или размещение носоглоточного дыхательного пути или трубки), младенцу надевают хирургическую трубку с тупым концом (или эндотрахеальную трубку, подогнанную к ребенку), которая помещается под прямую визуализацию с помощью ларингоскопа и вводится в нос и вниз по глотке (или горлу), заканчиваясь чуть выше голосовых связок. Хирургические нити, проходящие через отверстия во внешнем конце трубки, прикрепляются к щеке с помощью специального кожевенного адгезива, называемого стомагезивом, который также наматывается вокруг внешнего конца трубки (но не поверх отверстия на конце. ), чтобы трубка оставалась на месте. Эта трубка или канюля, которая сама по себе действует как дыхательные пути, в первую очередь действует как своего рода «шина», которая поддерживает проходимость дыхательных путей, удерживая язык от падения на заднюю стенку глотки и закупорки дыхательных путей, тем самым предотвращая обструкцию дыхательных путей, гипоксию. и асфиксия. Назофарингеальные дыхательные пути доступны не во всех центрах; тем не менее, если это возможно, назофарингеальную канюляцию следует отдать предпочтение перед другими видами лечения, упомянутыми в этой статье, так как это гораздо менее инвазивно; это позволяет ребенку кормить без дальнейшего введения назогастрального зонда. Это лечение можно использовать в течение нескольких месяцев, пока челюсть не вырастет достаточно, чтобы язык принял более нормальное положение во рту и дыхательных путях (при рождении челюсти у некоторых младенцев настолько недоразвиты, что только кончик языка может быть видно при осмотре в горле). Некоторые учреждения выписывают ребенка домой с установленным носоглоточным зондом. [13]
Дистракционный остеогенез (DO), также называемый «дистракцией нижней челюсти», можно использовать для коррекции аномального маленького размера одной или обеих челюстей, наблюдаемого у пациентов с PRS. Увеличение нижней челюсти выдвигает язык вперед, предотвращая закупорку верхних дыхательных путей. Процесс DO начинается с предоперационной оценки. Врачи используют трехмерную визуализацию для выявления частей лицевого скелета пациента, которые необходимо изменить, и определения степени и направления отвлечения. Затем они могут выбрать наиболее подходящее отвлекающее устройство или иногда могут изготовить специальные устройства. По возможности используются внутриротовые устройства.
Операция DO начинается с остеотомии (хирургического разделения или рассечения кости), после чего устройство для отвлечения внимания помещается под кожу и поперек остеотомии. Через несколько дней два конца кости очень постепенно разъединяются за счет постоянной регулировки устройства, которую родители вносят в устройство дома. Регулировка осуществляется поворотом небольшого винта, который выступает через кожу, обычно со скоростью 1 мм в день. Это постепенное отвлечение приводит к образованию новой кости между двумя концами. После завершения процесса остеотомии дают зажить в течение шести-восьми недель. Затем выполняется небольшая вторая операция по удалению устройства.
Расщелина неба обычно лечит в возрасте от 6,5 месяцев до 2 лет пластическим хирургом, челюстно-лицевой хирург, или оториноларинголог (ЛОР-хирург). Во многих центрах сейчас есть бригада специалистов по лечению заячьей губы и неба, состоящая из этих специалистов, а также координатор, логопед, ортодонт, иногда психолог или другой специалист в области психического здоровья, аудиолог и медперсонал. В глоссоптоз и микрогнатизм обычно не требуют хирургического вмешательства, так как они улучшаются до некоторой степени без посторонней помощи, хотя нижнечелюстная дуга остается значительно меньше средней. В некоторых случаях требуется отвлечение челюсти для облегчения дыхания и кормления. Прикрепление губ к языку выполняется в некоторых центрах, хотя его эффективность недавно была поставлена под сомнение.
Расщелина неба (PRS или нет) мешает людям произносить звуки речи, что может быть связано с физической природой волчьей пасти или с потерей слуха, связанной с этим заболеванием. [14] Обычно именно поэтому с пациентом привлекается речевой патолог и / или аудиолог. Слух должен регулярно проверяться аудиологом, и его можно лечить с помощью усилителей слуха, например, слуховых аппаратов. Поскольку выпот в среднем ухе обнаруживается у многих пациентов с PRS, тимпаностомические (вентиляционные) трубки часто являются вариантом лечения. [15]
Одно исследование с участием детей показало, что пациенты с PRS чаще всего демонстрировали умеренную и тяжелую потерю слуха. [15] Планиграфы височных костей у этих пациентов показали слаборазвитую пневматизацию сосцевидный отросток кости у всех пациентов с PRS и у большинства пациентов с расщелиной неба (без PRS). [15] У пациентов с PRS аномалий анатомии внутреннего или среднего уха не было. [15]
Прогноз
Дети, пораженные PRS, обычно достигают полного развития и роста. Тем не менее, на международном уровне было установлено, что дети с ССН часто немного ниже среднего роста, что вызывает опасения по поводу неполного развития из-за хронических заболеваний. гипоксия связанные с обструкцией верхних дыхательных путей, а также с недостатком питания из-за трудностей с кормлением в раннем возрасте или развития отвращения к ротовой полости. Однако общий прогноз неплохой, если начальные трудности с дыханием и кормлением будут преодолены в младенчестве. Большинство младенцев PRS растут и ведут здоровую и нормальную взрослую жизнь.
Самыми серьезными проблемами со здоровьем являются затруднения дыхания и кормления. Больные младенцы очень часто нуждаются в помощи при кормлении, например, им необходимо оставаться в боковом (на боку) или животном (на животе) положении, которое помогает вывести язык вперед и открыть дыхательные пути. Младенцам с волчьей пастью потребуется специальное устройство для кормления заячьей губы (например, Хаберман Фидер ). Младенцы, которые не могут получать достаточно калорий через рот для обеспечения роста, могут нуждаться в добавках с назогастральный зонд. Это связано с трудностью создания вакуума в полости рта, связанной с волчья пасть, а также затрудненное дыхание, связанное с задним положением языка. Учитывая трудности с дыханием, с которыми сталкиваются некоторые дети с PRS, им может потребоваться больше калорий для роста (поскольку работа с дыханием в чем-то похожа на упражнения для младенца). Младенцы с умеренным или тяжелым поражением могут иногда нуждаться в носоглотке. канюляция, или размещение носоглоточная трубка чтобы обойти обструкцию дыхательных путей у основания языка. в некоторых местах детей выписывают домой с назофарингеальной трубкой на определенный период времени, а родителей учат, как правильно поддерживать трубку. Иногда эндотрахеальный интубация или же трахеостомия может быть показан для преодоления обструкции верхних дыхательных путей. В некоторых центрах язык губ выполняется, чтобы выдвинуть язык вперед, эффективно открывая дыхательные пути. Отвлечение нижней челюсти может быть эффективным путем перемещения челюсти вперед, чтобы преодолеть обструкцию верхних дыхательных путей, вызванную задним расположением языка. Учитывая, что у некоторых детей с PRS будет Синдром Стиклера, важно, чтобы дети с PRS были осмотрены окулистом или офтальмологом. Поскольку отслоение сетчатки, которое иногда сопровождает синдром Стиклера, является основной причиной слепоты у детей, очень важно распознать этот диагноз. [ нужна цитата ]
Эпидемиология
Распространенность PRS оценивается в 1 из 8 500–14 000 человек. [2]
Потеря слуха чаще встречается у людей с волчьей пастью, чем у пациентов без волчьей пасти. Одно исследование показало, что потеря слуха при PRS составляет в среднем 83% по сравнению с 60% людей с расщелиной без PRS. [16] Другое исследование с участием детей показало, что потеря слуха чаще выявлялась при PRS (73,3%) по сравнению с пациентами с расщелиной и без PRS (58,1%). [15] Потеря слуха при PRS обычно представляет собой двустороннюю кондуктивную потерю (затрагивающую внешнюю / среднюю часть уха). [16]
История
Состояние названо в честь французского хирурга-стоматолога. Пьер Робен. [17] [18]
Похоже, что Ноэль Роза, один из самых известных и влиятельных художников в истории Бразильская музыка, имел ССБ. [19]
Читайте также: