Синдром Ретта - этиология, клиника, диагностика
Добавил пользователь Владимир З. Обновлено: 29.01.2026
Синдром Дреcслера (СД) – это одно из многочисленных осложнений инфаркта миокарда (ИМ), проявляющееся перикардитом, плевритом, пневмонитом, артритом, лихорадкой, лейкоцитозом, увеличением скорости оседания эритроцитов и титров антимиокардиальных антител
Этиология и патогенез
Основная причина СД – ИМ. Считается, что СД чаще развивается после крупноочаговых и осложненных инфарктов, а также после кровотечений в полость перикарда. СД, точнее синдром постповреждения сердца, может развиваться после кардиохирургических вмешательств (постперикардиотомический синдром, посткомиссуротомический синдром). Помимо этого, типичные признаки СД могут появляться после других повреждений сердца (ранение, контузия, непроникающий удар в область грудной клетки, катетерная абляция).
В настоящее время СД рассматривается как аутоиммунный процесс, обусловленный аутосенсибилизацией к миокардиальным и перикардиальным антигенам. Определенное значение придается также антигенным свойствам крови, попавшей в полость перикарда . При постинфарктном синдроме антитела к тканям сердца обнаруживаются постоянно, хотя их нередко находят и у больных с ИМ без каких-либо признаков этого синдрома, правда в меньшем количестве
Этиологическим фактором СД может быть инфекция, в частности вирусная, поскольку у больных, у которых этот синдром развился после кардиохирургических вмешательств, часто регистрируют повышение титра противовирусных антител
Патологическая анатомия
Даже при тяжелом течении постинфарктный синдром не ведет к летальному исходу. Если же такие больные умирают от других осложнений острого ИМ, патологоанатомы обычно обнаруживают фибринозный, серозный или серозно-геморрагический перикардит. В отличие от эпистенокардического перикардита, воспаление перикарда при СД имеет диффузный характер
Эпидемиология
Признак распространенности: Редко
Постинфарктный синдром развивается обычно на 2 — 6-й неделе ИМ, иногда в более ранние и в более отдаленные сроки. Частота типичного постинфарктного синдрома составляет 3 — 5,8%, при учете же атипичных или малосимптомных форм увеличивается до 14,7 — 22,7%.
Клиническая картина
Клинические критерии диагностики
Cимптомы, течение
Классически синдром развивается на 2–4-й неделе ИМ, однако эти сроки могут уменьшаться – «ранний СД» и увеличиваться до нескольких месяцев, «поздний СД». Иногда течение СД принимает агрессивный и затяжной характер, он может длиться месяцы и годы, протекать с ремиссиями и обострениями
Основные клинические проявления синдрома: лихорадка, перикардит, плеврит, пневмонит и поражение суставов. Одновременное поражение перикарда, плевры и легких при постинфарктном синдроме наблюдается не часто. Чаще перикардит сочетается с плевритом или с пневмонитом. В ряде случаев имеет место только перикардит или плеврит, либо пневмонит.
Лихорадка при СД не имеет какой-либо строгой закономерности. Как правило, она бывает субфебрильной, хотя в отдельных случаях может быть фебрильной или вообще отсутствовать .
Перикардит является обязательным элементом СД. Клинически он проявляется болью в перикардиальной зоне, которая может иррадиировать в шею, плечо, спину, брюшную полость. Боль может быть острой приступообразной (плевритическая) или давящей, сжимающей (ишемической). Она может усиливаться при дыхании, кашле, глотании и ослабевать в вертикальном положении или лежа на животе . Как правило, она длительная и исчезает или ослабевает после появления в полости перикарда воспалительного экссудата. Главный аускультативный признак перикардита – шум трения перикарда: в первый день болезни при внимательной аускультации он определяется у абсолютного большинства (до 85 %) больных. Шум лучше всего выслушивается у левого края грудины, при задержке дыхания и наклоне туловища пациента вперед. В классическом варианте он состоит из трех компонентов – предсердного (определяется в систолу) и желудочкового (систолического и диастолического). Как и боль, шум трения перикарда уменьшается или исчезает вовсе после появления в полости перикарда выпота, раздвигающего трущиеся листки перикарда . Обычно перикардит протекает нетяжело: уже через несколько дней боли стихают, а экссудат в полости перикарда почти никогда не накапливается в таком количестве, чтобы ухудшить кровообращение, хотя иногда могут появиться признаки тяжелой тампонады сердца . Иногда воспалительный процесс в перикарде при СД принимает затяжной рецидивирующий характер и заканчивается развитием констриктивного перикардита.
При применении антикоагулянтов на фоне СД возможно также развитие геморрагического перикардита, хотя подобное осложнение может быть и при отсутствии антикоагулянтной терапии .
Плеврит. Проявляется болью в боковых отделах грудной клетки, усиливающейся при дыхании, затруднением дыхания, шумом трения плевры, притуплением перкуторного звука. Он может быть сухим и экссудативным, односторонним и двусторонним. Нередко плеврит носит междолевой характер и не сопровождается типичными физикальными симптомами .
Пневмонит. Пневмонит при СД выявляется реже, чем перикардит и плеврит. Если очаг воспаления достаточно велик, также отмечается притупление перкуторного звука, ослабленное или жесткое дыхание, появление фокуса мелкопузырчатых хрипов. Возможен кашель и выделение мокроты, иногда с примесью крови, что всегда вызывает определенные диагностические трудности .
Поражение суставов. Для СД характерно появление так называемого «синдрома плеча»: болезненных ощущений в области плечелопаточных суставов, чаще слева, ограничение подвижности этих суставов. Вовлечение в процесс синовиальных оболочек нередко приводит к возникновению болей и в крупных суставах конечностей .
Другие проявления. Проявлением постинфарктного синдрома может быть сердечная недостаточность вследствие диастолической дисфункции, геморрагический васкулит и острый гломерулонефрит .
Диагностика
Электрокардиография (ЭКГ). При наличии перикардита на ЭКГ определяются диффузный подъем сегмента ST и, периодически, депрессия сегмента PR, за исключением отведения aVR, в котором наблюдаются депрессия ST и подъем PR. По мере накопления экссудата в полости перикарда может снизиться амплитуда комплекса QRS .
Эхокардиография. При накоплении жидкости в полости перикарда выявляется сепарация его листков и могут появиться признаки тампонады сердца . Следует подчеркнуть, что для СД не характерен большой объем жидкости в полости перикарда – как правило, сепарация листков перикарда не достигает 10 мм в диастолу.
Рентгенография. Обнаруживают скопление жидкости в плевральной полости, междолевой плеврит, расширение границ сердечной тени, очаговые тени в легких.
Компьютерная или магнитнорезонансная томография также выявляют жидкость в полости плевры или перикарда и легочную инфильтрацию .
Плевральная и перикардиальная пункция. Извлеченный из полости плевры или перикарда экссудат может быть серозным или серозно-геморрагическим. При лабораторном исследовании в нем определяется эозинофилия, лейкоцитоз и высокий уровень С-реактивного белка .
Лабораторная диагностика
Часто (но не всегда!) отмечается повышение СОЭ и лейкоцитоз, а также эозинофилия . Весьма характерно резкое повышение уровня С-реактивного белка.
У больных с СД регистрируются нормальные уровни маркеров повреждения миокарда (МВ-фракция креатинфосфокиназы (МВ-КФК), миоглобин, тропонины), хотя иногда отмечается их незначительное повышение, что требует проведения дифференциальной диагностики с рецидивом ИМ
Дифференциальный диагноз
СД дифференцируют с рецидивирующим или повторным ИМ, бактериальной пневмонией, тромбоэмболией легочной артерии, перикардитом или плевритом другой этиологии и некоторыми другими более редкими заболеваниями. Помимо клинических и инструментальных данных, большое значение в дифференциальной диагностике имеют лабораторные исследования: определение маркеров повреждения миокарда (миоглобин, МВ-КФК, тропонины) и продуктов деградации фибрина (D-димер).
Осложнения
Тампонада сердца, геморрагический или констриктивный перикардит, окклюзия (сдавление) коронарного шунта и редко – анемия .
Лечение
Нестероидные противовоспалительные препараты (НПВС). Препаратом выбора при СД традиционно считается ибупрофен (400– 800 мг/сут). Реже используют аспирин. Другие НПВС не применяют из-за их негативного влияния на периинфарктную зону .
Глюкокортикоиды обычно используют при СД, рефрактерном к терапии НПВС. Как правило, применяют преднизолон , хотя можно использовать и другие препараты .
После достижения клинического эффекта глюкокортикоиды отменяют постепенно в течение 6–8 нед, так как при быстрой отмене возможен рецидив СД .
Иногда используют малые дозы глюкокортикоидов (преднизолон в дозе 15 мг/сут) в комбинации с НПВС. В тяжелых случаях глюкокортикоиды вводят парентерально. Быстрое исчезновение проявлений СД при назначении глюкокортикоидов столь характерно, что имеет определенное дифференциально-диагностическое значение .
Длительное применение глюкокортикостероидов нежелательно, так как, кроме характерных для этой группы препаратов осложнений (гастродуоденальные язвы, задержка жидкости, остеопороз), они могут способствовать формированию аневризмы и разрыву миокарда, поскольку тормозят процесс его рубцевания и вызывают истончение формирующегося рубца . Естественно, при терапии НПВС и глюкокортикоидами необходима гастропротекция.
Другие препараты. При тяжелом рефрактерном к терапии НПВС и глюкокортикоидами рецидивирующем СД могут использоваться другие препараты, такие как колхицин и метотрексат . В частности, колхицин (1,0 мг/сут) особенно эффективен при постперикардиотомическом синдроме
Антикоагулянты
В связи с угрозой развития гемоперикарда и тампонады сердца от применения антикоагулянтов при СД следует воздержаться. Если же это невозможно, то их назначают в субтерапевтических дозах .
Перикардиоцентез используют при тампонаде сердца
Перикардэктомия. Из-за угрозы усугубления или рецидива СД перикардэктомия применяется редко (при констриктивном перикардите)
Прогноз
Прогноз при СД, как правило, благоприятный. Вместе с тем его течение иногда принимает затяжной рецидивирующий характер. Кроме того, имеются данные о том, что выживаемость в течение 5 лет среди перенесших этот синдром, хотя и незначительно, но снижается.
Профилактика
Использование НПВС и глюкокортикоидов для профилактики развития СД неэффективно. В настоящее время проводится исследование COPPS (COlchicine for the Prevention of Post-pericardiotomy Syndrome) для изучения эффективности применения колхицина с целью предупреждения развития данного осложнения после кардиохирургических вмешательст в
Синдром Мартина-Белл ( Синдром ломкой X-хромосомы )
Синдром Мартина-Белл – это наследственная болезнь, которая характеризуется стойким интеллектуальным снижением, расстройствами аутистического спектра и специфическими фенотипическими особенностями. Ключевой симптом – недостаточность познавательных функций. Отмечается гиперактивность, дефицит коммуникативных способностей, замкнутость. Лицо удлиненное, ушные раковины большие, лоб выступающий, кончик носа загнутый. Диагностика основывается на клинико-анамнестических данных и результатах биогенетического анализа. Лечение симптоматическое, включает использование медикаментов и психолого-педагогическую коррекцию.
МКБ-10
Общие сведения
Причины
Синдром Мартина-Белл является результатом дефекта гена FMR1, расположенного в X-хромосоме. Наследование происходит по доминантному сцепленному с полом типу с неполной пенетрантностью. У мужчин присутствует одна X-хромосома, поэтому мутантный аллель всегда провоцирует болезнь. У женщин есть две половые хромосомы типа X: одна активная, другая – резервная, инактивированная. Таким образом, при наличии мутации в одном из двух генов FMR1 заболевание проявляется или нет в зависимости от активности измененной хромосомы. Мужчины с ломкой хромосомой X не могут передать ее сыновьям, но передают всем дочерям, которые либо болеют, либо остаются здоровыми носителями мутации. Женщины с дефектной хромосомой передают ее детям обоих полов с вероятностью 50%. Наследование синдрома учащается от поколения к поколению, этот феномен называется парадоксом Шермана.
Патогенез
При секвенировании FMR1-гена было выявлено, что основой симптоматики и цитогенетически определяемой ломкости хромосомы X является многократное увеличение количества единичных тринуклеотидов ЦГГ. Это приводит к подавлению транскрипции и последующему недостаточному производству белка FMR1, ответственного за развитие центральной нервной системы, а именно – за формирование аксонов и синапсов, появление и усложнение нейронных связей, успешность процессов обучения и запоминания.
Участок хромосом, подверженный структурным изменениям при наследственном синдроме Мартина-Белл, может находиться в четырех состояниях, характеризующихся различным удлинением повторяющихся последовательностей тринуклеотидов. При отсутствии болезни и носительства определяется нормальное количество повторов – от 6 до 39. В промежуточном состоянии диагностируется 40-60 повторов, в состоянии премутации – 55-200. В обоих случаях заболевание отсутствует. Поскольку экспансия тринуклеотидов возможна лишь в период гаметогенеза, премутация способна превратиться в полную мутацию. Это происходит при передаче измененного материнского гена, аллель «утяжеляется» во время овогенеза. При полной мутации выявляется больше 200 повторов ЦГГ, чаще всего – от 230 до 4 000.
Симптомы
Дети рождаются с увеличенной массой тела, в среднем – 3,5-4 кг. Первыми обращают на себя внимание фенотипические особенности младенцев. Характерен макроорхизм – увеличение яичек без эндокринного заболевания. Окружность головы больше нормы или соответствует ее верхним границам. Лоб высокий и широкий, лицо вытянутое с уплощенной средней частью. Нос имеет слегка клювовидный загиб, ушные раковины крупные, располагаются низко. Суставы отличаются хорошей подвижностью, кости кистей и стоп широкие. Кожа зачастую гиперэластичная, волосы и радужные оболочки глаз светлого оттенка. Фенотипические признаки могут быть выражены по-разному, от одного-двух едва определяемых до полного комплекса.
Ключевое клиническое проявление заболевания – умственная отсталость. Стойкое интеллектуальное снижение проявляется слабым развитием сложных форм мышления и памяти. Пациентам недоступно понимание абстрактно-логических высказываний и явлений, использование категорий, установление аналогий. Сравнение, анализ и обобщение могут осуществляться на простом уровне, например, в конкретных бытовых ситуациях. Словарный запас обеднен. У многих мальчиков IQ равен 40-50 баллам, реже достигает 70-79. Относительно сохранна номинативная речь и зрительное восприятие. У девочек когнитивное снижение менее выраженное, соответствует легкой степени олигофрении или пограничному уровню интеллектуального развития.
Другой типичный симптом заболевания – своеобразие речи. Она ускоренная, сбивчивая, изобилует повторами, эхолалиями и персеверациями. Аутистические расстройства представлены трудностями коммуникации и поведенческими нарушениями. Дети часто проявляют агрессивность и замкнутость при попытке установления контакта. В тяжелых случаях развивается мутизм – полное отсутствие речи как средства общения. В поведении преобладает двигательная расторможенность, гиперактивность, стереотипии, самопровреждения. Пациенты избегают смотреть в глаза, не допускают прикосновений, но по сравнению с больными аутизмом интерес к общению присутствует. Стереотипные движения включают хлопки руками, прыжки, вращения вокруг своей оси, встряхивания руками, бег по кругу, гримасничанье и однообразное хныканье. Имеются трудности планирования и контроля поведения, переключения внимания и пространственной координации.
Неврологические симптомы неспецифичны. Определяется легкое снижение мышечного тонуса, двигательная дискоординация. Недостаточное развитие мелкой моторики затрудняет освоение письма, некоторых игровых и бытовых навыков (сборки конструктора, рисования, шитья и др.). У части больных имеются глазодвигательные нарушения, усиление сухожильных рефлексов, экстрапирамидные паракинезы, например, зажмуривание глаз, нахмуривание бровей, гримасничанье. При тяжелых формах синдрома возникают эпилептические припадки. У 25% пациенток с премутационным состоянием развивается первичная недостаточность яичников.
При выраженных фенотипических изменениях заболевание может быть обнаружено с первых месяцев жизни ребенка – неонатологи и врачи-педиатры обращают внимание на увеличенные размеры яичек и характерные особенности лица. В иных случаях подозрение на умственную отсталость возникает в возрасте от полугода до 2-3 лет. В этот период прослеживается отставание умственного развития, поведенческие и речевые нарушения. Дифференциальная диагностика нацелена на исключение РАС, в частности раннего детского аутизма, а также умственной отсталости другого происхождения (не связанной с ломкостью хромосомы Х). Обследование проводится психиатрами, неврологами и врачами-генетиками, включает:
- Клинический опрос, осмотр. В беседе с ребенком на первый план выходит снижение интеллекта, гиперактивность и расторможенность поведения, нарушение коммуникативных навыков. Уровень психического развития не соответствует возрасту, методики исследования интеллекта выявляют олигофрению (IQ – 40-79 баллов). Внешне наблюдаются характерные фенотипические признаки, при неврологическом осмотре выявляется мышечный гипотонус, усиленные сухожильные рефлексы, паракинезы.
- Генеалогический анализ. В отличие от других форм олигофрении при синдроме Мартина-Белл прослеживается наследственная передача болезни. Как правило, у пациента имеются родственники с данным заболеванием, чаще – мужчины (дед, дядя, брат). Иногда признаки легкого интеллектуального снижения обнаруживаются у матери, но диагноз у нее часто не установлен (не подтвержден).
- Генетическое исследование. В лабораторных условиях исследуется строение ДНК: определяется количество ЦГГ-повторов и статус метилирования. Применяется ПЦР и цитогенетический метод. Диагноз подтверждается, если количество триплетных повторов составляет более 200. При результате 60-199 возможны легкие фенотипические проявления болезни, риск развития патологии в следующем поколении (если показатель диагностирован у женщины).
Лечение синдрома Мартина-Белл
Методы специфической терапии синдрома в настоящее время отсутствуют. Проводится симптоматическое медикаментозное лечение и психолого-педагогическая коррекция. Усилия врачей и специальных психологов направлены на минимизацию эмоционально-поведенческих отклонений, овладение навыками ходьбы, речи и общения, чтения и письма. Медикаментозная терапия включает прием психостимуляторов, антидепрессантов, ноотропов, противоэпилептических средств и гормональных препаратов (при первичной недостаточности яичников). Обучение пациентов проводится по специальным коррекционно-развивающим программам. Для улучшения социальных навыков используются методы когнитивно-поведенческой терапии, групповые тренинги.
Прогноз и профилактика
Синдром Мартина-Белла не имеет осложнений и не сокращает продолжительность жизни больных, поэтому при своевременной и адекватной медико-психолого-педагогической помощи прогноз достаточно благоприятный: пациенты осваивают навыки общения и самообслуживания, обучаются в специальных школах, иногда овладевают рабочими профессиями. Профилактика основана на медико-генетическом консультировании пар из групп риска и пренатальной диагностике синдрома. Эти меры необходимы женщинам с синдромом преждевременного истощения яичников, семьям, в которых диагностированы премутационные состояния FMR1 или выявлены случаи интеллектуальной недостаточности у мальчиков и мужчин.
Синдром Ретта - этиология, клиника, диагностика
Синдром Ретта: причины, диагностика, лечение
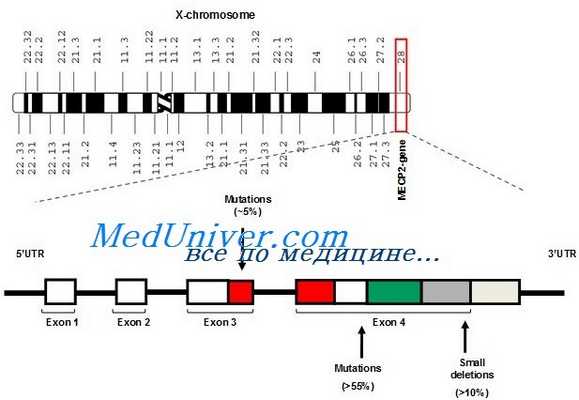
Этиология и встречаемость синдрома Ретта. Синдром Ретта (MIM № 312750) — панэтническое Х-сцепленное доминантное заболевание с распространением среди девочек 1 на 10 000-15 000.
Вызывается мутациями с утратой функции в гене МЕСР2. Описано несколько мальчиков с выраженными нарушениями развития и неврологическими аномалиями с мутациями, вызывающими частичную потерю функции МЕСР2, но обычно у мужчин типичного синдрома Ретта не бывает, кроме случаев кариотипа 47.XXY или соматического мозаицизма.
У нескольких пациентов с атипическим синдромом Ретта найдены мутации в одном, также Х-сцепленном, аллеле гена CDKL5. Белок CDKL5 — киназа треонина и серина, но о его функции мало известно.
Патогенез синдрома Ретта
Ген МЕСР2 кодирует ядерный белок, связанный с метилированием ДНК и переносящий гистоновую деацетилазу в область метилирования ДНК. Точная функция МеСР2 полностью не определена, но существует гипотеза, что он связан с транскрипционным молчанием и эпигенетической регуляцией генов в областях метилированной ДНК. Соответственно дисфункция или утрата МеСР2, наблюдаемая при синдроме Ретта, должна вызывать неправильную активизацию гена.
Мозг у пациентов с синдромом Ретта небольшого размера, с атрофией коры и мозжечка, но без потери нейронов; синдром Ретта, следовательно, не относится к типичным нейродегенеративным заболеваниям. В коре и гиппокампе нейроны пациентов с синдромом Ретта имеют меньшие размеры и более плотно упакованы, чем в норме, и имеют упрощенное ветвление дендритов.
Эти наблюдения указывают, что белок МеСР2 важен для возникновения и поддержки межнейронного взаимодействия, а не для пролиферации предшественников нейронов или их дифференцировки.
Фенотип и развитие синдрома Ретта
Впоследствии они быстро теряют речь и приобретенные двигательные навыки, особенно целенаправленного использования рук. В ходе непрерывного протекания болезни у них развиваются стереотипные движения рук, нерегулярное дыхание, атаксия и судороги.
После краткого периода псевдостабилизации, обычно в дошкольном или ранним школьном возрасте, состояние пациентов вновь ухудшается, появляется выраженная умственная отсталость, прогрессирующая спастичность, ригидность и сколиоз. Больные обычно доживают до взрослого возраста, однако продолжительность жизни уменьшена из-за повышения встречаемости необъяснимой внезапной смерти.
Кроме синдрома Ретта, мутации в гене МЕСР2 вызывают широкий спектр болезней, поражающих как мальчиков, так и девочек. Среди девочек симптоматика колеблется от сильно пораженных пациентов, не способных говорить, поворачиваться, сидеть или ходить, имеющих выраженную эпилепсию, до слабо пораженных пациентов, которые говорят, имеют сохранные двигательные функции, а также сравнительно хорошо сохранившуюся функцию рук.
У мальчиков колебания симптоматики — от внутриутробной гибели, врожденной энцефалопатии до умственной отсталости с различными неврологическими симптомами, или изолированной легкой умственной задержки; классический синдром Ретта описан только у мальчиков с соматическим мозаицизмом по мутации МЕСР2 или с дополнительной Х-хромосомой.
Лечение синдрома Ретта
Заподозренный на основе клинических признаков, диагноз синдрома Ретта обычно подтверждается ДНК-тестированием; тем не менее в настоящее время такое тестирование обнаруживает мутации в гене МЕСР2 только у 80-90% пациентов с типичным синдромом Ретта.
Клинические критерии диагноза для типичного синдрома Ретта включают нормальный пренатальный и перинатальный период, нормальную окружность головы при рождении, сравнительно нормальное развитие до 6-месячного возраста, задержку роста в возрасте между 6 и 48 мес, утрату приобретенных способностей и целенаправленных движений руками к 5-30 мес жизни, и последующее развитие стереотипных движений руками, потерю речевых навыков, выраженную психомоторную отсталость и развитие апраксическои походки и атаксии в возрасте между 12 и 48 мес жизни.
К настоящему времени эффективного лечения синдрома Ретта нет, и помощь сосредоточена на уходе и симптоматическом лечении. Медицинская помощь включает антиконвульсанты при судорогах, прием ингибиторов серотонина, карбидопы или леводопы при ригидности и мелатонина для улучшения сна. Семьи часто нуждаются в социальной поддержке, во взаимодействии с аналогичными семьями через группы взаимопомощи, а в некоторых случаях и в профессиональном консультировании.
Риски наследования синдрома Ретта
Приблизительно 99% случаев синдрома Ретта спорадические; большинство мутаций МЕСР2 возникают вновь, хотя в редких случаях они могут наследоваться от здоровой или мало пораженной матери со смещенной инактивацией Х-хромосомы. По крайней мере, 70% новых мутаций возникают в половых клетках отцов.
Если пара имеет больного ребенка, но мутация в гене МЕСР2 у родителей не выявлена, риск для будущих детей низкий, хотя и выше, чем в общей популяции, из-за возможности необнаруженного полового мозаицизма. Если же мать несет мутацию гена МЕСР2, каждый ребенок, независимо от пола, имеет 50% риск унаследовать мутацию.
Тем не менее недостаточная корреляция между генотипом и фенотипом у пациентов с мутациями в гене МЕСР2 обычно не позволяет давать прогнозы, разовьется ли у женского плода с мутацией МЕСР2 классический синдром Ретта или другая патология. Аналогично, идентификация мутации МЕСР2 у плода мужского пола также не позволяет предсказать внутриутробную гибель, развитие врожденной энцефалопатии или другой патологии.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Ретта
Синдром Ретта – генетическое заболевание, характеризующееся нарушением развития нервной системы по причине отсутствия ингибирования определенных генов. Проявлениями этого состояния являются прогрессирующая умственная отсталость у девочек (при крайне редких атипичных формах – и у мальчиков), мышечная гипотония, атаксия, искривления позвоночника. Диагностика синдрома Ретта основывается на данных общего и неврологического осмотра, магнитно-резонансной томографии, электроэнцефалографии и молекулярно-генетических анализов. Специфического лечения не существует (имеются лишь определенные наработки с обнадеживающими результатами при опытах на животных), применяют симптоматическую терапию для облегчения состояния больного.
Синдром Ретта – генетическое заболевание психоневрологического характера, практически всегда развивающееся у девочек и проявляющееся тяжелой степенью умственной отсталости. Эта патология была впервые выявлена еще в 1954 году австрийским неврологом А. Реттом, однако в качестве отдельной нозологической единицы он выделил данное заболевание лишь в 1966 году. Широкую известность в научном мире синдром Ретта получил в 1983 году после исследований Б. Хагберга. Это состояние является довольно распространенным – его встречаемость составляет примерно 1:10-15 тысяч новорожденных девочек, всего на сегодняшний день описано несколько десятков тысяч случаев патологии. Механизм наследования синдрома Ретта – доминантный, сцепленный с Х-хромосомой, именно поэтому он встречается практически всегда у девочек. У мальчиков из-за отсутствия парной Х-хромосомы генетические повреждения, приводящие к такому заболеванию, почти всегда являются летальными. Однако существует несколько атипичных форм синдрома Ретта, характеризующихся более сглаженной клинической картиной и поэтому поражающих лиц мужского пола. Кроме того, у мальчиков такая патология может развиться при наличии дополнительной Х-хромосомы – синдроме Клайнфельтера.
Причины синдрома Ретта
Этиология и патогенез синдрома Ретта достаточно сложны и обусловлены взаимодействием различных генов и их влиянием на развитие головного мозга человека. Первопричиной заболевания является нонсенс-мутация (по некоторым данным, к аналогичным нарушениям приводят и миссенс-мутации) гена MECP2, локализованного на Х-хромосоме, в результате чего его экспрессия полностью прекращается. Он кодирует специфический протеин под названием метил-CpG-связывающий белок 2, участвующий в регуляции транскрипции определенных участков ДНК. Данный белок содержит два домена, один из которых способствует его присоединению к метилированным участкам хромосом (которые расположены вблизи генов, регулирующих развитие головного мозга), а второй выступает как репрессор транскрипции. Причина синдрома Ретта как раз и заключается в отсутствии ингибирования некоторых генов, что приводит к нарушению формирования нервной ткани.
При этом синдром Ретта нельзя рассматривать как нейродегенеративное заболевание, так как при нем не наблюдается разрушения нейронов или нейроглии. Гистологические исследования тканей головного мозга больных выявляют нарушение ультраструктуры нервных клеток – уменьшение размеров, изменение количества дендритов, затрудненное образование нервных тканей. Объем нейроглии при синдроме Ретта снижен, в результате этого на макроскопическом уровне размер головного мозга тоже уменьшается на 20-30% по сравнению с возрастной нормой. Одной из причин вышеперечисленных процессов является отсутствие торможения выделения фермента GAD67 (ингибирование гена этого энзима осуществляется метил-CpG-связывающим белком 2), что, в свою очередь, приводит к увеличению концентрации тормозных трансмиттеров из группы ГАМК. В результате этого у больных синдромом Ретта наблюдается значительное превалирование процессов торможения в головном мозге, что отражается не только на физиологии центральной нервной системы, но и на ее морфологическом строении.
Врачами-генетиками было установлено, что полное отсутствие в геноме нормального гена MECP2 в подавляющем большинстве случаев является летальным состоянием и нередко приводит к внутриутробной смерти плода. Такое состояние имеет место у мальчиков (по причине наличия только одной Х-хромосомы) или у девочек-гомозигот, что встречается крайне редко. Из-за этого в половом распределении синдрома Ретта наблюдается абсолютное превалирование больных женского пола. Мутации гена MECP2 в большинстве случаев являются спонтанными или герминативными – предположительно, 70% случаев этого заболевания обусловлено генетическим дефектом Х-хромосомы в половых клетках отца. Дефекты этого гена приводят и другим патологиям центральной нервной системы – варианту Запела, синдрому Луба (Х-сцепленная умственная отсталость у мальчиков), врожденной энцефалопатии. Некоторые исследователи относят эти состояния к атипичным формам синдрома Ретта.
Симптомы синдрома Ретта
У новорожденных девочек синдром Ретта поначалу никак не проявляется, первые 6-12 месяцев развитие ребенка происходит обычными темпами без каких-либо отклонений. В дальнейшем прогрессирование заболевания характеризуется определенной стадийностью. Первая стадия синдрома Ретта, чаще всего возникающая в возрасте от 6-ти месяцев до 2,5 лет, характеризуется появлением у ребенка мышечной гипотонии, замедления психомоторного развития с последующим отставанием от сверстников, потерей интереса к играм и окружающим людям. Врачи-педиатры отмечают более медленный, нежели в норме, рост стоп и кистей в длину и замедление роста окружности головы. Иногда помимо неврологических проявлений может отмечаться нарушение работы печени, сердца, желудочно-кишечного тракта.
Вторая стадия синдрома Ретта характеризуется более выраженными клиническими проявлениями. Она развивается на протяжении 1-2 лет после появления первых симптомов заболевания, при этом у ребенка сначала наблюдается беспокойство, нарушения сна. Затем довольно быстро, всего за несколько недель, больные синдромом Ретта теряют практически все приобретенные до этого времени навыки – утрачивается речь, исчезает способность к ходьбе. Также для этой стадии развития патологии характерны расстройства дыхания – периоды апноэ по 1-2 минуты могут перемежаться с приступами учащенных и глубоких дыхательных движений (гипервентиляция). Дыхательные нарушения при синдроме Ретта отличаются наличием только при бодрствовании больного и отсутствием во время сна. Часто возникают многочисленные неврологические нарушения: атаксия, эпилептические припадки, часто повторяющиеся стереотипные движения.
Третья стадия синдрома Ретта называется псевдостационарной, так как при ней мало заметны признаки прогрессирования заболевания. Обычно она длится от 4 до 15 лет, состояние больных стабильно, однако наблюдаются судорожные приступы, глубокая умственная отсталость, гиперкинезы. В большинстве случаев синдрома Ретта этот этап оканчивается в пубертатном периоде. Четвертая стадия синдрома Ретта характеризуется уменьшением частоты эпилептических припадков вплоть до их исчезновения при прогрессировании двигательных расстройств. Большинство больных полностью теряют подвижность, возникает атрофия мышц, сосудистые нарушения в нижних конечностях, что может привести к развитию трофических язв. Из-за слабости мышечного корсета спины при синдроме Ретта возникает сколиоз или другие формы искривления позвоночника.
Диагностика синдрома Ретта производится на основании изучения анамнеза больного, его настоящего статуса, магнитно-резонансной томографии и энцефалографии, молекулярно-генетических анализов. Изучение наследственного анамнеза, как правило, не имеет особого смысла по причине спорадического характера мутаций гена MECP2. Характерными для синдрома Ретта являются нормальное развитие ребенка до 6-12 месяцев, возникновение мышечной гипотонии и беспокойства в раннем детстве, появление в дальнейшем атаксии и частых эпилептических припадков, стремительное утрачивание приобретенных навыков. В дальнейшем у больных регистрируется тяжелая умственная отсталость, мышечная слабость (вплоть до атрофии), искривление позвоночника, судорожные припадки.
При осмотре больных синдромом Ретта выявляется отставание в росте и его остановка, резкое уменьшение окружности головы, отсутствие речи (на начальных этапах патологии характерна эхолалия). На магнитно-резонансной томографии головного мозга обнаруживается уменьшение размера органа, нечеткая дифференциация серого и белого вещества, базальных ганглиев, снижение складчатости коры больших полушарий. Электроэнцефалограмма подтверждает снижение фоновой активности головного мозга и резко ослабленную реакцию на внешние раздражители.
Наиболее точную диагностическую информацию дают методы современной генетики – поиск делеций в локусе гена MECP2 или прямое секвенирование его последовательности для определения мутаций. Такое подтверждение синдрома Ретта возможно и в рамках пренатальной диагностики генетических заболеваний. Вспомогательную роль в установлении этого состояния может играть обследование внутренних органов (например, методами УЗИ) – у 20-30% больных выявляется недоразвитие печени или селезенки.
Лечение синдрома Ретта
Специфического лечения синдрома Ретта на сегодняшний день не существует. Имеются обнадеживающие данные некоторых исследовательских лабораторий, сотрудникам которых удалось «включить» ген MECP2 у мышей и тем самым добиться исчезновения симптомов заболевания. В сфере практической медицины пока доступна только симптоматическая терапия, однако и она сопряжена с рядом трудностей – в частности, эпилептические припадки при этом заболевании плохо поддаются устранению противосудорожными средствами. Также для лечения синдрома Ретта применяют ноотропные препараты, нарушения сна корректируют снотворными препаратами из группы барбитуратов или мелатонином.
Прогноз синдрома Ретта неблагоприятный, так как это заболевание неуклонно ведет к тяжелой умственной отсталости, а также к ряду двигательных и неврологических нарушений. Пациенты с этой патологией при соответствующем уходе и симптоматическом лечении способны доживать до 40-50 лет, однако риск внезапной смерти у них довольно высокий. Значительно ухудшает прогноз синдрома Ретта и снижает продолжительность жизни больных наличие пороков развития внутренних органов, что имеет место примерно в трети случаев. Основная причина летального исхода – дыхательная или полиорганная недостаточность и внезапная смерть, у взрослых больных также велик риск инсульта. Профилактика синдрома Ретта возможна только в виде пренатальной диагностики этого заболевания генетическими методами. При наличии дефектной формы гена MECP2 у мальчика нарушение формирования головного мозга и внутренних органов можно заметить при профилактических ультразвуковых исследованиях во время беременности.
Синдром Туретта
Синдром Туретта – нервно-психическое расстройство, манифестирующее в детском возрасте и характеризующееся неконтролируемыми двигательными, вокальными тиками и нарушениями поведения. Синдром Туретта проявляется гиперкинезами, выкриками, эхолалиями, эхопраксиями, гиперактивностью, которые периодически, самопроизвольно возникают и не поддаются контролю со стороны больного. Синдром Туретта диагностируется на основании клинических критериев; с целью дифдиагностики проводится неврологическое и психиатрическое обследование. В лечении синдрома Туретта используется фармакотерапия нейролептиками, психотерапия, акупунктура, БОС-терапия; иногда - глубокая стимуляция мозга (DBS).
Причины синдрома Туретта
Точные причины патологии неизвестны, однако установлено, что в подавляющем большинстве случаев в развитии синдрома Туретта прослеживается роль генетического фактора. Описаны семейные случаи заболевания у братьев, сестер (в т. ч. близнецов), отцов; у родителей и близких родственников больных детей нередко имеются гиперкинезы. По наблюдениям, преобладает аутосомно-доминантный тип наследования с неполной пенетрантностью, хотя возможен аутосомно-рецессивный путь передачи и полигенное наследование.
Нейрорадиологическими (МРТ и ПЭТ головного мозга) и биохимическими исследованиями доказано, что наследственный дефект, обусловливающий возникновение синдрома Туретта, связан с нарушением структуры и функций базальных ганглиев, изменениями нейротрансмиттерных и нейромедиаторных систем. Среди теорий патогенеза синдрома Туретта наиболее популярна дофаминергическая гипотеза, исходящая из того, что при данном заболевании имеет место либо увеличение секреции дофамина, либо повышение чувствительности рецепторов к нему. Клинические наблюдения показывают, что назначение антагонистов дофаминовых рецепторов, приводит к подавлению моторных и голосовых тиков.
Среди возможных пренатальных факторов, увеличивающих риск развития синдрома Туретта у ребенка, называются токсикозы и стрессы беременной; прием во время беременности лекарственных препаратов (анаболических стероидов), наркотических веществ, алкоголя; внутриутробная гипоксия, недоношенность, внутричерепные родовые травмы.
На манифестацию и тяжесть течения синдрома Туретта оказывают влияние инфекционные, экологические и психосоциальные факторы. В ряде случаев возникновение и обострение тиков отмечено в связи с перенесенной стрептококковой инфекцией, интоксикацией, гипертермией, назначением психостимуляторов детям с синдромом гиперактивности и дефицита внимания, эмоциональными нагрузками.
Симптомы синдрома Туретта
Первые проявления синдрома Туретта чаще всего относятся к возрасту 5-6 лет, когда родители начинают замечать странности в поведении ребенка: подмигивание, гримасничанье, высовывание языка, частое моргание, хлопки ладонями, непроизвольные сплевывания и т. д. В дальнейшем, по мере прогрессирования заболевания гиперкинезы распространяются на мышцы туловища и нижних конечностей и становятся более сложными (прыжки, приседания, выбрасывание ног, прикосновения к частям тела и пр.). Могут иметь место явления эхопраксии (повторения движений других людей) и копропраксии (воспроизведения оскорбительных жестов). Иногда тики носят опасный характер (удары головой, прикусывание губ, надавливание на глазные яблоки и пр.), в результате чего пациенты с синдромом Туретта могут наносить себе серьезные травмы.
Вокальные (голосовые) тики при синдроме Туретта так же многообразны, как и двигательные. Простые вокальные тики могут проявляться повторением бессмысленных звуков и слогов, свистом, пыхтением, вскрикиванием, мычанием, шипением. Вплетаясь в речевой поток, вокальные тики могут создавать ложное впечатление запинок, заикания и других нарушений речи. Навязчивый кашель, сопение носом нередко ошибочно воспринимаются как проявления аллергического ринита, синусита, трахеита. К звуковым феноменам, сопровождающим течение синдрома Туретта, также относятся эхолалии (повторение услышанных слов), палилалии (многократное повторение одного и того же собственного слова), копролалии (выкрикивание нецензурных, бранных слов). Вокальные тики также проявляются изменением ритма, тона, акцента, громкости, скорости речи.
Пациенты с синдромом Туретта отмечают, что перед началом тика они испытывают нарастающие сенсорные феномены (ощущение инородного тела в горле, зуд кожи, резь в глазах и пр.), вынуждающие их издать звук либо совершить то или иное действие. После окончания тика напряженность спадает. Эмоциональные переживания оказывают индивидуальное влияние на частоту и выраженность моторных и вокальных тиков (уменьшают или увеличивают).
В большинстве случаев при синдроме Туретта интеллектуальное развитие ребенка не страдает, однако отмечаются трудности в обучении и поведении, связанные, главным образом, с СГДВ. Другими поведенческими нарушениями могут служить импульсивность, эмоциональная лабильность, агрессия, обсессивно-компульсивный синдром.
Те или иные проявления синдрома Туретта могут быть выражены в различной степени, на основании чего выделяю 4 степени заболевания:
- (легкая) степень – пациентам удается хорошо контролировать проявления болезни, поэтому внешние признаки синдрома Туретта не заметны для окружающих. В течение заболевания имеются короткие асимптомные периоды.
- (умеренно выраженная) степень – гиперкинезы и вокальные нарушения заметны для окружающих, однако относительная способность к самоконтролю сохраняется. «Светлый» промежуток в течение заболевания отсутствует.
- (выраженная) степень – проявления синдрома Туретта очевидны для окружающих и практически не поддаются контролю.
- (тяжелая) степень – вокальные и моторные тики преимущественно сложные, ярко выраженные, их контроль невозможен.
Проявления синдрома Туретта обычно достигают наивысшего пика в подростковом возрасте, затем, по мере взросления, могут уменьшаться или прекращаться совсем. Однако у части больных они сохраняются в течение всей жизни, усиливая социальную дезадаптацию.
Диагностика синдрома Туретта
Диагностическими критериями, позволяющими говорить о наличии синдрома Туретта, служат дебют заболевания в молодом возрасте (до 20 лет); повторяющиеся, непроизвольные, стереотипные движения нескольких мышечных групп (моторные тики); не менее одного вокального (голосового) тика; волнообразный характер течения и длительность заболевания более года.
Проявления синдрома Туретта требуют дифференциации с пароксизмальными гиперкинезами, характерными для ювенильной формы хореи Гентингтона, малой хореи, болезни Вильсона, торсионной дистонии, постинфекционного энцефалита, аутизма, эпилепсии, шизофрении. Для исключения данных заболеваний необходимо обследование ребенка детским неврологом, детским психиатром; динамическое наблюдение, проведение КТ или МРТ головного мозга, ЭЭГ.
Определенную помощь в диагностике синдрома Туретта может оказать определение уровня катехоламинов и метаболитов в моче (увеличение экскреции норадреналина, дофамина, гомованилиновой кислоты), данные электромиографии и электронейрографии (увеличение скорости проведения нервных импульсов).
Лечение синдрома Туретта
Вопрос о методах лечения синдрома Туретта решается в индивидуальном порядке, исходя из возраста пациента и степени выраженности проявлений. Хорошим эффектом при легких и умеренно выраженных проявлениях синдрома Туретта обладает детская арт-терапия, музыкотерапия, анималотерапия. Одним из ключевых звеньев терапии является психологическая поддержка и создание благоприятной эмоциональной атмосферы, окружающей ребенка.
Фармакологическая терапия показана в тех случаях, когда проявления синдрома Туретта препятствуют нормальной жизнедеятельности больного. Основными применяемыми препаратами являются нейролептики (галоперидол, пимозид, рисперидон), бензодиазепины (феназепам, диазепам, лоразепам), адреномиметики (клонидин) и др., однако их прием может быть сопряжен с длительными и кратковременными побочными эффектами.
Течение и прогноз синдрома Туретта
На фоне лечения синдрома Туретта у половины пациентов отмечается улучшение или стабилизация состояния в позднем подростковом или взрослом возрасте. При сохранении постоянных генерализованных тиков и невозможности их контроля требуется пожизненная лекарственная терапия.
Несмотря на хроническое течение, синдром Туретта не оказывает влияния на продолжительность жизни, но может значительно ухудшать ее качество. Пациенты с синдром Туретта склонны к депрессии, паническим атакам, антисоциальному поведению, поэтому нуждаются в понимании и психологической поддержке со стороны окружающих.
Читайте также:
- Локализация опухолей индуцированных радиацией. Особенности радиационного канцерогенеза
- Вспомогательные репродуктивные технологии (ВРТ). ЭКО, перенос гамет и зигот
- Левый желудочек. Клапан аорты. Синусы Вальсальвы. Клапаны сердца.
- Вегетативные расстройства при контузиях. Изменения крови при контузиях
- Лучевые признаки аномалии Эбштейна сердца плода