Синдром персистирующих мюллеровых протоков. Дисгенезия гонад
Добавил пользователь Евгений Кузнецов Обновлено: 29.01.2026
В работе представлены варианты врожденного первичного гипогонадизма у мальчиков, показаны основные виды генетических нарушений полового развития, отмечены основные критерии дифференциальной диагностики различных вариантов врожденного первичного гипогонадизма, основанные на данных кариотипа, гормональных показателей, фенотипических особенностей при наиболее часто встречающихся нозологических формах данной патологии.
Clinical variants of congenital primary hypogonadism at boys
The paper presents variants of congenital primary hypogonadism at boys, shows the main types of genetic disorders of sexual development, marked the basic criteria of differential diagnosis of different variants of congenital primary hypogonadism based on data the karyotype, hormonal parameters, phenotypic characteristics with the most frequent nosological forms of this disease.
К гипогонадизму относят комплекс симптомов, отражающих недостаточность стероидогенной функции половых желез. Различают первичный (гипергонадотропный) и вторичный (гипогонадотропный) гипогонадизм. Первичные нарушения протекают с компенсаторной гиперсекрецией гонадотропных гормонов, вторичные — в условиях дефицита гонадотропной стимуляции. Такое разделение, основанное на содержании тестостерона и гонадотропных гормонов в крови, весьма условно, ибо в обоих случаях клинические проявления гипогонадизма не различаются. Вместе с тем картина гормональной недостаточности яичек, вплоть до формирования женского фенотипа у мужчины, может быть при нормальном биосинтезе андрогенов, если их рецепция нарушена. Дефицит гонадотропных гормонов может быть не только при заболеваниях гипофизарного уровня, но быть следствием избыточной продукции половых стероидов вне гонад либо экзогенного происхождения. В то же время разделение на первичный и вторичный гипогонадизм удобно в плане топической диагностики патологии.
Данные о распространенности синдрома гипогонадизма основаны на частоте выявления основных причин его развития. Так, анорхию наблюдают у 3-5% мальчиков с отсутствием яичек в мошонке. Синдром Клайнфельтера диагностируют у 1 из 500 мальчиков, а синдром Каллмана — у 1 из 5000. Гипогонадизм, связанный с другими причинами, встречается гораздо реже.
Классификация
В зависимости от уровня поражения гипоталамо-гипофизарной системы, различают следующие формы заболевания:
— гипергонадотропный (первичный) гипогонадизм;
— гипогонадотропный (вторичный) гипогонадизм;
— гипогонадизм, обусловленный резистентностью органов-мишеней.
В зависимости от времени возникновения:
В зависимости от этиологии:
• Первичный врожденный гипогонадизм
I. Нарушение развития гонад
Дисгенез семенных канальцев — синдром Клайнфельтера и его варианты
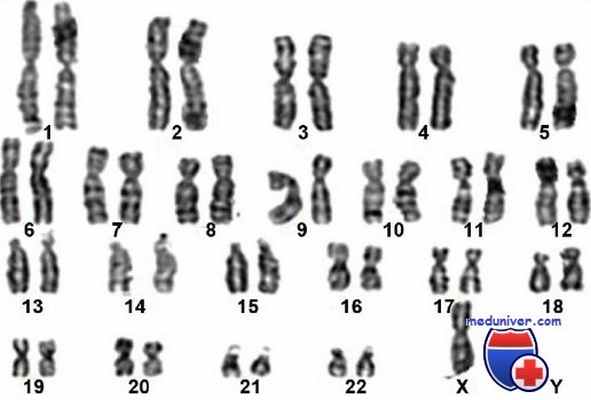
Синдром Клайнфельтера — распространенная патология (частота в мужской популяции 1 на 660 новорожденных мальчиков), является частой причиной гипогонадизма и бесплодия. Заболевание обусловлено наличием дополнительной Х хромосомы в кариотипе. В 90% случаев в кариотипе пациентов с синдромом Клайнфельтера выявляется одна дополнительная Х хромосома — 47, XXY. В остальных 10% имеются мозаицизм 46,XY/47,XXY, более выраженные X-хромосомные анэуплоидии (48,XXXY; 49,XXXXY, 47,XXY/48,XXXY) или структурно аномальные X-хромосомы.
Ключевым звеном в патогенезе синдрома Клайнфельтера является недостаточная продукция тестостерона клетками Лейдига (первичный или гипергонадотропный гипогонадизм). Основные клинические проявления данной патологии (и соответственно основные жалобы пациентов) — бесплодие, эректильная дисфункция, гинекомастия и избыточная масса тела. Фенотип таких пациентов разнообразный, как правило, они высокого роста, с евнухоидными пропорциями тела, маленькими плотными яичками. Отмечают также нарушения речи и снижение интеллекта.
Несмотря на высокую распространенность, эта патология более чем у половины больных на протяжении всей жизни остается нераспознанной. Более того, лишь у 10% пациентов диагноз устанавливают до периода полового созревания. Причины столь поздней диагностики могут быть обусловлены вариабельностью фенотипа, поздним клиническим проявлением симптомов заболевания, а также недостаточной осведомленностью врачей. Последствиями поздней постановки диагноза является несвоевременное назначение заместительной терапии препаратами тестостерона и как следствие того прогрессирование симптомов гипогонадизма и развитие осложнений, ассоциированных с гипогонадизмом. Наличие длительного персистирующего гипогонадизма у таких пациентов является моделью для изучения влияния дефицита андрогенов на половую функцию, предстательную железу, костную ткань, углеводный и жировой обмен, когнитивную функцию.
Аплазия герминативных клеток (синдром наличия только клеток Сертоли), или синдром Дель Кастильо
Этот синдром известен также как синдром «одних клеток Сертоли». Он обусловлен врожденным отсутствием зародышевых клеток — гоноцитов, возникает в результате гибели герминативных клеток в нормально сформированной гонаде. Подобные состояния описаны в литературе при радиоактивном облучении и некоторых других заболеваниях. Основные признаки синдрома: малые размеры яичек, нормальные размеры полового члена и мошонки, достаточное развитие мужских вторичных половых признаков, нормальный физический статус, в эякуляте — азооспермия.
При гистологическом исследовании: малые размеры семенных канальцев, полное отсутствие сперматогенного эпителия при наличии сустентоцитов, нормальная базальная мембрана, клетки Лейдига.
При описываемом синдроме в отличие от синдрома Клайнфельтера отсутствует гинекомастия, отмечается нормальный мужской генотип 46 XY, проба на половой хроматин отрицательная. Копулятивные функции, спонтанные и адекватные эрекции, эякуляции, оргазм, половое влечение сохранены, больные не испытывают затруднений в половой жизни, жалуются только на бесплодие. В лечении гормонами не нуждаются. Прогноз в отношении способности к оплодотворению неблагоприятный. Лечение таким больным назначают только при наличии признаков недостаточной андрогенизации.
Анорхизм (Синдром исчезнувших яичек)
Этот синдром характеризуется отсутствием яичек. Мальчики рождаются с мужским фенотипом, со сформированными мужскими половыми органами, но яички в мошонке отсутствуют, что может расцениваться как крипторхизм. Кариотип 46 XY. Рост и развитие до полового созревания нормальные. В период полового созревания не развиваются вторичные половые признаки. Не происходит рост волос на лобке, в подмышечной области, на лице. Формируется евнухоидный тип телосложения. Может развиться остеопороз, иногда очень тяжелый. Половой член маленький, мошонка не развита, яичек в мошонке нет. При оперативном вмешательстве по поводу крипторхизма яичек не находят.
Полагают, что причиной исчезновения яичек является мутация гена тестис-стимулирующего фактора в коротком плече Y-хромосомы. Прекращение выработки этого фактора ведет к регрессии и исчезновению яичек. Предполагается, что регрессия и исчезновение яичек происходит после того, как у плода сформируется мужской фенотип, то есть спустя 70 и более дней после зачатия.
Крипторхизм
Крипторхизм означает отсутствие двух или одного яичка в мошонке. Он встречается у 5% мальчиков при рождении. У большинства из них яички опускаются в мошонку в течение первого года жизни. У 1% мальчиков крипторхизм сохраняется и в дальнейшем. В норме яички опускаются в мошонку между 20-й недели эмбрионального развития и родами.
Считается, что причиной крипторхизма может быть дефект в синтезе тестостерона и дегидротестостерона. Это подтверждено экспериментами, в которых было продемонстрировано, что дегидротестостерон необходим для нормального опускания яичек в мошонку. Яичко или яички могут находиться в паховом канале, и их нередко удается пальпировать. В других случаях оба или одно яичко находятся в брюшной полости.
В яичках уменьшено количество семенных канальцев и сперматогониев. Позже может развиться склероз ткани яичка. Если яички не опустились в мошонку до 4-5 лет. Возникает риск бесплодия. В 12% случаев у лиц с крипторхизмом, если яички находятся в брюшной полости, развиваются злокачественные опухоли неопустившегося яичка, чаще всего семиномы. Считают, что этому способствует необычная для яичек среда, более высокая температура в брюшной полости, чем в мошонке, которая является фактором риска развития новообразований яичка.
Синдром Ульриха-Нунан, или синдром Тернера у мужчин
Мужской синдром Тернера не имеет в отличие от женского хромосомных нарушений. Кариотип 46 XY, фенотип мужской. Сходство с женским синдромом Тернера состоит в наличии целого ряда клинических появлений, характерных для обоих заболеваний: маленький рост, «кубитус вальгус», складки на шее («голова сфинкса»), нередко врожденные пороки сердца, в основном правых отделов. Яички маленькие, в них уменьшено количество семенных канальцев, иногда развивается склероз и уменьшается количество, вплоть до полного исчезновения, зародышевых клеток. Клетки Лейдига гиперплазированы. Часто встречается крипторхизм. Потенция и либидо снижены. У некоторых больных размеры яичек нормальные и может сохраниться фертильность.
Синдром рудиментарных яичек
Синдром рудиментарных яичек возникает в связи с тем, что внутренние половые органы образуются сразу из двух зачатков — мюллеровых и вольфовых протоков, причем первые образуют резко недоразвитую матку и маточные трубы, а вторые — рудиментарные яички и их придатки. Больные имеют евнухоидный вид, наружные половые органы — мужские, гипотрофированы; вторичные половые признаки недоразвиты. Рудиментарные яички нередко служат почвой для злокачественных опухолей.
Дисгенез гонад, или синдром Свайера
Дисгенезия гонад при 46XY («чистая» дисгенезия гонад, или синдром Свайера) описана G.I.M. Swyer в 1955 году как случай мужского псевдогермафродитизма. Больные характеризуются женским фенотипом, нормальным или слегка повышенным ростом, недостаточным развитием вторичных половых признаков и первичной аменореей, которая сочетается
с гипергонадотропным половым инфантилизмом. При обследовании определяется кариотип 46XY, а при лапаротомии выявляются «стрековые гонады», которые не секретируют ни тестостерон, ни антимюллеров гормон, что и создает условия для развития протоков Мюллера в период внутриутробной жизни. Молекулярно-генетические исследования показывают, что в таких случаях имеется делеция на коротком плече Y хромосомы (Yp) или имеется 46Yxp как результат транслокации фрагмента ХР на нормальную Y-хромосому (T. Ogata и соавт., 1992). Лишь у небольшого числа больных с данной патологией выявляется делеция короткого плеча Y-хромосомы, где локализуется ген SRY (ген, определяющий развитие яичек). При проведении молекулярно-генетических исследований семейных случаев частичной 46XY дисгенезии гонад P. Fechner и соавт. (1993) пришли к заключению, что, вероятно, при этой патологии в патогенез заболевания вовлечены не гены, определяющие пол и расположенные на Y-хромосоме, а какие-то другие определяющие пол гены, но локализованные на Х-хромосоме. Для заболевания характерен низкий уровень половых стероидов в сыворотке крови и повышенное содержание гонадотропинов. В пубертатный период и позже рекомендуется заместительная терапия эстрогенами. В связи с большим риском перерождения «стрековых гонад» в гонадобластому, дисгерминому или семиному рекомендуется их хирургическое удаление.
Аплазия клеток Лейдига
Причины аплазии клеток Лейдига не установлены. При этом дефекте яички сохранены, но в них отсутствуют клетки Лейдига, в которых синтезируется тестостерон. В отсутствии тестостерона не происходит должного развития семенных канальцев, и клетки Сертоли недостаточно синтезируют мюллеров — канал ингибирующий фактор. В связи с этим нарушается мужская дифференцировка и мальчик рождается с женским фенотипом — мужской псевдогермафродитизм. При рождении мальчика принимают за девочку. Однако кариотип мужской 46 XY.
Если гибель клеток Лейдига произошла после 10-12 недель эмбрионального развития, когда мужские органы уже сформировались, ребенок рождается с мужским фенотипом. В этих случаях заболевание диагностируется только в период полового созревания, когда не происходит развития вторичных половых признаков — отсутствует рост волос на лобке, в аксиллярных областях, на лице. Яички и половой член маленькие, мошонка не развита. Отсутствуют либидо и потенция. У лиц с мужским псевдогермафродитизмом, принятых за девочку, в период полового созревания имеет место первичная аменорея, не развиваются молочные железы, не происходит феминизация фигуры. При обследовании обнаруживается отсутствие матки и яичников. В паховом канале или в брюшной полости обнаруживают неразвитые яички. Кариотип XY.
Синдром 46ХХ у мужчин, или синдром де ля Шапеля
XX — нарушение или синдром половой реверсии — вариант синдрома Клайнфельтера. Клинические симптомы похожи, кроме более низкого роста и гипоспадии; случаи умственной неполноценности встречаются реже. Пациенты имеют кариотип 46ХХ. Такой парадокс объясняется экспрессией на клетках Н-Y антигена и предположительно наличием в составе генома структур Y-хромосомы.
Синдром ХYY
Фенотипические проявления XYY синдрома сходны с синдромом Клайнфельтера, но более вариабельны. Показатели спермы у этих мужчин варьируют от нормальных до азооспермии. Больные с наличием XXY-cиндрома отличаются высоким ростом, нередко мышечной силой, наличием угревой сыпи, а также снижением умственного развития и наклонностью к совершению криминальных действий. Симптомы гипогонадизма выражены умеренно (гипоплазированные тестикулы при нормальном половом члене). Оволосение на лице скудное. В спермограмме — олигозооспермия или у незначительной части больных — нормальный сперматогенез. У многих больных отмечается гипотония и замедленная речь. Содержание гонадотропинов умеренно повышено, а тестостерона в плазме — снижено. Метода лечения бесплодия не существует.
А.О. Поздняк
Казанская государственная медицинская академия
Поздняк Александр Олегович — доктор медицинских наук, профессор кафедры эндокринологии
Литературa:
1. Андрология. Мужское здоровье и дисфункция репродуктивной системы. Под общ. ред. Нишлага Э., Бере Г.М. М.: 2005. 554 с.
2. Благосклонная Я.В. Эндокринология. ООО « Издательство СпецЛит», 2004. с. 223-226.
3. Вакс В.В. Гипогонадотропный гипогонадизм у мужчин: Материалы конференции «Актуальные проблемы нейроэндокринологии». 13-14 дек. 2001. С. 73-84.
4. Васильченко Г.С . Общая сексопатология. М.: Медицина, 2005. с. 512.
5. Гусакова Д.А., Калинченко С.Ю., Курило Л.Ф. Первичный гипогонадизм как причина метаболического синдрома (на примере синдрома Клайнфельтера). Журнал «Проблемы Репродукции» 2008; 3.
6. Дедов И.И. и др. Половое развитие детей: норма и патология. М., 2002. 232 с.
7. Лавин Л. Эндокринология. Медицинский справочник. Москва, 1999. Глава 15. Врожденная гиперплазия коры надпочечников. с. 222-242.
8. Никулин Б.А. Мужское бесплодие. Оценка, диагностика, лечение. 14 с.
9. Рациональная фармакотерапия заболеваний эндокринной системы и нарушений обмена веществ. Под ред. И.И. Дедова, Г.А. Мельниченко. Москва, 2006. Глава 47. с. 716-747.
10. Устинова Т.И. Эндокринология мужской половой системы. С.- Петербург, 2007. с. 113-128.
11. Morales A., Lunenfeld B. Investigation, treatment and monitoring of late-onset hypogonadism in males. Aging Male. 2002; 5: 74-86.
Синдром персистирующих мюллеровых протоков. Дисгенезия гонад
Синдром персистирующих мюллеровых протоков. Дисгенезия гонад
Синдром персистирующих мюллеровых протоков характеризуется наличием полностью развитых производных мюллеровых протоков у нормально маскулинизированного в остальном генетического мужчины (46.XY). Всего описано менее 200 случаев синдрома. Отсутствие редукции мюллеровых протоков приводит к развитию внутренних женских половых органов у пораженных мужчин при наличии в той или иной степени развитых производных вольфо-вых протоков. Развитие гениталий по промежуточному типу встречается при этом синдроме нечасто. Возможен одно- или двусторонний крипторхизм.
Синдром персистирующих мюллеровых протоков бывает вызван мутациями генов АМГ или рецепторов к АМГ II типа. Для выявления мюллеровых структур используют УЗИ и МРТ.
Дисгенезия гонад — описательный термин, относящийся к характеристике разнородной группы лиц, при рождении имеющих гениталии женского типа, структуры-производные мюллеровых протоков и дисгенетичные или полосковидные гонады. Этот термин используют, как правило, при описании синдрома Тернера, хотя существуют и другие формы дисгенезии гонад. У пациенток с синдромом Тернера выявляют не только дисгенетичные гонады, но и другие аномалии, в том числе низкий рост, бочкообразную грудную клетку, коарктацию аорты. При хромосомном анализе обнаруживают аномалии строения или полное отсутствие одной Х-хромосомы. Синдром Тернера является причиной дисгенезии гонад в 50% случаев.
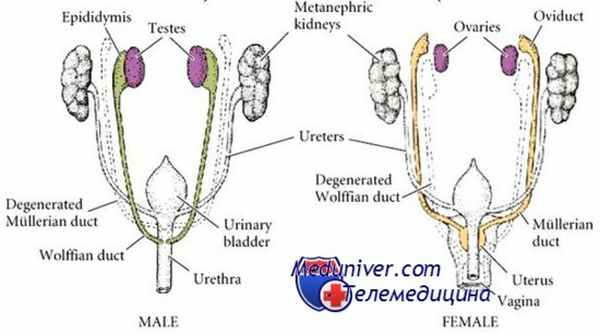
Две остальные разновидности дисгенезии гонад представляют собой формы при кариотипе больных — 46.ХХ (частая форма) или 46.XY (синдром Свайера). Из-за отсутствия необходимой гормональной стимуляции у больных часто встречаются аномалии развития вторичных половых признаков. У них нет фенотипиче-ских проявлений, характерных для синдрома Тернера.
Чистая дисгенезия гонад
Характерны дисгенетичные гонады на фоне нормальных внутренних и наружных половых путей. Иногда встречается гипоплазия гениталий. В большинстве случаев кариотип 46.XY, иногда 46XХ. Состояние может быть спорадическим либо наследоваться по аутосомно-рецессивному или Х-сцепленному типу (в некоторых случаях XY-дисгенезии гонад). При синдроме Свайера лица с кариотипом 46.XY имеют женский фенотип, достигают среднего или выше среднего в популяции роста, у них нет ни одного симптома, характерного для синдрома Тернера. Концентрация ФСГ повышается в раннем подростковом возрасте из-за того, что полосковидные гонады не синтезируют стероидные гормоны и ингибин.
Микроскопически дисгенетичные гонады, как правило, представлены стромой и фиброзной тканью первичных фолликулов. Диагноз обычно устанавливают в подростковом возрасте при обследовании по поводу первичной аменореи. В одной работе описана девочка-подросток с синдромом Свайера, у которой развилась вторичная аменорея. Пациенткам с XY-дисгенезией гонад показана гонадэктомия, так как у них высок риск развития гонадобластом.
Смешанная дисгенезия гонад
Большинство пациентов со смешанной дисгенезией гонад имеют мозаичный кариотип 45,XO/46,XY. Состояние характеризуется асимметрией развития гонад и персистированием структур-производных мюллеровых протоков. С одной стороны, как правило, имеется неразвитое яичко или опухоль из Сертоли или Лейдига клеток, с другой — дисгенетичная гонада или ее отсутствие. Развитие мюллеровых протоков обусловлено функциональной недостаточностью неразвитого яичка. Как было указано выше, таким пациентам показана гонадэктомия.
Дисгенезия яичников — зачастую первый предположительный диагноз у девочки-подростка, которая имеет задержку или отсутствие признаков полового развития. При УЗИ может подтвердиться наличие дисгенетичных яичников, выглядящих как небольшие атрофичные образования. Хотя и редко, но в дисгенетичных гонадах могут развиваться стероид-синтезирующие гонадобластомы, из-за которых УЗ-картина яичников может напоминать норму. К другим ценным методам диагностики относятся МРТ и КТ. Пациентам как с кариотипом 46,ХХ, так и с кариотипом 46,XY назначают с заместительной целью эстроген- и гестогенсодержащие препараты, а проблему деторождения решают с помощью ВРТ, обычно используя донорские яйцеклетки.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром персистирующих мюллеровых протоков. Дисгенезия гонад
Эндокринологический научный центр, Москва
ФГБУ «Национальный медицинский исследовательский центр эндокринологии» Минздрава России, Москва, Россия
ГБУ «Детская республиканская клиническая больница им. Н.М. Кураева», Махачкала, Россия
ФГБУ «Эндокринологический научный центр» Минздрава России, Москва, Россия
ФГБУ «Национальный медицинский исследовательский центр эндокринологии» Минздрава России, Москва, Россия
Эндокринологический научный центр, Москва
Случай поздней диагностики синдрома персистирующих мюллеровых протоков I типа
Журнал: Проблемы эндокринологии. 2018;64(1): 50‑53
Эндокринологический научный центр, Москва
Синдром персистирующих мюллеровых протоков — один из редких вариантов нарушения формирования пола (НФП), характеризующийся наличием у лиц мужского пола с кариотипом 46,XY дериватов мюллеровых протоков (ДМП). Так как единственным внешним проявлением синдрома персистирующих мюллеровых протоков (СПМП) является крипторхизм, до недавнего времени диагноз СПМП у пациентов оставался неустановленным, а обнаружение ДМП во время оперативного лечения крипторхизма при кариотипе 46,ХУ расценивалось как вариант дисгенезии гонад, что приводило к ошибочной тактике ведения пациентов. Лишь с появлением методов молекулярной диагностики, в частности — секвенирования нового поколения, позволяющего одномоментно исследовать несколько генов-кандидатов, появилась возможность точной дифференциальной диагностики СПМП. В данной статье описан клинический случай поздней диагностики семейной формы СПМП, обусловленной мутацией в гене AMH. Обнаружение персистирующих ДМП в сочетании с агенезией тестикула у пробанда в раннем возрасте привело к установлению диагноза дисгенезия гонад. Учитывая высокий риск малигнизации дисгенетичных гонад, расположенных вне мошонки, пациенту было произведено удаление единственной гонады, расположенной в паховом канале. Установить верный диагноз удалось после проведения комплексного молекулярно-генетического исследования методом секвенирования нового поколения с использованием панели Нарушение формирования пола. При исследовании выявлена гомозиготная мутация в гене AMH.
Эндокринологический научный центр, Москва
ФГБУ «Национальный медицинский исследовательский центр эндокринологии» Минздрава России, Москва, Россия
ГБУ «Детская республиканская клиническая больница им. Н.М. Кураева», Махачкала, Россия
ФГБУ «Эндокринологический научный центр» Минздрава России, Москва, Россия
ФГБУ «Национальный медицинский исследовательский центр эндокринологии» Минздрава России, Москва, Россия
Эндокринологический научный центр, Москва
Нарушения формирования пола (НФП), связанные с дефектом биосинтеза или действия антимюллерового гормона (АМГ), относятся к редким вариантам НФП, которые характеризуются наличием у лиц мужского пола с кариотипом 46, XY дериватов мюллеровых протоков (ДМП): матки, маточных труб и влагалищного отростка урогенитального синуса [1].
В мужском организме одной из основных биологических функций АМГ является инициация регрессии мюллеровых протоков в период внутриутробного развития плода. Поскольку развитие наружных половых органов и дериватов вольфовых протоков происходит под влиянием тестостерона, продукция которого у лиц мужского пола с дефицитом АМГ или нарушением его действия не нарушена, в таких случаях ДМП сохраняются на фоне правильно сформированных по мужскому типу наружных половых органах, поэтому сомнений в выборе мужского паспортного пола при рождении не возникает. Единственным клиническим проявлением синдрома персистирующих мюллеровых протоков (СПМП) может быть наличие двустороннего (реже одностороннего) крипторхизма. Однако ввиду высокой распространенности крипторхизма среди новорожденных мальчиков без видимых других патологических нарушений (1:300—600) [2], СПМП нередко остается недиагностированным. ДМП обнаруживаются случайно во время хирургического вмешательства по поводу крипторхизма [3—8]. Таким образом, истинная частота СПМП остается неизвестной. Между тем правильность и своевременность выявления данного состояния могут влиять на выбор тактики хирургического ведения таких пациентов, объем вмешательства и необходимость медико-генетического консультирования в семье.
СПМП является аутосомно-рецессивным заболеванием, обусловленным дефектами гена AMH, следствием чего является дефицит АМГ (СПМП тип I), или мутациями в гене рецептора АМГ (AMHR2), следствием чего является резистентность к АМГ (СПМП тип II) [1, 9].
До появления методов молекулярной диагностики, позволяющих точно установить правильный диагноз, обнаружение ДМП в детском возрасте у пациентов с крипторхизмом нередко приводило к ошибочной диагностики других форм НФП и соответственно неправильной тактике ведения.
Описание случая
Пробанд — мальчик, рожденный в близкородственном браке (родители двоюродные брат и сестра). При рождении на основании двустороннего отсутствия яичек в мошонке при правильном мужском строении наружных половых органов установлен диагноз «двусторонний крипторхизм». В возрасте 5 лет проведена ревизия пахового канала слева — яичко не обнаружено. В возрасте 6 лет при УЗИ малого таза визуализировалась простата 17×12×16 мм, матка 29×7×12 мм в виде тяжа; справа от мочевого пузыря — однородное овальное образование 18×13× 19 мм, по эхоструктуре напоминающее яичко.
При ревизии пахового канала справа и брюшной полости обнаружен семенной канатик, слепо заканчивающийся у входа в мошонку округлым влагалищным отростком, в брюшной полости обнаружена недоразвитая матка с маточной трубой и широкой связкой справа. В возрасте 9 лет впервые обследован в ФГБУ ЭНЦ: кариотип 46, XY, обнаружен ген SRY, уровень ЛГ и ФСГ соответствовал допубертатным значениям, при УЗИ гонады в паховых каналах и брюшной полости не визуализировались. Однако при проведении теста с хорионическим гонадотропином был получен удовлетворительный подъем тестостерона до 4,5 нмоль/л, что свидетельствовало в пользу наличия тестикулярной ткани, расположенной, вероятнее всего, в брюшной полости. В связи с этим пациент направлен на повторную диагностическую лапароскопию с направительным диагнозом «дисгенезия гонад», установленным на основании отсутствия яичка слева (подтвержденного диагностической лапароскопией), возможной брюшной формой крипторхизма справа и наличия ДМП. При ревизии брюшной полости произведено удаление маточной трубы и недифференцированной гонады справа. В возрасте 13 лет пациент повторно поступил в ФГБУ ЭНЦ. Учитывая предположительный диагноз НФП 46, XY, дисгенезию гонад, пациенту было проведено комплексное молекулярно-генетическое исследование методом секвенирования нового поколения с использованием панели Нарушение формирования пола. В гене AMH выявлена гомозиготная мутация: замена гуанина на аденин в акцепторном сайте сплайсинга интрона 3 (c.665−1G>A, референсный транскрипт — NM_000479). Данная мутация ранее не описана. Родители пациента оказались гетерозиготными носителями этой мутации. У пробанда есть старший брат, у которого также с рождения установлен двусторонний крипторхизм; в возрасте 1,5 года и 5 лет проведено хирургическое низведение обоих яичек, однако в течение 5 лет 2 раза фиксировались рецидивы крипторхизма, приведшие к повторным хирургическим вмешательствам. В возрасте 20 лет брат пробанда обратился в нашу клинику с жалобами на снижение либидо, бесплодие в браке. При обследовании выявлено значительное повышение уровня гонадотропинов [ЛГ — 21,6 мМЕ/мл (норма 2,5—10 мМЕ/мл), ФСГ —59 мМЕ/мл (1,4—8,0 мМЕ/мл)], при сниженном уровне тестостерона [7,6 нмоль/л (12—33 нмоль/л)], на основании чего диагностирован гипергонадотропный гипогонадизм. Учитывая анамнез (двусторонний крипторхизм) и подтвержденный диагноз СПМП у пробанда, брату было проведено УЗИ малого таза: выявлены персистирующие ДМП (матка, маточные трубы). Для подтверждения семейной формы СПМП старшему брату также было проведено секвенирование гена AMH и выявлена аналогичная мутация.
АМГ, или ингибитор мюллеровых протоков, — гомодимерный гликопротеин, принадлежащий к группе трансформирующих ростовых факторов β (TGF-β). Ген AMH картирован на участке короткого плеча 19 хромосомы (19р13.3), состоит из 5 экзонов и кодирует белок, состоящий из 560 аминокислот. Изначально АМГ синтезируется в виде прогормона и подвергается последующему расщеплению до N-концевого продомена и активной COOH-молекулы размером 25 кДа.
В мужском организме незрелые клетки Сертоли являются основным источником синтеза АМГ. Внутриутробно экспрессия АМГ начинается примерно с конца 7-й недели после связывания транскрипционного фактора SOX9 с промотером AMH. Затем, под влиянием других транскрипционных факторов — SF1, GATA4, WT1 [10, 11], его продукция усиливается. В настоящее время считается, что АМГ приводит к регрессии дериватов мюллеровых протоков, вызывая апоптоз клеток, экспрессирующих рецептор АМГ (AMHR). В постнатальном и препубертатном периодах продукция АМГ сохраняется на достаточно высоком уровне, тогда как с наступлением пубертата, повышением уровня тестостерона и созреванием клеток Сертоли, его синтез резко снижается. Высокая продукция АМГ незрелыми клетками Сертоли позволяет использовать АМГ как специфический маркер их функционального состояния в допубертатном периоде.
Наиболее частой причиной нарушения продукции АМГ у плода с кариотипом 46, XY является дисгенезия гонад, при которой нарушается функция как клеток Сертоли, так и клеток Лейдига, синтезирующих тестостерон, что приводит к сочетанию персистенции ДМП с нарушением строения половых органов, обусловленного внутриутробным дефицитом тестостерона. Поэтому одним из критериев дифференциальной диагностики дисгенезии гонад при кариотипе 46, XY является наличие или отсутствие ДМП.
Именно обнаружение ДМП в сочетании с агенезией левого яичка (подтвержденного лапароскопически) привело к ошибочной диагностике дисгенезии гонад в вышеописанной семье.
Впервые СПМП был описан в 1927 г. V. Seemen [12], обнаружившим ДМП во время оперативного лечения ребенка с крипторхизмом. К настоящему времени в литературе описано более 400 случаев СПМП, основным внешним клиническим проявлением которого являлся крипторхизм с разной степенью локализации. Наличие крипторхизма обусловлено тесной анатомической связью яичка с ДМП, поэтому степень миграции яичка и его локализация определяются подвижностью мюллеровых структур. Чаще всего яички остаются в брюшной полости (брюшная форма крипторхизма), но нередко недоразвитая широкая связка, не прикрепленная к брюшной стенке [13], «низводит» ДМП вместе с яичком в мошонку, приводя к возникновению грыжи (при СПМП до 30% случаев). Таким образом, клиническая картина крипторхизма при СПМП может варьировать от односторонней паховой формы до двусторонней брюшной формы.
В середине 80-х годов с появлением методов определения уровня АМГ в плазме было установлено, что у части пациентов с СПМП уровень АМГ значительно снижен, тогда как у других его уровень находится в пределах нормальных значений. На основании чего СПМП разделили на две группы: АМГ-позитивный и АМГ-негативный.
С развитием молекулярно-генетических технологий появилась возможность установить и патогенетическую основу СПМП. В 1991 г. B. Knebelmann и соавт. [1] впервые описали нонсенс-мутацию в экзоне 5 гена AMH среди нескольких членов мороканской семьи с СПМП и сниженным уровнем АМГ, установив таким образом причину возникновения АМГ-негативных форм. К настоящему времени описано более 150 семей с мутациями в гене АМГ, расположенных в различных участках кодирующей области гена. Так называемых «горячих точек» для мутаций, приводящих к возникновению СПМП, в гене AMH обнаружено не было.
В 1994 г. был клонирован ген рецептора АМГ II типа (AMHR2) [14]. Белок АМГ, как и другие члены семейства TGF-β, использует для передачи сигнала два рецептора. Изначально АМГ формирует комплекс с рецептором АМГ II типа, который в свою очередь активирует рецептор АМГ I типа, отвечающий за фосфорилирование внутриклеточных белков.
Первая мутация в гене AMHR2 была описана S. Imbeaud и соавт. в 1995 г. [9]. К настоящему времени описано более 150 семей с мутациями в этом гене. Несмотря на выявление мутаций на протяжении всей кодирующей области гена AMHR2, обнаружено 2 наиболее часто встречающиеся мутации с разной популяционной распространенностью, что обусловлено эффектом основателя. Так, среди пациентов северной Европы наиболее частой является делеция 27 bp (от 6331до 6357 нуклеотида) в экзоне 10, составляя до 65% всех причин СПМП II типа. Данная мутация в гомозиготном состоянии была описана и в России [15]. Другая частая мутация, R407X в экзоне 9, чаще встречается среди пациентов Средиземноморья и стран Ближнего Востока.
Мутации в генах AMH или AMHR2 с одинаковой частотой приводят к возникновению СПМП, однако примерно в 10—12% случаев этого синдрома мутации в данных генах не выявляются.
Заключение
Несмотря на наличие методов точной диагностики СПМП, поиск мутаций в генах AMH и AMHR2 у всех пациентов с крипторхизмом, учитывая высокую распространенность последнего (0,1—0,2% к 1-му году жизни), рекомендовать нецелесообразно. Между тем при наличии таких сопутствующих факторов, как отягощенный анамнез по крипторхизму в семье, отсутствие яичек с двух сторон, особенно брюшные формы крипторхизма, обследование должно быть максимально расширено и включать кариотипирование, УЗИ малого таза, органов мошонки и паховых каналов, определение уровня АМГ, ингибина В, ЛГ, ФСГ и тестостерона в период минипубертата, либо (в более старшем возрасте) определение тестостерона на пробе с хорионическим гонадотропином и лапароскопию. При подозрении на СПМП обязательно проведение молекулярно-генетического исследования генов AMH и AMHR2 до операции. Данный алгоритм обследования позволит своевременно установить диагноз и соответственно выбрать правильную тактику ведения пациента.
Дополнительная информация
Источник финансирования. Молекулярно-генетическое исследование проводилось в рамках программы «Альфа-Эндо» при финансовой поддержке «Альфа-Групп» и фонда «КАФ».
Согласие пациента. Законные представители (родители) пациента добровольно подписали информированное согласие на публикацию персональной медицинской информации в обезличенной форме.
Авторы декларируют отсутствие явных и потенциальных конфликтов интересов, связанных с публикацией настоящей статьи.
Сведения об авторах
НФП. ПРОИЗВОДНЫЕ МЮЛЛЕРОВЫХ ПРОТОКОВ У РЕБЕНКА С МУЖСКИМ СТРОЕНИЕМ НАРУЖНЫХ ПОЛОВЫХ ОРГАНОВ

Введение. Нарушение формирования пола - состояние, связанное с клинико-биохимиче- ским проявлением несоответствия между генетическим, гонадным и фенотипическим полом ребенка требующего детального обследования для окончательного выбора половой принад- лежности. Материалы и методы. Мальчик П. в возрасте 1 г, поступил в ДГКБ им. Н.Ф. Филатова с направляющим диагнозом «Крипторхизм слева, абдоминальная форма». При осмотре по- ловые органы сформированы по мужскому типу. Правое яичко в мошонке, левое яичко в мо- шонке, паховом канале не определяется. При проведении УЗИ левое яичко достоверно не ви- зуализировалось. Ребенку с мужским строением наружных половых органов была проведена диагностическая лапароскопия, на которой обнаружено образование, напоминающее матку, и гонада, напоминающая яичник, расположенная в маточной трубе. Выполнена биопсия гона- ды, принято решение о дообследовании ребенка. При гистологическом исследовании гонады морфологическая картина соответствовала streak (гонада со стромой яичника без фолликулов). Кариотипирование методом FISH по- казало хромосомные нарушения - мозаицизм 46ХУ /45ХО (83 % и 17 %). По результатам гормонального обследования основные показатели соответствовали нормальным значениям. Антимюллеров гормон - 140 нг/мл. Отмечалось некоторое повышение эстрадиола до 73,4 (0,0-51,0). Проведена трехдневная проба с ХГЧ, на котором отмечалось повышение тестосте- рона до 14,95 нмоль/л. Результаты. По результатам проведенного обследования у ребенка был сформулирован диагноз: хромосомное НФП 45ХО/46ХУ, смешанная дисгенезия гонад. Учитывая риск гонадо- бластомы у ребенка с описанным диагнозом, принято решение об удалении streak и дериватов мюллеровых протоков. Было проведено хирургическое лечение: лапароскопическое удаление стрек-гонады, дериватов мюллеровых протоков. Выводы. При обнаружении производных мюллеровых протоков у детей с мужским стро- ением наружных половых органов необходимо проведение детального обследования: биопсия гонад, консультация генетика, эндокринолога, кариотипирование, гормональное обследование. При выявлении хромосомного НФП 45ХО/46ХУ, смешанной дисгенезии гонад рекомендовано проведение удаление streak и производных мюллеровых протоков, учитывая риск их малигни- зации.
Об авторах
Ю. В Петрухина
С. Л Коварский
Л. Б Меновщикова
А. И Захаров
С. П Блох
А. А Бебенина
З. З Соттаева
А. Н Текотов
К. А Струянский
З. В Бетанов
Н. А Агеева
Т. А Склярова
Список литературы
© Петрухина Ю.В., Коварский С.Л., Меновщикова Л.Б., Захаров А.И., Блох С.П., Бебенина А.А., Соттаева З.З., Текотов А.Н., Струянский К.А., Бетанов З.В., Агеева Н.А., Склярова Т.А., 2020
Эта статья доступна по лицензии Creative Commons Attribution-NonCommercial-NoDerivatives 4.0 International License.
ЖУРНАЛЫ
ПРАВОВАЯ ИНФОРМАЦИЯ
ИНФОРМАЦИЯ
СЕРВИСЫ
Реклама на сайте
Реклама в журналах
КОНТАКТЫ
191186, Санкт-Петербург, Аптекарский переулок, д.3, литера А, помещение 1Н
Синдром персистирующих мюллеровых протоков. Дисгенезия гонад

новая философия пола
НФП (или интерсекс) вариация — это комплекс врожденных половых признаков, имеющих общую причину, и которые не вписываются в привычные представления о мужском и женском организме.
Половые признаки включают в себя:
— кариотип (XX у типичной женщины, XY у типичного мужчины)
— гонады (яичники и женщин, яички у мужчин)
— внутренние половые органы (фаллопиевы трубы, матка и влагалище у женщин; придаток яичка, семявыносящий проток, семенные пузырьки и простата у мужчин)
— наружные половые органы (половые губы и клитор у женщин, пенис и мошонка у мужчин)
— вторичные половые признаки (молочные железы и отложение жира на бедрах у женщин, низкий голос и борода у мужчин)
— уровень половых гормонов в крови (уровень эстрадиола и прогестерона в крови у взрослой женщины колеблется в зависимости от фазы менструального цикла, уровень тестостерона у взрослого мужчины в 10-15 раз выше уровня тестостерона у взрослой женщины)
Ниже приведен список и описание наиболее распространенных НФП вариаций.

Клайнфельтерова вариация
При этой вариации имеет место кариотип XXY. Ребенок рождается с мужскими гонадами и мужскими внутренними и наружными половыми органами и вплоть до периода полового созревания ничем не отличается от обычного мальчика. Во время полового созревания имеет место очень слабое развитие вторичных мужских половых признаков, уровень тестостерона обычно ниже того, что считается нормой для мужчин, часто происходит развитие молочных желез. Чаще всего они бесплодны, но в некоторых случаях способны стать отцами.
Вариация Шерешевского-Тернера
При этой вариации имеет место кариотип X0. Вместо гонад развиваются соединительнотканные тяжи. Внутренние и наружные половые органы развиваются по женскому типу. Характерными особенностями являются низкий рост и наличие складок кожи по бокам шеи. Полового созревания как такового не происходит. Во взрослом возрасте выглядят как женщины очень низкого роста.
Свайерова вариация
При синдроме Свайера у плода с кариотипом XY не происходит развития мужских гонад, на месте которых развиваются соединительнотканные тяжи. Внутренние и наружные половые органы развиваются по женскому типу. Люди с такой вариацией вплоть до подросткового возраста внешне ничем не отличаются от обычных девочек. В подростковом возрасте у них не происходит полового созревания, рост груди и менструации отсутствуют. Отсутствие функционирующих гонад и, как следствие, отсутствие секреции половых гормонов приводит к тому, что не происходит закрытия зон роста костей. Во взрослом возрасте люди с такой вариацией выглядят как женщины очень высокого роста.
Дефицит 5-альфа-редуктазы
5-альфа-редуктаза — это фермент, превращающий тестостерон в более мощный андроген — дигидротестостерон. При дефиците 5-альфа-редуктазы у плода мужского пола не происходит достаточной маскулинизации наружных половых органов, которые при этой вариации чаще всего имеют промежуточное строение. Во время полового созревания происходит развитие мужских вторичных половых признаков (развитая мускулатура, низкий голос) и увеличение клитора/полового члена.
Полная нечувствительность к андрогенам
При этой вариации мужской XY кариотип и мужские гонады сочетаются с женскими наружными половыми органами и коротким, слепо заканчивающимся влагалищем. Яички могут располагаться как внутри брюшной полости, так и в паху, а иногда даже внутри больших половых губ.
Вторичные половые признаки развиваются по женскому типу, и взрослые люди с полной нечувствительностью к андрогенам внешне выглядят как обычные женщины, но в отличие от обычных женщин у них не растут волосы на лобке и подмышками. Причиной вариации является полная нечувствительностью тканей организма к мужским половым гормонам — андрогенам, результатом чего становится развитие наружных половых органов по женскому типу. В период полового созревания яички начинают вырабатывать мужской половой гормон — тестостерон, который превращается в жировой ткани в женский половой гормон — эстрадиол, вызывающий развитие вторичных женских половых признаков — груди, широкого таза, отложения жира по женскому типу.
Частичная нечувствительность к андрогенам
В данном случае у человека с мужским XY кариотипом и мужскими гонадами имеет место сильно сниженная чувствительность тканей к мужским половым гормонам — андрогенам. При минимальной степени снижения чувствительности к андрогенам наружные половые органы развиваются по мужскому типу, при более сильной степени снижения чувствительности к андрогенам наружные половые органы имеют строение, промежуточное между мужским и женским, а при почти полной нечувствительности к андрогенам наружные половые органы развиваются по женскому типу.
В период полового созревания начинают развиваться вторичные половые признаки. В случае почти полной нечувствительности к андрогенам развиваются женские вторичные половые признаки и обычная женская внешность, включая рост волос на лобке и подмышками, а в остальных случаях характер развития вторичных половых признаков смешанный, и внешность в той или иной степени промежуточная между обычной женской внешностью и обычной мужской внешностью.
Овотестис
При овотестисе (истинном гермафродитизме) в организме присутствуют как мужская, так и женская гонадальная ткань. Гонада, которая состоит одновременно из мужской, и из женской гонадальных тканей, называется овотестисом. Чаще всего в овотестисе мужская и женская гонадальные ткани разделены перегородкой.
При истинном гермафродитизме могут быть два овотестиса, либо один овотестис и один яичник, либо один овотестис и одно яичко, либо же одна из гонад может быть яичником, а другая — яичком.
Смешанная дисгенезия гонад/Мозаичность 45X0/46XY
При смешанной дисгенезии гонад на одной половине тела имеется яичко, а на другой половине тела — соединительнотканный тяж. Наружные половые органы могут развиваться как по мужскому, так и по женскому типу, но чаще всего они имеют промежуточное строение. Кариотип чаще всего мозаичный X0/XY
Майера-Рокитанского-Кюстнера-Хаузера вариация
Данная вариация состоит в отсутствии влагалища, матки и фаллопиевых труб у обычной во всех остальных отношениях женщины с кариотипом XX, яичниками и женскими наружными половыми органами. Процесс полового созревания ни чем не отличается от такового у обычной женщины. У взрослых женщин с этим синдромом в организме происходят точно такие же месячные циклические изменения, как и у обычных женщин, но из-за отсутствия матки у нет менструаций.
Врожденная гиперплазия коры надпочечников (ВГКН)
При врожденной гиперплазии коры надпочечников нарушен синтез кортизола — основного гормона коры надпочечников. Есть множество вариантов ВГКН, некоторые из которых не совместимы с жизнью, но при самых распространенных из них кора надпочечников вырабатывает большие количества мужских половых гормонов — андрогенов. У девочек ВГКН приводит к маскулинизации наружных половых органов, которые имеют строение, промежуточное между мужским и женским, а в случае очень сильной маскулинизации становятся почти неотличимыми от обычных мужских гениталий.
Вырабатываемые корой надпочечников андрогены приводят к раннему закрытию зон роста костей, поэтому люди с ВГКН имеют низкий рост.
Список вариаций
Андроген-индуцированный гермафродитизм
Анорхизм
Аплазия клеток Лейдига
Ассоциация MURCS
Афалия
Вирилизация индуцированная прогестогенами
Врождённая гиперплазия коры надпочечников
Врождённые нарушения метаболизма стероидов
Гипоспадия
Дефицит 5-альфа-редуктазы
Дефицит 17β-гидроксистероиддегидрогеназы III
Дефицит ароматазы
Дефицит цитохрома P450 оксидоредуктазы
Дефицит WNT4
Дисгенезия гонад
Изолированная недостаточность 17,20-лиазы
Истинный гермафродитизм
Клиторомегалия
Клоакальная экстрофия
Липоидная врождённая гиперплазия надпочечников
Микропенис
Недостаточность цитохрома b5
Овотестис
Псевдогермафродитизм
Раздвоенный пенис
Синдром 48, XXXY
Синдром 48, XXYY
Синдром 48, XYYY
Синдром 49, XXXXY
Синдром 49, XXXYY
Синдром 49, XYYYY
Синдром де ля Шапеля
Синдром избытка ароматазы
Синдром Клайнфельтера
Синдром лёгкой нечувствительности к андрогенам
Синдром Майера — Рокитанского — Кустера — Хаузера
Синдром нечувствительности к андрогенам
Синдром нечувствительности к эстрогенам
Синдром Перро
Синдром персистирующих мюллеровых протоков
Синдром полной нечувствительности к андрогенам
Синдром Рейфенштейна
Синдром Свайера
Синдром Шерешевского — Тёрнера
Синдром SERKAL
Смешанная дисгенезия гонад
46,XX/46,XY
Читайте также: