Синдром Крузона. Черепно-лицевой дизостоз
Добавил пользователь Валентин П. Обновлено: 27.01.2026
Черепно-лицевой дизостоз, или синдром Крузона является генетически обусловленной врожденной аномалией развития, которая характеризуется ранним закрытием черепных швов (синостозированием черепа), окулярным гипертелоризмом, экзофтальмом и гипоплазией (недоразвитием) части лица с наличием относительного нижнечелюстного прогнатизма.
Точные причины синдрома Крузона специалистами пока еще не выявлены, однако предположительно в его возникновении играют роль такие факторы, как:
- отягощенная наследственность (аналогичное заболевание у родителей);
- выявление у здоровых родителей гена, вызывающего черепно-лицевой дизостоз;
- пожилой возраст будущего отца на момент зачатия.
Внешне дети, страдающие синдромом Крузона, имеют весьма специфический вид.
Во-первых, у них преждевременно, еще до того, как полностью сформируется мозг, окостеневают швы, которые соединяют кости лица и черепной коробки. Из-за этого череп и лицо приобретают неправильную форму: задняя и верхняя части головы, а также лоб и виски уплощаются, средняя часть лица более вытянута, чем в норме, и при этом имеет относительно небольшие размеры, а нос приобретает остроконечную, клювовидную форму. Нижняя челюсть на этом фоне выглядит массивно и значительно выступает вперед (нижнечелюстной прогнатизм). Кроме того, обычно наблюдается смещение зубов и окулярный гипертелоризм − несоразмерно большое расстояние между глазницами. Может обнаруживаться волчья пасть или аномально узкое небо.
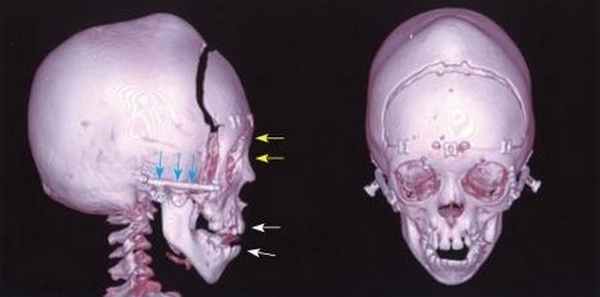
Однако внешними дефектами рассматриваемая врожденная патология, к сожалению, не ограничивается. Вследствие того, что ноздри у ребенка с синдромом Крузона сжаты, поток воздуха через нос недостаточен. Отклонения в формировании внутреннего уха приводят к потере слуха. Возможно присоединение гидроцефалии (водянки мозга) со всеми вытекающими последствиями. Нередко такие дети жалуются на головные боли, звон в ушах и головокружения (синдром Меньера). У них могут быть расстройства зрения, косоглазие (страбизм), нистагм (колебательные непроизвольные движения глаз). И, наконец, страдает умственное развитие: интеллектуальный дефект может быть разной степени выраженности.
Диагностика черепно-лицевого дизостоза предполагает комплексное обследование, которое непременно включает в себя рентгенографию, компьютерную и/или магнитно-резонансную томографию. Для окончательного подтверждения диагноза проводится генетическое тестирование. Чтобы составить полную клиническую картину, доктора назначают различные дополнительные исследования.
Тактика лечения синдрома Крузона всегда индивидуальна и зависит от степени тяжести заболевания и общего самочувствия пациента. Нужно понимать, что целиком исцелиться от этого недуга пока еще невозможно, современные методы лечения способны только устранить или компенсировать уже имеющиеся симптомы: костные деформации, нарушения слуха и зрения, искривленный прикус.
Чем раньше, начато адекватное лечение, тем, соответственно лучше прогноз и выше шансы на то, что удастся избежать тяжелых осложнений в виде полной потери зрения, изъязвлений роговицы, сохранения умственного дефекта на всю жизнь, социальной и трудовой дезадаптации и т.д.
Хирургические вмешательства при челюстно-лицевом дизостозе следует выполнять как можно раньше. Основные цели операций при этом заболевании:
Оформить заявку на лечение
Больница Красного Креста в Касселе
В отделении ЧЛХ и дентальной имплантологии больницы Красного Креста в Касселе проводится лечение пороков развития, приобретенных заболеваний и повреждений височно-нижнечелюстного сустава, включая артроз, артриты, анкилоз, вывихи и подвывихи, опухоли.
Челюстно-лицевая хирургия в Израиле
В ведущем государственном медицинском комплексе страны − в тель-авивском медцентре им. Сураски функционирует великолепно оснащенное отделение ЧЛХ и дентальной имплантологии, где опытнейшие специалисты занимаются вопросами диагностики и лечения широкого спектра профильных заболеваний и патологий.
Синдром Крузона. Черепно-лицевой дизостоз
Синдром Крузона. Черепно-лицевой дизостоз
Черепно-лицевой дизостоз характеризуется преждевременным синостозированием черепа, гипоплазией средней части лица с относительным нижнечелюстным прогматизмом, экзофтальмом и гипертелоризмом.
Клинические данные. Данные осмотра. Клинические изменения ограничены черепом и лицом. Нос может иметь несколько необычную форму в связи с вертикальной гипоплазией средней трети лица. Часто наблюдаются относительный нижнечелюстной прогматизм, опущенная нижняя губа, короткая верхняя губа и торчащий язык. Постоянной чертой является неполное смыкание зубов.
Костная система. Форма черепа зависит от того, какие швы вовлечены в процесс. Могут наблюдаться брахицефалия, скафоцефалия, тригоноцефалия и изредка аномалия Kleeblattschadel. Обычно отчетливо пальпируются утолщенные края швов. Около брегмы (места соединения венечного и стрелковидного швов черепа) могут наблюдаться экзостозы (Bertelsen). Преждевременный краниосиностоз варьирует по времени начала, но чаще всего он развивается в течение первого года и заканчивается к 2—3 годам жизни. В некоторых случаях синостоза может не быть до 10-летнего возраста.
Орган зрения. Экзофтальм является вторичным. Он обусловлен уменьшением глубины орбит. Отмечаются расходящееся косоглазие и нистагм. Постоянной чертой является гипертелоризм. Bertelsen отметил у 80% больных с черепно-лицевым дизостозом поражение зрительного нерва. Иногда могут также наблюдаться спонтанный вывих глазного яблока, мегалокорнеа, эктопия хрусталика, колобома радужной оболочки и эктопия зрачка.
Орган слуха. Boedts установил, что у 1/3 больных с синдромом Крузона имеется глухота, чаще проводящего типа. При хирургическом и посмертном исследовании он обнаружил деформацию слуховых косточек и фиксацию стремени в овальном окне. Aubrey, Nager и de Reynier, Wiegand и Baldwin отметили двустороннюю атрезию наружного слухового прохода и смешанную или проводящую глухоту. Schurmans и Hariga выявили снижение костной проводимости, отсутствие восстановления и посредством томографии деформацию внутреннего слухового прохода.
Вестибулярная система. Aubrey описал нормальную вестибулярную функцию.
Лабораторные данные. Рентгенограммы. Чаще всего преждевременным синостозированием охвачены коронарный, сагиттальный и лямбдовидный швы. Кроме этого, могут наблюдаться и другие рентгенологические находки, такие, как пальцевые вдавления, уплощение глазниц, базилярный кифоз, расширение ямки гипофиза и маленькие параназальные синусы (Bertelsen). Как отметили Schurmans и Hariga, при томографическом исследовании наблюдается деформация внутреннего слухового прохода. Рентгенография показала нормально развитые лабиринты (Wiegand). Изящное томографическое исследование височной кости, проведенное Terrahe, обнаружило наружную ротацию каменистой части пирамиды, вторичную по отношению к дисплазии основания черепа.
В результате этого слуховые каналы имели косое направление, неправильным было и направление лицевого нерва, кроме того, наблюдались гиперостозы. Terrahe подчеркнул, что первичными изменениями являлись фиксация слуховых косточек со скоплением костных масс внутри барабанной полости, аномалии слуховых косточек и закрытие овального окна.
Патология. Томография височной кости обнаружила стеноз или атрезию наружного слухового прохода, отсутствие барабанной полости, деформацию стремени и костное синостозирование его с мысом, анкилоз молоточка с наружной стенкой верхней части барабанной полости, искривление и сужение среднего уха, а также воздухоносных пространств сосцевидного отростка (Nager, de Reynier). Baldwin обнаружил также недоразвитие периостальной части лабиринта. Молоточек и наковальня были анкилознрованы к боковой стенке пиши эпитимпанума. Ножки стремени были косо направлены к основанию, сустав между стременем и наковальней соприкасался с мысом. И круглое, и овальное окна были сужены. Барабанная перепонка отсутствовала.
Наследственность. Наследование аутосомно-доминантное с полной пенетрантностью (Schiller, Vulliamy, Normandale). Почти 1/3 случаев — новые мутации.
Диагноз. Черепно-лицевой дизостоз должен быть отграничен от изолированного крапиостеноза, синдрома Апера, синдрома Пфейффера и синдрома Сетре—Чотцена (Gorlin et al.).
Лечение. Могут быть произведены косметическая и функциональная коррекция посредством удаления по частям преждевременно синостозированных швов и хирургическое создание блефарофимоза с целью предотвращения вывиха глазного яблока. Tessier описал превосходную радикальную хирургическую операцию для исправления деформаций лица.
Прогноз. С возрастом часто становится невозможным бинокулярное зрение и отмечается расходящееся, альтернирующее и содружественное косоглазие. Нередко наблюдается атрофия зрительного нерва и потеря зрения. Иногда встречается полное выпадение одного или обоих глазных яблок, происходящее вследствие уплощения глазниц. Преждевременное синостозирование швов ведет к образованию экзостозов в области брегмы. С возрастом гипоплазия средней части лица может становиться все более выраженной при нормальном росте нижней челюсти.
Выводы. Характеристика синдрома Крузона включает:
1) аутосомно-доминантное наследование;
2) преждевременный варьирующий краниосиностоз;
3) гппертелоризм, уплощение глазниц и экзофтальм;
4) клювовидный нос;
5) гипоплазию верхней челюсти с относительным нижнечелюстным прогнатизмом;
6) иногда двустороннюю атрезию наружных слуховых проходов, аномалии слуховых косточек и смешанную глухоту.
Глазные проявления у ребенка с синдромом Крузона (cлучай из практики)
Синдром Крузона – черепно-лицевой дизостоз или черепно-лицевая дисплазия (Dysostosis craniofacialis hereditaria). Это редко встречающееся генетическое заболевание, проявляющееся неправильным формированием мозгового и лицевого черепа, вследствие преждевременного заращения швов (между костями лица и черепа). Частота заболевания оценивается как 16,5: 1000000 новорожденных [3, 5].
В 1912 г. впервые данный синдром был описан французским врачом L.E. Ostave Crouzon [2]. Заболевание расценивается, как семейно-наследственная аномалия черепа (его лицевой и мозговой частей) типа оксицефалии с доминантной передачей и высокой пенетрантностью [6]. Генетическое нарушение связано с изменением гена FGF R2 в десятой хромосоме [6]. Факторами, вызывающими мутацию гена, являются: рентгеновское излучение, различные интоксикации, заболевание краснухой матери во время беременности и др. Одинаково часто синдром Крузона наблюдается у мужчин и женщин.
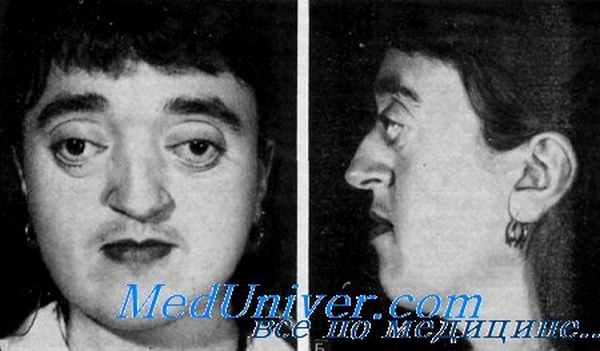
Общими симптомами черепно-лицевого дизостоза Крузона является деформация черепа и лица: широко расставленные глаза, дивергирующее косоглазие, экзофтальм, недоразвитая верхняя челюсть, выступающие нижняя губа и нижняя челюсть, расщелина неба и язычка, клювовидный нос («клюв попугая»), редкие шиповидные зубы. Характерна задержка умственного развития с поздним развитием речи и снижением интеллекта, возникновение эпилептических припадков. Описаны двухфаланговые пальцы, полидактилия, параличи черепно-мозговых нервов, гипофункция гипофиза с адипозо-генитальной дистрофией [4].
Глазные проявления, характерные для синдрома Крузона, заключаются в экзофтальме (чаще двустороннем), уменьшении объема орбит, связанном с их недоразвитием, что часто приводит к вывиху глазных яблок; расходящемся косоглазии, гипертелоризме. Часто встречаются астигматизм, нистагм, подвывих хрусталика, глаукома, катаракта. На глазном дне нередко диагностируются застойные диски зрительного нерва в связи с повышением внутричерепного давления [1].
Для диагностики заболевания рекомендуется проведение рентгенографии черепа, компьютерной и магнитно-резонансной томографии. Необходима консультация невролога или нейрохирурга и генетическое обследование. Проводится хирургическое лечение с целью декомпрессии (снижения внутричерепного давления), коррекция костно-мышечных деформаций, исправляющих форму лица [5].
Представляем клинический случай синдрома Крузона. Ребенок У., в возрасте 2 мес. находился на лечении в нейрохирургическом отделении Республиканской детской клинической больницы г. Уфы с диагнозом: синдром Крузона, гипертензионно-гидроцефальный синдром, бронхопневмония. Из анамнеза: ребенок родился от первой беременности. Возраст отца – 40 лет (это второй для него брак), возраст матери – 35 лет. Как известно, одним из факторов риска развития синдрома Крузона является весьма зрелый или пожилой возраст отца на момент зачатия [2]. Генетическая консультация до рождения ребенка не проводилась, т.к., со слов родителей, наследственность не была отягощена. В частности, у отца от первого брака имеются здоровые дети.
Необходимость в консультации офтальмолога возникла в связи с развитием у ребенка в стационаре спонтанного вывиха правого глазного яблока.
При осмотре ребенок беспокойный, взгляд не фиксирует, имеется заметная асимметрия лицевого черепа с уменьшением орбит, особенно правой, гипертелоризм. Со стороны правого глаза наблюдается выраженный экзофтальм с вывихом глазного яблока из орбиты. Отмечаются также несмыкание век, расходящееся косоглазие, умеренная иньекция сосудов коньюнктивы глазного яблока, небольшое отделяемое слизистого характера в нижнем коньюнктивальном своде. Имеются явления ксероза роговицы и коньюнктивы (отсутствие глянцевого блеска, легкое помутнение роговицы и т.п.) передняя камера средней глубины, влага ее прозрачная, зрачок центрирован, рефлекс с глазного дна ослаблен из-за ксеротического изменения роговицы. Левый глаз: умеренный экзофтальм, неполное смыкание век, расходящееся косоглазие, легкая иньекция коньюнктивальных сосудов, незначительное слизистое отделяемое, роговица достаточно прозрачная, передняя камера средней глубины, влага прозрачная, зрачок в центре, глубжележащие среды прозрачные, рефлекс с глазного дна розовый. Офтальмоскопия оказалась затруднена из-за беспокойного состояния ребенка.
После инстилляций анестетика, антибактериальных капель и аппликации корнеопротектора было проведено вправление правого глазного яблока в орбиту с наложением фиксирующей повязки. При повторном осмотре ребенка на следующий день отмечено заметное улучшение состояния роговицы и конъюнктивы (уменьшение ксеротических изменений), при сохранении, однако, тенденции к вывиху правого глазного яблока при беспокойстве ребенка. Поэтому было принято решение о проведении блефарорафии под местным обезболиванием.
В итоге продолжительная (в течение месяца) блефарорафия способствовала предотвращению повторных вывихов глазного яблока и дальнейшему ксеротическому повреждению глазной поверхности. Кроме того, она позволила подготовить и провести успешное вентрикулоперикардиальное шунтирование с целью снижения внутричерепного давления. При осмотре ребенка через 2 мес. после операции отмечено улучшение общего соматического состояния ребенка и его офтальмологического статуса (с сохранением умеренного экзофтальма, неполного смыкания век, незначительным ксерозом роговицы на фоне постоянной слезозаместительной терапии и отсутствием тенденции к повторному вывиху глазного яблока из орбиты).
Выводы. Синдром Крузона – редкое наследственное заболевание, связанное с развитием деформаций лицевого скелета, что обуславливает развитие экзофтальма, роговично-коньюнктивального ксероза и может привести к спонтанному вывиху глазных яблок из орбит. В приведенном случае своевременно выполненная временная блефарорафия позволила избежать повторного вывиха глазного яблока, предотвратив тем самым серьезные ксеротические изменения со стороны роговицы, и успешно провести жизненно необходимое хирургическое лечение, чтобы избежать тяжелых осложнений, угрожающих здоровью пациента.
Черепно-лицевой дизостоз Крузона
Черепно-лицевой дизостоз Крузона – это наследственное заболевание, проявляющееся неправильным формированием мозгового и лицевого черепа вследствие преждевременного заращения швов (в основном венечного, отделяющего лобную кость от теменных и височных).
Симптомы черепно-лицевого дизостоза Крузона
- Характерная деформация черепа и лица:
- широко расставленные глаза;
- расходящееся косоглазие (глаза направлены в разные стороны);
- экзофтальм (выстояние глазных яблок) — глаза словно выпучены;
- короткая верхняя губа, недоразвитая верхняя челюсть;
- выступающие нижняя губа и нижняя челюсть.
- клювовидный нос (“ клюв попугая”);
- редкие шиповидные зубы.
Причины
- Генетическое нарушение: нарушение функционирования гена FGFR2 в десятой хромосоме, результатом чего является внутриутробное или раннее (в первый год жизни) сращение венечных швов (отделяют лобную кость от височных и теменных костей) и недоразвитие костей лица (неглубокие широко расставленные глазницы, выступающая нижняя челюсть, недоразвитая верхняя челюсть).
- Тип наследования – аутосомно-доминантный, то есть в семье, где один из родителей имеет синдром Крузона, вероятность рождения больного ребенка составляет от 50 до 100%.
LookMedBook напоминает: что данный материал размещен исключительно в ознакомительных целях и не заменяет консультацию врача!
Врач невролог поможет при лечении заболевания
Диагностика
- Анализ жалоб и анамнеза заболевания: были ли в семье случаи подобного заболевания (с характерными особенностями лица (широко расставленные, выпученные глаза, выступающий подбородок) и умственными нарушениями).
- Рентгенография черепа: оценка формы черепа и наличия сращений швов черепа.
- КТ (компьютерная томография) и МРТ (магнитно-резонансная томография) головы: оценка формы черепа и наличия сращений швов черепа. Кроме того, у людей с синдромом Крузона отмечается опущение миндалин мозжечка (из-за уменьшения объема задней черепной ямки).
- Генетическое обследование: поиск мутаций в гене FGFR2 десятой хромосомы.
- Возможна также консультация нейрохирурга, генетика.
Лечение черепно-лицевого дизостоза Крузона
- декомпрессия (снижения давления внутри черепа) — для этого раскрываются рано заращенные швы черепа;
- исправление костно-мышечных деформаций — с помощью специальных устройств исправляется форма лица (глазницы, выступающая нижняя челюсть).
Осложнения и последствия
- Потеря зрения: из-за длительного повышения внутричерепного давления зрительный нерв сдавливается, и происходит его атрофия (истончение волокон и нарушение функции).
- Язвы роговицы: из-за выстояния глазных яблок из глазниц невозможно их полное закрытие, роговица полностью не прикрывается веками, сохнет, что приводит к образованию на ней дефектов – язв.
- Сохранение умственного дефекта при отсутствии своевременного хирургического лечения.
- Нарушение социальной и трудовой адаптации: умственный дефект в сочетании с косметическими особенностями лица (широко расставленные выпученные глаза, выступающая нижняя челюсть) может нарушать взаимодействие больного с другими членами общества.
Профилактика черепно-лицевого дизостоза Крузона
- Проводить профилактику развития синдрома Крузона невозможно в связи с наследственным характером этого заболевания.
- В семьях, где уже были случаи этого заболевания, целесообразно проводить генетическое обследование (поиск мутаций в гене FGFR2).
ИНФОРМАЦИЯ ДЛЯ ОЗНАКОМЛЕНИЯ
Необходима консультация с врачом
Дяченко Алексей Олегович, врач-невролог.
Певцова Анастасия Владимировна, врач-методист, акушер-гинеколог, медицинский редактор.
Подоляк Анжелика Алексеевна, редактор.
Что делать при черепно-лицевом дизостозе Крузона?
- Выбрать подходящего врача невролог
- Сдать анализы
- Получить от врача схему лечения
- Выполнить все рекомендации
Синдромом Крузона
Синдромом Крузона называют аутосомно-доминантное заболевание, которое проявляется врожденным окостенением швов черепа ребенка и характерными изменениями в чертах лица. Второе название патологии – черепно-лицевой дизостоз.
Синдром Крузона сопровождается деформацией черепа, аномально большим расстоянием между парными органами, пучеглазием и неправильным развитием средней трети лица.
Этиология:
Заболевание относится к группе синдромальных краниосиностозов, но отличается от остальных форм этой группы отсутствием грубых пороков развития ступней и кистей рук. Синдромом Крузона страдает один новорожденный на 65 тысяч малышей. Заболевание спровоцировано генетическими мутациями. «Поломка» происходит в гене FGFR2, расположенном на 10-ой хромосоме.
Ген FGFR2 кодирует рецепторы фактора роста фибробластов-2. Он содержит 20 экзонов. Факторы роста фибробластов – белки, связывающие гепарин. Эти вещества играют ключевую роль в процессе пролиферации и дифференциации клеток в момент эмбрионального развития плода. Кроме того, факторы, кодируемые геном, в котором происходит «поломка» на фоне патологии, принимают участие в образовании новых сосудов и заживлении ран.
Существует отдельная форма заболевания – синдром Крузона с черным акантозом. Причины такой формы генетической мутации немного отличаются. В этом случае «поломка» происходит на 4-ой хромосоме в гене FGFR3, кодирующим другую группу факторов роста фибробластов. Механизм развития схож, единственным отличием становится появление участков утолщения эпидермиса кожи и разрастание родинок.
Проявления болезни
Наличие симптомов синдрома Крузона врачи могут определить сразу после появления малыша на свет, однако выраженными они становятся после достижения 3-летнего возраста. Наиболее явный симптом – раннее зарастание швов черепа ребенка с его выраженной деформацией. Порок затрагивает коронарный, сагиттальный и лямбдовидный швы, мешая костям черепа нормально расти и развиваться.
В скором времени отчетливо становятся видны и другие симптомы заболевания:
большое расстояние между глазами;
нарушение прикуса вследствие патологии смыкания зубных рядов (нижние зубы имеют заднее расположение по отношению к верхним);
изменение формы носа;
уменьшение размеров глазницы, что подчеркивает пучеглазие;
низкая локализация наружного слухового прохода.
Пациент 6 лет, страдающий синдромом Крузона. Определяется экзофтальм, гипоплазия средней зоны лица. На КТ визуализируется панкраниосиностоз и открытый прикус.
В некоторых случаях симптомом может быть сращение двух или более пальцев на кистях или стопах, но в этом случае синдром Крузона нужно дифференцировать с другими краниосиностозами.
У некоторых детей, имеющих генетическую патологию, может наблюдаться частичное или полное заращение соединительнотканных элементов или костей полости носа, а также скопление жидкости в головном мозге, затрудняющее лечение пациентов.
Из-за того, что швы окостенели, а череп продолжает расти, форма головы больного ребенка постоянно меняется. На поверхности костей могут появляться доброкачественные костно-хрящевые наросты. Подобные изменения провоцируют усугубление поражения зрительного анализатора. Пучеглазие продолжает увеличиваться, возникает значительное косоглазие. При тяжелых формах синдрома Крузона возможен даже вывих глазных яблок.
Заболевание затрагивает и слуховой анализатор, что проявляется в прогрессирующем снижении уровня слуха вплоть до абсолютной глухоты. Практически все дети с синдромом Крузона имеют умственные отклонения, судорожные приступы, головные боли.Синдром Крузона с черным акантозом характеризуется теми же изменениями со стороны черепа, швов и костей, однако эта форма генетических нарушений имеет более тяжелое течение и значительное количество осложнений.
полное сращение костных элементов носовой полости у 50% пациентов;
разрастание бородавок и родинок;
гиперкератоз на значительных участках кожи;
появление грубых рубцов и шрамов после малейших повреждений.
Проявления при синдроме Крузона затрагивают область передней брюшной стенки, шею, коленные и локтевые сгибы, лицо.
Диагностика синдрома Крузона
Наличие синдрома Крузона у ребенка можно определить еще на этапе его внутриутробного развития, сразу после появления на свет или на протяжении первых лет жизни. Диагностика синдрома Крузона включает следующие моменты:
оценка психического состояния;
исследование функций слуха;
На рентгене черепа больного определяют раннее окостенение швов, наличие костно-хрящевых наростов, уплощение формы глазниц.
Лечение синдрома Крузона
Оказание помощи больным детям может начинаться уже в первые дни после рождения, что связано с расстройствами дыхания на фоне врожденной атрезии хоан. Такие случаи требуют проведения трахеостомии. Иногда лечение синдрома Крузона начинают в первые полгода жизни. Специфической терапии, которая могла бы устранить заболевание, не существует. Проводится исключительно поддерживающее лечение.
Главное хирургическое вмешательство, которое показано больным детям с синдромом Крузона, – краниопластика. Такие операции требуют повторных проведений, поскольку малыш растет, а соответственно, его череп тоже.
В случае патологического скопления жидкости в системе головного мозга, устанавливается вентрикулоперитонеальная шунтирующая система, позволяющая нормализовать показатели давления внутри черепа. Гипоплазию верхней челюсти устраняют примерно на 7-9 году жизни ребенка. До этого периода нарушение функции дыхания компенсируют следующими методами лечения:
дыхание под повышенным давлением;
На современных этапах лечение синдрома Крузона предполагает использование аппаратов Илизарова. Они позволяют одновременно выдвинуть верхнюю челюсть вперед, что обеспечит функцию дыхания, и увеличить объем черепа, выдвинув лобную кость.
Синдром Крузона имеет неблагоприятный исход , что связано с развитием чрезмерного экзофтальма, глухоты, невозможностью адаптироваться к условиям современного социума. Однако известны случаи, когда больные люди доживали до преклонного возраста.
Читайте также: