Синдром Рефсума: наследственная атаксическая полинейропатия с глухотой
Добавил пользователь Евгений Кузнецов Обновлено: 27.01.2026
Это большая группа невропатий включает симметричное, диффузное поражение ПНС. Полиневропатии подразделяют на аксонопатии и миелинопатии, что базируется на анатомо-физиологических представлениях о функционировании ПНС.L. Cavanagh предложил классификацию приобретенных аксональных полиневропатий. В первую группу включены энергозависимые невропатий с предположительным дефектом окислительных метаболических процессов - ограниченная дистальная аксонопатия при дефиците тиамина, рибофлавина, отравлении мышьяком, таллием.
Вторую группу составляют аксональные невропатий - дистальные, преимущественно сенсорные, с более проксимальным повреждением нерва, чем при болезнях в первой группе: изониазидная полиневропатия.
В третью группу вошли полиневропатий с вовлечением длинных проводников спинного мозга и периферических нервов: при отравлении акриламидом и фосфорорганическими соединениями.
Под сегментарной демиелинизацией обычно подразумевают первичную деструкцию миелиновой оболочки при интактном аксоне (в отличие от вторичной демиеяинизации при дегенерации аксона). При этом избирательно поражаются шванновские клетки и разрушается миелин, процесс часто начинается в области перехватов Ранвье. После повторных эпизодов демиелинизации и ремиелинизации появляются “луковичные головки”. Этот термин обозначает круговые листочки шванновских клеток, окружающих аксональный стержень. Результатом сегментарной демиелинизации является блокада проведения импульса или выраженное его замедление. Мышца при этом не денервируется, но может развиться ее атрофия от длительного бездействия. При активной ремиелинизации восстановление может наступить быстро, возможно полное выздоровление, например при дифтерийной острой демиелинизирующей полиневропатии.
В большинстве случаев заболевание проявляется симметричными сенсорными или моторными расстройствами или чаще их сочетанием. Дистальные сухожильные рефлексы, особенно ахилловы, обычно отсутствуют. Чувствительные нарушения имеют тип “носков” и “перчаток”. Изолированная двигательная или чувствительная полиневропатия возникает редко. В равной мере вегетативная невропатия обычно является частью генерализованной полиневропатии и как изолированный синдром встречается очень редко. В случае вовлечения в процесс, помимо периферических нервов, спинномозговых корешков более адекватен термин “полирадикулоневропатия”. В этих случаях обычно доминирует поражение проксимальной мускулатуры, часто встречается краниальная невропатия, а в цереброспинальной жидкости обнаруживается повышенное содержание белка.
Как правило, при полиневропатии доминирует поражение ног. Начало заболевания с рук, и их преимущественное повреждение иногда наблюдается при свинцовой и порфирийной невропатии, при В12-дефицитной невропатии и синдроме Гийена-Барре. Поражение вегетативной нервной системы приводит к ортостатической гипотензии, нарушению сердечного ритма, нарушению потоотделения, дисфункции тазовых органов (в целом - около 30 синдромов). Прогрессирующая вегетативная недостаточность наблюдается при диабете, амилоидозе, порфирии, алкоголизме, карциноматозной сенсорной невропатии, острой воспалительной демиелинизирующей полиневропатии, некоторых наследственных формах и др.
Утолщение периферических нервов, выявляемое при их пальпации, встречается при лепре, амилоидозе, болезни Рефсума, хронической воспалительной демиелинизирующей полирадикулоневропатии (ХВДП), гипертрофической форме болезни Шарко - Мари - Тута.
Различают четыре типа течения полиневропатии: острый (симптомы развиваются быстрее, чем за неделю), подострый (не более 1 мес), хронический (более месяца) и рецидивирующий, когда повторные обострения возникают на протяжении многих лет.
Ниже приводится описание наиболее актуальных для практики форм невропатии. Представляется целесообразным, помимо выделения особенностей каждого полиневропатического синдрома, указывать на сопутствующие этой форме полиневропатии другие формы невропатии, а также поражения ЦНС. Попытка жесткой фиксации только на картине полиневропатии нецелесообразна.
При анализе клинических симптомов полиневропатии можно использовать данные о функции тонких и толстых волокон, составляющих периферический нерв. Все двигательные волокна являются толстыми миелинизированными волокнами. Проприоцептивная и вибрационная чувствительность также проводятся по толстым миелинизированным волокнам. Волокна, передающие болевую и температурную чувствительность, относятся к немиелинизированным и тонким миелинизированным; вегетативные волокна - к тонким немиелинизированным, тогда как в передаче тактильной чувствительности участвуют толстые и тонкие волокна. Поражение тонких волокон может привести к избирательной потере болевой и температурной чувствительности, жгучей боли и дизестезии при отсутствии парезов и при нормальных рефлексах. Невропатия толстых волокон вызывает мышечную слабость, арефлексию, сенситивную атаксию и легкое нарушение поверхностной чувствительности. Вовлечение всех волокон приводит к смешанной (сенсомоторной и вегетативной) полиневропатии. Следует учитывать, что эти взаимоотношения между характером поражения и клинической картиной не являются абсолютными.
Боли при полиневропатиях зависят в основном от остроты процесса, а также от типа и величины пораженных волокон. Хроническая идиопатическая сенсорная невропатия сопровождается атаксией, обусловленной поражением толстых миелиновых волокон. Больных беспокоят парестезия, нарушение проприоцептивной чувствительности, но боли полностью отсутствуют. Очень сложен и неясен генез боли при некоторых формах диабетической невропатии, также протекающей с поражением тонких нервных волокон. Известно, что гипергликемия может снижать болевой порог и уменьшать антиноцицептивный эффект анальгетиков, поэтому при диабетических невропатиях нормализация глюкозы крови может привести к существенному уменьшению боли. Гиперпатия, дизестезия и аллодиния при невропатиях обычно связаны с регенерацией аксональных отростков в поврежденном сегменте нерва. Возникающие в процессе регенеративного спраутинга отростки могут быть источником эктопической спонтанной импульсной активности. В них также возникают условия для эфаптической трансмиссии электрической активности (передача нервного импульса при прямом контакте аксонов без участия медиатора), приводя к спонтанным болевым ощущениям.
Двигательные нарушения при полиневропатиях чаще локализуются дистально. Однако при некоторых видах полиневропатий (порфирийной, синдроме Гийена - Барре) проксимальные группы мышц могут страдать сильнее, чем дистальные. Фасцикуляции могут появляться при поражении корешков.
Важным в диагностике является исследование цереброспинальной жидкости. При острой и хронической воспалительной демиелинизирующей полирадикулоневропатии повышение содержания белка является одним из диагностических критериев. Умеренное повышение содержания белка характерно также для других демиелинизирующих полиневропатий, включая диабетическую. Клеточно-белковая диссоциация характерна для менингополиневрита Баннварта при боррелиозе Лайма и при СПИДе.
Наряду с клиническим обследованием для оценки нервно-мышечных нарушений при полиневропатиях большое значение имеет электрофизиологическое исследование.
Существенную помощь в диагностике оказывает биопсия кожного нерва. Обычно для изучения берут икроножный нерв или поверхностную ветвь лучевого. Биопсия помогает диагностике амилоидоза, лепры, метахроматической лейкодистрофии, метаболических болезней, некоторых случаев ХВДП. Биопсия показана только в тех случаях, когда нельзя установить диагноз, используя неинвазивные методы.
Следует подчеркнуть, что, несмотря на исчерпывающее обследование, примерно у 1/3 больных с синдромом полиневропатии этиологический диагноз установить не удается.
Синдром Рефсума: наследственная атаксическая полинейропатия с глухотой
Синдром Рефсума: наследственная атаксическая полинейропатия с глухотой
Клинические данные. Данные осмотра. Больные обычно выглядят здоровыми до второго десятилетня жизни, когда становятся заметными мышечные атрофии и слабость, а также нарушения зрения. Эти расстройства медленно прогрессируют у взрослых. Состояние может резко ухудшиться в связи с беременностью (Fryer et al.).
Орган зрения. Потеря зрения является, по-видимому, одним из первых проявлений синдрома. Впервые отмечающаяся в начале второго десятилетия жизни ночная слепота медленно прогрессирует, приводя к концу этого периода к серьезным нарушениям зрения. Поля зрения медленно сужаются. Реакция зрачков становится замедленной. Постоянными симптомами являются миоз и гемералопия.
Офтальмологическое исследование выявляет бледность диска зрительного нерва и умеренное увеличение пигмента, более заметное в макулярной области и на периферии сетчатки. Сосуды сетчатки сужены. В 80% случаев наблюдаются задние катаракты (Richterich et al.), относительно часты также нистагм и анизокория (Fryer et al.).
Нервная система. В детстве или в молодости появляются атаксия и общая мышечная слабость, поражающие главным образом ноги, а позже и руки. Мышечные атрофии и параличи медленно прогрессируют и более заметны в дистальпых отделах конечностей. В детстве нарушение чувствительности к булавочным уколам и прикосновениям отмечается только в дистальных отделах конечностей, впоследствии оно, прогрессируя, ведет к расстройствам вибрационной и позиционной чувствительности. Сухожильные рефлексы снижены.
Постепенно развивается полная арефлексия. У большей части больных отмечается патологическая пальце-носовая проба и положительная проба Ромберга.
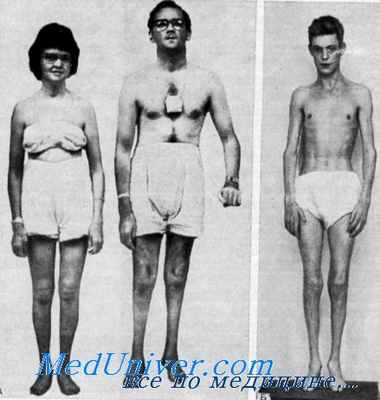
При обзоре данных о 37 случаях публикации Richterich с сотр. нашли следующие симптомы в порядке убывающей частоты: аносмия, парестезия, боль, утрата поверхностных рефлексов, наружная врожденная офтальмоплегия, иальпируемость периферических нервов.
Сердечно-сосудистая система. У 20 из 25 обследованных больных отмечались заболевания сердца. Клинические данные включали тахикардию, ритм галопа, систолический шум, расширение сердца и сердечную недостаточность (Richterich et al.).
Электрокардиографическая патология, такая, как увеличение интервала Р — Q, узловые или предсердные экстрасистолы и изменения комплекса QRS, отмечалась почти у 35% больных.
Костная система. Почти у 75% больных были выявлены некоторые виды костной патологии. Наиболее часто встречались спондилиты, экзостозы грудины, кифосколиоз, молоткообразное искривление пальцев ног и полая стопа. Были случайно обнаружены и такие аномалии, как укорочение нескольких пястных н плюсневых костей или клешнеобразная рука (Richterich et al.).
Кожные покровы. Почти у 60% молодых больных наблюдается легкий ихтиоз.
Орган слуха. У 34 из 44 больных с синдромом Рефсума Bergsmark и Djupesland обнаружили нейросенсорную глухоту. Часто снижение слуха на одной стороне было более глубоким, чем на другой. Степень глухоты у разных больных слегка варьировала. Потеря слуха наиболее часто начиналась во втором и третьем десятилетиях жизни и медленно прогрессировала, захватывая главным образом высокие частоты, tone-decay-тест и тест на различение речи были нормальными.
Вестибулярная система. Колорические вестибулярные пробы были нормальными (Fleming, Bergsmark, Djupesland).
Международный неврологический журнал 5 (51) 2012
Клинический случай позднего дебюта недифференцированной спинальной амиотрофии

Авторы: Гончарова Я.А., Симонян В.А., Евтушенко С.К., Белякова М.С., Евтушенко И.С. - ГУ «Институт неотложной и восстановительной хирургии им. В.К. Гусака НАМН Украины», Донецкий национальный медицинский университет им. М. Горького
Версия для печати
В данной статье описан случай спинальной амиотрофии с поздним дебютом, его клинические проявления, критерии диагностики, а также многообразие проявлений данной нозологии.
У цій статті описано випадок спінальной аміотрофії з пізднім дебютом, його кліничні прояви, критерії діагностики, а також різноманіття проявів даної нозології.
In this article there was described a case of spinal amyotrophy with late onset, its clinical manifestations, diagnostic criteria and also variety of this nosologic manifestations.
спинальная мышечная атрофия, электронейромиографический мониторинг, фармакологическая проба (прозериновая) и проба на истощение, магнитно-резонансная томография.
спінальна м’язова атрофія, електронейроміографічний моніторинг, фармакологічна проба (прозерінова) і проба на виснаження, магнітно-резонансна томографія.
spinal muscular atrophy, electroneuromyography monitoring, pharmacological test (proserin), exhaustion test, magnetic resonance imaging.
Спинальные амиотрофии представляют собой гетерогенную группу наследственных заболеваний периферической нервной системы, характеризующихся выраженным клиническим полиморфизмом.
Спинальная мышечная атрофия — разнородная группа наследственных заболеваний, протекающих с поражением и потерей моторных нейронов передних рогов спинного мозга. Амиотрофия — нарушение трофики мышц, сопровождающееся истончением мышечных волокон и уменьшением их сократительной способности, обусловленное поражением нервной системы: мотонейронов (на различных уровнях ЦНС — нейроны двигательной коры, ядер ствола мозга, передних рогов спинного мозга) или периферических нервных волокон. Болезнь считается наследственной в результате генных мутаций [6], хотя если мы посмотрим истории болезни, то у многих больных семейный анамнез не выявляется.
Различают наследственные и симптоматические амиотрофии. Нейрогенные наследственные амиотрофии делятся на две большие группы — спинальные и невральные амиотрофии. В большинстве случаев более тяжелыми являются спинальные формы. К ним относятся: спинальная амиотрофия (болезнь Верднига — Гоффманна), псевдомиопатическая прогрессирующая спинальная амиотрофия Кугельберга — Веландер, редкие формы спинальных амиотрофий и недифференцированные формы. Невральные амиотрофии: болезнь Шарко — Мари — Тута, гипертрофическая невропатия Дежерина — Сотта, синдром Русси — Леви, атактическая полинейропатия или болезнь Рефсума, а также недифференцированные формы. Спинальные амиотрофии делятся также на взрослые и детские. К проксимальным спинальным амиотрофиям детского возраста относятся: острая злокачественная инфантильная спинальная амиотрофия Верднига — Гоффманна (спинальная амиотрофия 1го типа), хроническая инфантильная спинальная амиотрофия (спинальная амиотрофия 2го типа), ювенильная спинальная амиотрофия (болезнь Кугельберга — Веландер), редкие формы спинальных амиотрофий в детском возрасте: инфантильная нейрональная дегенерация, врожденная форма болезни Пелицеуса — Мерцбахера, врожденная цервикальная спинальная амиотрофия, атипичный вариант GMганглиозидоза, детский прогрессирующий бульбарный паралич (синдром Фацио — Лонде), понтобульбарный паралич с глухотой (синдром Виалетто — Ван Лэре). Спинальные амиотрофии взрослых: бульбоспинальная амиотрофия Кеннеди, дистальная спинальная амиотрофия, сегментарная спинальная амиотрофия, мономиелическая спинальная амиотрофия, скапулоперонеальная спинальная амиотрофия Старка — Кайзера, лицелопаточноплечевая спинальная амиотрофия Феничела, окулофарингеальная спинальная амиотрофия [1]. Существуют также недифференцированные формы спинальных амиотрофий с быстропрогрессирующим, медленнопрогрессирующим и непрогрессирующим течением.
Согласно рекомендации Европейского консорциума по изучению нервномышечных заболеваний, клиническими критериями спинальной мышечной амиотрофии являются: симметричная мышечная гипотония и гипотрофия, фасцикуляции различных мышечных групп, гипо или арефлексия мышц конечностей, отсутствие чувствительных, мозжечковых и интеллектуальных расстройств. Патогномоничных изменений при спинальной мышечной амиотрофии нет. Однако важным является определение активности креатинкиназы сыворотки крови: считается, что превышение ее нормы более чем в 10 раз характерно для миодистрофии и противоречит диагнозу спинальной мышечной амиотрофии. При электронейромиографии выявляются симптомы поражения периферических моторных нейронов: спонтанная мышечная активность, увеличение длительности и амплитуды потенциалов действия двигательных единиц при нормальной скорости проведения импульсов по афферентным и эфферентным волокнам периферических нервов. При гистологическом исследовании биоптатов мышц обнаруживаются признаки денервационной мышечной атрофии [3].
Взрослые формы амиотрофий чаще всего развиваются в возрасте 15–60 лет, в среднем 35 лет, и имеют чаще всего доброкачественный характер. При выявлении клинического синдрома спинальной амиотрофии следует проводить дифференциальную диагностику между псевдогипертрофическими формами прогрессирующих мышечных дистрофий, боковым амиотрофическим склерозом и другими спинальными амиотрофиями.
Приводим описание клинического случая спинальной амиотрофии, недифференцированной формы, с миастеническим синдромом, полимиалгией, медленным прогрессированием у взрослого пациента.
Больной К., 40 лет, находился на лечении в отделении ангионеврологии ГУ «ИНВХ им. В.К. Гусака НАМНУ» с жалобами на чувство выраженной усталости, слабости в руках и ногах после незначительной физической работы, прохождения 2–3 трамвайных остановок, пекущие боли в поясничной области («ощущение кипятка»), скованность и боли в шее, ощущение онемения в руках.
Анамнез заболевания: считает себя больным с 1999 г., когда стали беспокоить боли в шее, прогрессивное ухудшение состояния около 2 лет, когда появились вышеперечисленные жалобы. Лечился у невролога по поводу шейного остеохондроза с цервикобрахиалгией. Обращался к вертебрологу, занимался лечебной гимнастикой с незначительным улучшением. В апреле 2010 г. консультирован неврологом ИНВХ им. В.К. Гусака — установлен синдром спинальной амиотрофии с полифибромиалгией, мышечной утомляемостью неясного генеза. Направлен на госпитализацию для уточнения диагноза и выбора тактики лечения. Из анамнеза жизни: со слов больного, у отца наблюдалась незначительная мышечная слабость, которая беспокоила его в возрасте 40 лет.
Неврологический статус: гинекомастия, сознание ясное. Снижение фона настроения, на вопросы отвечает правильно. При проведении нейропсихологического тестирования по шкале ММSE 30 баллов. Глазные щели D = S, зрачки равны. Движения глазных яблок в полном объеме. Небольшие атрофии и фибрилляции языка. Фибриллярных мышечных подергиваний в верхних и нижних конечностях, а также в мышцах туловища не обнаружено. Язык по средней линии. Сухожильные рефлексы с рук снижены, D = S, коленные снижены, D = S, ахилловы снижены, D = S. Слабость и атрофия мышц проксимальных отделов верхних конечностей, ограничение объема активных движений в руках, атрофия икроножных мышц.
Обследование: общеклинические и биохимические показатели у пациента в пределах возрастной нормы. Уровень креатинфосфокиназы — 59 нг/л (норма 14–190), лактат — 1,93 ммоль/л (норма 0,50–2,20), кальций — 2,72 ммоль/л (норма 2,20–2,75), магний — 0,75 ммоль/л (норма 0,65–1,14), калий — 3,50 (норма 3,50–5,40), натрий — 137,8 ммоль/л (норма 135,0–155,0), что соответствовало пределам нормативных показателей.
Также выполнялось такое исследование, как функциональная спондилография шейного и грудного отделов позвоночника. В шейном отделе ось позвоночника удовлетворительная, спондилоартроз унковертербральных и дугоотростчатых сочленений, смежные и замыкательные пластины склерозированы, снижена высота межпозвонковых щелей, ограничение функции сгибания и разгибания. В грудном отделе — ось позвоночник Sобразно искривлена, позвонки ротированы, смежные замыкательные пластинки склерозированы, омечается множество грыж Шморля, снижена высота межпозвоночных щелей на уровне Т4–Т10, передние остеофиты грудных позвонков.
Выполнялась магнитнорезонансная томография поясничного отдела позвоночника на аппарате Phillips c разрешающей способностью 0,23 Тесла — при исследовании в сагиттальной и аксиальной проекциях в режимах Т1 и Т2 определяются дегенеративные изменения дисков (за счет их дегидратации) со снижением их высоты. Определяются протрузии дисков L2–L3 до 0,3 см кзади, L3–L4 до 0,35 см кзади, L4–L5 до 0,4 см кзади, L5–S1 до 0,4 см парамедиально влево. Определяется утолщение задней продольной связки, на уровне замыкательных пластинок визуализируются остеофиты в дугоотростчатых сочленениях, артрозные изменения в виде их гипертрофии с наличием остеофитов на уровне суставных поверхностей. Дополнительных образований и очагов патологически измененного МРсигнала в позвоночном канале не выявлено. Учитывая характер жалоб и проводимую дифференциальную диагностику, выполнена спиральная компьютерная томография органов грудной клетки на аппарате Phillips с дозой 10 мЗв — легкие обычной пневматизации, без очагов патологической плотности и дополнительных образований. Корни структурны и не расширены, сердце и аорта в пределах возрастной нормы, костнодеструктивных изменений не выявлено.
Электрокардиография особых изменений не выявила, ритм был синусовый регулярный, ЧСС составляла 63 удара в минуту, отклонение электрической оси сердца вправо.
Электронейромиографический мониторинг проводился трижды (перед поступлением, через 10 дней после начала лечения и в день перед выпиской) на четырехканальном аппарате Biomedica (Reporter, Италия). При игольчатом исследовании биконцентрическим электродом была получена спонтанная активность в виде редких потенциалов фибрилляций с удлиненной длительностью потенциала двигательных единиц (ПДЕ) до 15–16 мс, амплитудой высоких ПДЕ до 2–2,5 тыс. mV, полифазия 30–40 %, при втором исследовании у пациента был выявлен отрицательный декремент Мответа при низкочастотной стимуляции 3 имп/с = –28 % (допустимый декремент = –16 %). Для исключения патологии нервномышечной передачи больному проводились фармакологическая проба (прозериновая) и проба на истощение, которые были слабоположительные.
В отделении больной прошел курс лечения карнитина хлоридом, витаминами группы В, паравертебральными блокадами с убихиноном композитум и цель Т, а также электрофорезом с прозерином и никотиновой кислотой, ЛФК и массажем.
За время пребывания в стационаре состояние больного практически не изменилось, попрежнему сохранялись жалобы на слабость, утомляемость, мышечные боли в конечностях. Таким образом, сочетание характерного клинического и электронейромиографического паттерна при отсутствии признаков вторичной симптоматической амиотрофии, позволяет нам поставить диагноз наследственной спинальной амиотрофии, недифференцированной формы, с поздним дебютом у взрослого пациента.
Описанный клинический случай, надеемся, принесет пользу практикующим врачам в обеспечении своевременной диагностики, адекватной терапии и правильного прогноза у указанной категории пациентов.
1. Евтушенко С.К., Шаймурзин М.Р., Евтушенко О.С. и др. Ранняя клиникоинструментальная диагностика и терапия быстро и медленнопрогрессирующих мышечных дистрофий и амиотрофий // Международный неврологический журнал. — 2007. — № 4(14). — С. 1430.
2. Бадалян Л.О., Скворцов И.А. Клиническая электронейромиография. Руководство для врачей. — М.: Медицина, 1986. — 368 с.
3. Дубинская Е.Э., Вяткина С.Я. Атипичная спинальная амиотрофия взрослых // Журн. неврол. и психиат. — 1991. — № 3. — С. 1821.
4. Калмыкова Л.Г. Наследственная гетерогенность болезней нервной системы. — М.: Медицина, 1976. — 319 с.
5. Pyatt R.E., Prior T.W. A feasibility study for newborn screening of spinal muscular atrophy // Genet. Med. — 2006. — № 8. — Р. 428437.
6. Pearn J.H., Walton J.N. A clinical and genetic study of adultonset spinal muscular atrophy. The autosomal recessive form as a discrete disease entity // Brain. — 1978. — V. 101. — P. 591606.
7. Наследственные болезни нервной системы. Руководство для врачей / Под ред. Ю.Е. Вельтищева, П.А. Темина. — М.: Медицина, 2008. — 496 с.
Синдром Рефсума: наследственная атаксическая полинейропатия с глухотой
Facebook Если у вас не работает этот способ авторизации, сконвертируйте свой аккаунт по ссылке ВКонтакте Google RAMBLER&Co ID
Авторизуясь в LiveJournal с помощью стороннего сервиса вы принимаете условия Пользовательского соглашения LiveJournal
Спинальные амиотрофии (СА) взрослых

СА представляют собой гетерогенную группу наследственных заболеваний периферической нервной системы, которые характеризуются выраженным клиническим полиморфизмом.
Спинальная мышечная атрофия (или СА) - разнородная группа наследственных заболеваний, протекающих с поражением и потерей моторных нейронов передних рогов спинного мозга.
Амиотрофия - это нарушение трофики мышц, сопровождающееся истончением мышечных волокон и уменьшением их сократительной способности, обусловленное поражением нервной системы: мотонейронов (на различных уровнях ЦНС - нейроны двигательной коры, ядер ствола мозга, передних рогов спинного мозга) или периферических нервных волокон. Болезнь считается наследственной в результате генных мутаций, хотя если мы посмотрим истории болезни, то у многих больных семейный анамнез не выявляется.
Различают наследственные и симптоматические амиотрофии. Нейрогенные наследственные амиотрофии делятся на две большие группы - спинальные и невральные амиотрофии. В большинстве случаев более тяжелыми являются спинальные формы. К ним относятся: спинальная амиотрофия (болезнь Верднига - Гоффманна), псевдомиопатическая прогрессирующая спинальная амиотрофия Кугельберга - Веландер, редкие формы спинальных амиотрофий и недифференцированные формы. Невральные амиотрофии: болезнь Шарко - Мари - Тута, гипертрофическая невропатия Дежерина - Сотта, синдром Русси - Леви, атактическая полинейропатия или болезнь Рефсума, а также недифференцированные формы.
СА делятся также на взрослые и детские. К проксимальным СА детского возраста относятся: острая злокачественная инфантильная СА Верднига - Гоффманна (спинальная амиотрофия 1-го типа), хроническая инфантильная СА (спинальная амиотрофия 2-го типа), ювенильная СА (болезнь Кугельберга - Веландер), редкие формы СА в детском возрасте: инфантильная нейрональная дегенерация, врожденная форма болезни Пелицеуса - Мерцбахера, врожденная цервикальная СА, атипичный вариант GM-ганглиозидоза, детский прогрессирующий бульбарный паралич (синдром Фацио - Лонде), понтобульбарный паралич с глухотой (синдром Виалетто - Ван Лэре).
СА взрослых: бульбоспинальная амиотрофия Кеннеди, дистальная СА, сегментарная СА, мономиелическая СА, скапуло-перонеальная СА Старка - Кайзера, лицелопаточно-плечевая СА Феничела, окулофарингеальная СА. Существуют также недифференцированные формы СА с быстропрогрессирующим, медленнопрогрессирующим и непрогрессирующим течением.
Согласно рекомендации Европейского консорциума по изучению нервно-мышечных заболеваний, клиническими критериями спинальной мышечной амиотрофии являются: [ 1 ] симметричная мышечная гипотония и гипотрофия, [ 2 ] фасцикуляции различных мышечных групп, [ 3 ] гипо- или арефлексия мышц конечностей, [ 4 ] отсутствие чувствительных, мозжечковых и интеллектуальных расстройств.
Обратите внимание! Патогномоничных изменений при спинальной мышечной амиотрофии нет. Однако важным является определение активности креатинкиназы сыворотки крови: считается, что превышение ее нормы более чем в 10 раз характерно для миодистрофии и противоречит диагнозу спинальной мышечной амиотрофии.
При электронейромиографии (ЭНМГ) выявляются симптомы поражения периферических моторных нейронов: спонтанная мышечная активность, увеличение длительности и амплитуды потенциалов действия двигательных единиц при нормальной скорости проведения импульсов по афферентным и эфферентным волокнам периферических нервов. При гистологическом исследовании биоптатов мышц обнаруживаются признаки денервационной мышечной атрофии.
Классическая проксимальная СА взрослых начинается на 3-м десятилетии жизни и наследуется по аутосомно-рецессивному типу. СА обычно дебютирует в 40 – 50 лет , однако встречаются случаи с началом в подростковом возрасте. Распределение мышечной слабости при аутосомно-доминантном типе в ряде случаев значительно шире, чем при аутосомно-рецессивном типе. Проксимальные мышцы также поражаются тяжелее, чем дистальные. Симптомы прогрессируют медленно , двигательные функции и способность ходить у подавляющего числа больных сохраняются и в зрелом и даже в старческом возрасте. Слабость бульбарных мышц не характерна . Глазодвигательные мышцы не поражаются . Сухожильные рефлексы угнетены или отсутствуют. Контрактуры суставов редки . Уровень КФК нормальный или незначительно повышен. Будут рассмотрены следующие формы СА взрослых:
1. бульбоспинальная амиотрофия Кенеди;
2. дистальная СА;
3. сегментарная СА;
4. мономиелическая СА;
5. скапулоперониальная СА Старка-Кайзера;
6. лицелопаточноплечевая СА Феничела;
7. окулофарингеальная спинальная амиотрофия.
Дистальна СА. Аутос.-рецес. форма может начинаться в раннем детском возрасте, тогда как аутос.-домин. форма – в 23 – 25 лет. При обоих типах наследования могут быть представлены и тяжелые клинические формы, и формы средней тяжести. Заболевание начинается со слабости и атрофии передней группы мышц голеней, которым сопутствуют деформации стоп. Сухожильные рефлексы могут быть сохранены. Клиническая картина может напоминать НМСН Ι типа, однако при СА чувствительность не нарушается. При тяжелых аутос.-рецес. формах мышечная слабость постепенно распространяется на проксимальные мышцы ног, а иногда и рук. Степень слабости в руках варьирует между разными семьями, но практически одинакова у представителей одной семьи. Примерно у 25% больных обнаруживают сколиоз. В отдельных семьях у пораженных может выявляться псевдогипертрофия или атрофия икроножных мышц. Данные ЭНМГ позволяют отличить заболевание от периферической невропатии: скорость проведения по двигательным аксонам нормальная, несмотря на признаки тотальной денервации мелких мышц стопы. Вызванные сенсорные потенциалы также нормальны. Уровень КФК нормальный, иногда умеренно повышен.
Сегментарная СА: поражаются только руки или только стопы; заболевание характеризуется генетической гетерогенностью: аутос.-дом. наследование типично для формы с дебютом у взрослых; аутос.-рецес. - для формы, начинающейся у подростков, в основном мальчиков. Атрофии кистей, как правило, асимметричные, прогрессируют в течение 2 - 4 лет и иногда поражают и предплечья. Характерны фасцикуляции и крампи. Обычно нарастание артофий со временем прекращает, но в некоторых случаях вовлекаются мышцы ног.
Скапулоперонеальная СА Старка-Кайзера. Эта редкая форма СА является генертически гетерогенной. Случаи, наследуемые аутосомно-доминантно, дебютируют на 3-4-й декаде жизни и отличаются относительно доброкачественным течением, тогда как случаи с аутосомно-рецессивным наследованием – дебютируют в 3 – 5 лет. Предполагается сцепление с локусом 12q24. У некоторых больных определяется мутация в SMN гене 5-й хромосомы, что ставит под сомнение нозологическую самостоятельность ряда случаев скапулоперонеальной СА и свидетельствует о своеобразном варианте экспрессии гена проксимальной СА. Слабость и атрофии мышц превалируют в плечелопаточной группе мышц и разгибателях стопы. Возможно медленное распространение атрофий на проксимальные отделы ног и мускулатуру тазового пояса. Дифференциальную диагностику проводят со скапулоперонеальной миодистрофией.
Лицелопаточноплечевая СА Феничела. Редкая аутосомно-рецессивная форма СА, начинающейся на 2-м десятилетии жизни. Ген пока не картиован. Заболевание имитирует лицелопаточно-плечевую миодистрофию Ландузи-Дежерина, однако при нем сухожильные рефлексы обычно вызываются, а сила мышц снижается незначительно. На ЭМГ регистрируется нейронально-аксональный тип поражения. Активность КФК нормальная. Ряд исследователей оспаривает нозологическую самостоятельность данной формы и рассматривает ее в рамках болезни Ландузи-Дежерина.
Окулофарингеальная СА. Предполагается аутосомно-доминантный тип наследования. Заболевание обычно начинается на 4-м десятилетии жизни с наружной офтальмоплегии, дисфагии и дизартрии. В некоторых случаях присоединяется слабость в дистальных отделах конечностей и мышцах спины. Течение медленное, доброкачественное. Иногда болезнь рассматривается в рамках митохондриальных миопатий.
источник: использованы материалы руководства для врачей "Болезни нервной системы" под ред. Н.Н. Яхно, Д.Р. Штульмана, изд. 2-е, том 1; Москва, "Медицина", 2001 (а также указанных ниже статей).
читайте также:
статья «Клинический случай позднего дебюта спинальной амиотрофии у взрослого пациента - этап в развитии бокового амиотрофического склероза?» Т.Б. Бурнашева; Центр Израильской Медицины, г. Алматы, Казахстан (журнал «Medicine» №12, 2014) [читать];
статья «Клинический случай позднего дебюта недифференицированной спинальной амиотрофии» Гончарова Я.А., Симонян В.А., Евтушенко С.К., Белякова М.С., Евтушенко И.С.; ГУ «Институт неотложной и восстановительной хирургии им. В.К. Гусака НАМН Украины», Донецкий национальный медицинский университет им. М. Горького (Международный неврологический журнал, №5, 2012) [читать];
Синдромы, включающие пигментный ретинит и неврологические заболевания
Пигментная дегенерация сетчатки, прогрессирующий тетрапарез, умственная отсталость и умеренно глубокая нсйросенсорная глухота у 2 мальчиков сибсов описаны Gordon, Capute и Konigsmark.
Клинические данные. Данные осмотра. Братья отличались низким ростом, атрофичной мускулатурой всех конечностей и тупым, невыразительным лицом. Окружность головы, длина тела и вес были ниже физиологических норм на 30%. Пальцы, особенно средние фаланги, были короткими. Отмечалась клинодактилия V пальцев. Наблюдались умеренные сгибательные контрактуры на ногах.
Орган зрения. Произведенное братьям в возрасте 3 и 4 лет офтальмологическое исследование обнаружило по всей сетчатке диффузное распространение крупных гранул пигмента, маленький и бледный диск зрительного нерва и суженный калибр сосудов (артериол) сетчатки. С возрастом все эти изменения становились более резкими.
Нервная система. Уровень IQ.s, установленный при психометрическом тестировании, был равен 34 и 44. Наиболее постоянным неврологическим симптомом болезни являлся прогрессирующий спастический паралич ног со сгибательными контрактурами бедер и коленей. Отмечались повышение сухожильных рефлексов и сгибательный подошвенный рефлекс. Несмотря на то что масса мышц была резко уменьшена, мышечная сила оставалась вполне хорошей. Больные ходили на широко расставленных ногах. Их походка была неустойчивой. Постепенно они ходили все с большим трудом, и в конце концов передвижение стало совсем невозможным.
Патологических движений и нарушения чувствительности не обнаружено. Выражение лица становилось все более тупым. Нарастали угнетение рвотного и глотательного рефлексов, а также нарушения смыкания век.
Орган слуха. Проведение аудиометрического исследования было затруднено из-за умственной отсталости мальчиков. Отологическое исследование патологии не выявило. ЭЭГ-аудиометрия показала умеренно глубокую пейросенсорную глухоту, охватывающую главным образом высокие частоты. Другие аудиометрические пробы не производились. Речь у обоих братьев не развивалась.
Вестибулярная система. Калорические вестибулярные пробы были нормальными.
Пигментный ретинит, прогрессирующий тетрапарез, слабоумие и нейро-сенсорная глухота. Два брата маленького роста с невыразительным лицом и низко посаженными деформированными ушами
Лабораторные данные. Рентгенограммы. На краниограмме обнаружены малые размеры и асимметрия черепа. Отмечались укорочение средних фаланг и гипоплазия V пястной кости.
Другие данные. На ЭЭГ у одного из мальчиков была обнаружена чрезмерно медленная активность, в то время как у его брата ЭЭГ была нормальная. Электроретинографическое исследование зарегистрировало у старшего мальчика почти нормальную реакцию на свет.
Рутинные анализы мочи и крови, проба на электролиты и спинномозговая жидкость были нормальными.
Патология. Результаты гистопатологического исследования не представлены.
Наследственность. Родители больших детей состояли в отдаленном родстве. У тетки по линии матери был спастический церебральный паралич, но без ретинопатии. Тип наследования более всего похож на аутосомно-рецессивный, но нельзя исключить возможность Х-сцепленного наследования.
Диагноз. При синдроме Ушера моторика и психическая сфера ингактны. Другие заболевания, включающие пигментный ретинит и глухоту, такие как синдром Кокейна, синдром Рефсума и синдром Альстрёма, имеют другие сопутствующие аномалии, отличные от спастической параплегии и умственной отсталости, наблюдающихся при этом заболевании.
Существует несколько синдромов, включающих умственную отсталость и нарушения походки. Синдром Ричарде — Раидля (Richards — Rundle) характеризуется глухотой и медленно прогрессирующей атаксией, но мышечная сила остается вполне хорошей в течение многих лет. Синдром Тройера (Тгоуег) включает спастическую параплегию и дистальиые мышечные атрофии, которые начинаются в детстве и медленно прогрессируют, пока в третьем — четвертом десятилетии жизни ходьба становится совершенно невозможной. Хотя у некоторых больных и выявляют умственную отсталость, у них не доказаны пигментный ретинит или атрофия зрительных нервов (Cross, McKusick).
В одной семье была описана спастическая параплегия с дегенерацией сетчатки в сочетании с нейросеисорной глухотой, но интеллект у больных был нормальным (Louis-Bar, Pirot, Mahloudji, Chuke)
Лечение. Так как умственная отсталость у больных выражена резко, применение слуховых аппаратов ограничено.
Прогноз. Заболевание очень медленно прогрессирует, в результате развивается полная идиотия.
Выводы. Характеристика этого синдрома включает: 1) вероятное аутосомно-рецессивное наследование, 2) прогрессирующий пигментный ретинит, 3) прогрессирующий тетрапарез, 4) выраженное прогрессирующее слабоумие, 5) умеренную нейросенсорную глухоту.
- Читать далее "Синдром Кокейна: карлик со старческим видом, умственной отсталостью и тугоухостью"
Система восстановления зрения.
Система М. С. Норбекова.
Занятия проводит преподаватель Центра академика М.С. Норбекова Екатерина Зенина.
Бесплатное ознакомительное занятие состоится 17 марта 2014 г. в 18:00.
«Пусть во всех медицинских книгах вы прочтете, что ваша болезнь неизлечима, если что-то стоящее удерживает вас в этой жизни – вы обречены на успех».
Так утверждает человек, известный не только в нашей стране, но и далеко за ее пределами, Мирзакарим Санакулович Норбеков. В юности врачи признали его безнадежно больным. Но он сумел победить свой недуг. И теперь этот сильный человек показывает путь к здоровью другим.
Более 20 лет назад академик М.С. Норбеков создал оздоровительную систему, которая позволяет человеку любого возраста за десять занятий научиться самостоятельно наводить порядок в своем организме. В 1998 году по решению Международной ассоциации независимых медицинских экспертов она признана самой эффективной среди других альтернативных оздоровительных систем.
Система ускоренного обучения саморегуляции организма предлагает десять путей выхода из нездоровья. Это и суставная гимнастика, и гимнастика воли, и гимнастика воображения, и тренировка эмоций и многое другое. Но основа – в том, что человек из неудачника превращается в Победителя.
Формула Норбекова: волевое принуждение – мышечный корсет – настроение – вера – результат! Хотите – верьте, хотите – проверьте! Не забудьте, что проверять надо не менее сорока дней и обязательно создавать при этом внутреннее состояние того, что вы Победитель!
Но для получения результата надо потрудиться. Работа над собой требует преодоления лени, поэтому в аудитории только те, кто к этому готов. Это единственное условие выздоровления. Иначе любые теоретические знания бесполезны. И Наставник и Ученик, каждый – должен пройти свою половину пути, суть которого – настрой на добро и жизнь.
За 10 дней Вы сможете: восстановить зрение и слух; избавиться от боли в позвоночнике; нормализовать давление; укрепить середечно-сосудистую и нервную системы; избавиться от бессоницы; укрепить эндокринную и имунную системы; решить гинекологически-урологические проблемы; очистить и омолодить свой организм; устранить дефекты кожи; регулировать дыхание и питание; забыть об апатии и депрессии; наладить семейные отношения; создать личную модель успеха; раскрыть свои внутренние возможности; узнать о секретах древневосточной медицины. Мы учимся исцелять себя и поддерживать свое здоровье естественными методами. Мы освоим навыки: формирования внутренней уверенности; избавления от вредных привычек; создания положительных привычек; развития навыков по Вашему выбору; постановки и достижения целей.
Учебно-оздоровительный курс по методике М.С.Норбекова направлен на обучение наших слушателей базовым возможностям управления своим организмом, вплоть до клеточного уровня.
Результатом практического освоения материалов этого курса является:
- фактическое восстановление физического здоровья;
- повышение сопротивляемости иммунной и прочих жизненно важных систем к неблагоприятным воздействиям окружающей среды;
- ускоренная регенерация тканей и, как следствие, омоложение человека;
- качественное улучшение ежедневного самочувствия, повышение общего тонуса и работоспособности.
Многие вначале сомневаются, что можно быть здоровым при помощи всего лишь каких то физических и мысленных упражнений. Не глотая пачками таблетки и пилюли получать удовольствие от здоровой полноценной жизни. Конечно, Вы можете продолжать дальше жить, как привыкли: не изменяя своим стереотипам, не веря в собственные силы и не используя возможности, которые Природа заложила в нас с детства.
Для тех, кто желает изменять свою жизнь к лучшему, мы предлагаем начать со здоровья, используя проверенную эффективную методику с маленькими восточными хитростями, адаптированными к европейскому восприятию. Она позволит Вам легко и быстро получить ощутимые результаты по улучшению своего здоровья и самочувствия уже в течение первых дней занятий.
СКИДКА 20% на весь курс предоставляется студентам, пенсионерам, инвалидам, а также двум или более представителям одной семьи.
Читайте также: