Синдром Ретта: причины, признаки, фенотип
Добавил пользователь Владимир З. Обновлено: 10.01.2026
Синдром Ретта - этиология, клиника, диагностика
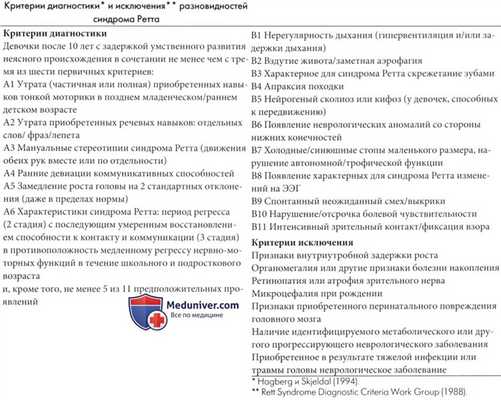
Синдром Ретта является необычным нарушением развития мозга, с напоминающими аутизм или нейродегенеративное заболевание признаками, но в действительности не является ни одним из них. Синдром Ретта представляет собой характерный комплекс клинических проявлений, включающих раннюю психомоторную регрессию с аутистическими проявлениями, замещение целенаправленных действий рук стереотипными движениями, атаксией и апраксией при ходьбе и приобретенной микроцефалией (Hagberg, 1989). В таблице 4.4 представлены международные критерии диагностики (Rett Syndrome Diagnostic Criteria Work Group, 1988; Trevathan и Naidu, 1988). За редким исключением данное заболевание встречается только у девочек (Zoghbi, 1988, Hagberg, 1989).
а) Распространенность. Распространенность синдрома Ретта в Швеции и западной Шотландии, составляет 1 на 10000 и 1 на 18000 девочек (Kerr и Stephenson, 1985; Hagberg, 1993; Bienvenu et al., 2006).
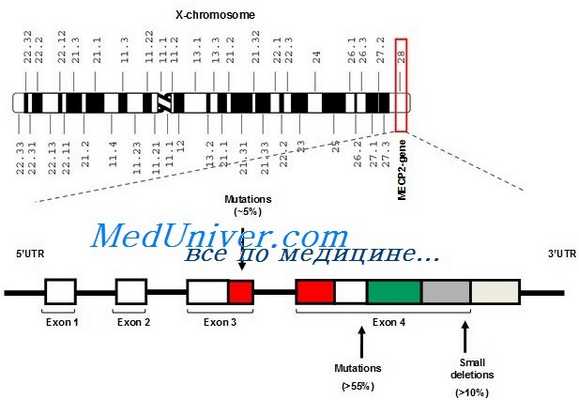
б) Патогенез. Синдром Ретта приблизительно в 80% случаев связан с мутациями гена МЕСР2, и длительное время считалось, что мутация данного гена является причиной синдрома Ретта. Тем не менее, фенотипические проявления мутаций гена МЕСР2 разнообразны и включают задержку умственного развития с припадками или без них, фенотип, подобный синдрому Ангельмана, и аутизм (Zoghbi, 2005). Как минимум еще один ген (CDKL5) связан с развитием судорожного варианта заболевания. Ген МЕСР2 является ингибитором фактора транскрипции, способным отключать несколько важных для развития головного мозга генов, а в связи с экспрессированием в различных типах клеток и органов— влиять на соматическое развитие в целом. В этой связи, представляется вероятным, что мутация гена МЕСР2 при синдроме Ретта является частью последовательной цепи событий, приводящих к развитию ряда сцепленных с Х-хромосомой нарушений развития нервной системы.
В настоящее время синдром Ретта представляется скорее патологией развития, а не дегенеративным заболеванием (Naidu, 1997). Структурные аномалии мозга выражены слабо и включают маленький размер мозга с плотно расположенными нейронами и снижением клеточных процессов. В подавляющем большинстве случаев заболевание не имеет наследственного характера, несмотря на то, что выборочное поражение девочек предполагает генетическое происхождение синдрома.

в) Клинические проявления. Течение синдрома Ретта имеет необычный характер. Заболевание начинается как прогрессирующее состояние с более или менее стремительной деградацией с утратой ранее полученных навыков.
Течение заболевания может быть разделено на четыре стадии. Дебют клинических симптомов приходится на возраст от шести месяцев до трех лет, в большинстве случаев заболевание манифестирует до 18 месяцев. Изначально развитие ребенка может не отличаться от нормы, но у больных девочек часто с рождения отмечается гипотония и слегка замедленное развитие (Einspieler et al., 2005).
Большинство девочек с синдромом Ретта развиваются нормально или почти нормально до 6-16 месяцев. Ретроспективно часто отмечается умеренная гипотония и минимальная задержка развития. Освоение целенаправленных движений рук является предварительным условием постановки диагноза. В дальнейшем у многих пациентов отмечается остановка развития или резкая утрата навыков (возможна потеря социальной улыбки, способности к взаимодействию и некоторых речевых навыков). Основным проявлением является утрата мануальных навыков. Данное проявление часто развивается стремительно в течение нескольких недель или носит «взрывной» характер, возникая в течение нескольких дней. Некоторые, но не все, дети становятся отчужденными, эмоционально отстраненными и описываются как «аутисты». У других медленно развивается малоэмоциональный стиль социального взаимодействия, иногда определяемый как аутистический.
Небольшое количество пациентов страдает от приступов ярости, тревоги, смущения и беспорядочной гиперактивности. При наличии аутистической или подобной аутистической фазы данное состояние может длиться от одного месяца до нескольких лет. Обычно к достижению школьного возраста (или не позднее пубертатного периода) аутистические симптомы начинают убывать, но не во всех случаях. По имеющимся данным, у большинства аутистов, вне зависимости от причины заболевания, присутствует одинаковый характерный тип развития.
Для девочек с синдромом Ретта характерны различные виды стереотипных движений рук, большая часть которых включают «движения в области средней линии», то есть обе руки «моются» или складываются по средней линии, обе руки засовываются в рот или шлепают по средней линии лба или шеи. На ранней стадии возможны более типичные для аутизма стереотипии с хлопаньем в ладоши.
Бруксизм и гипервентиляция являются типичными проявлениями синдрома Ретта и иногда интерпретируются как признаки чрезвычайной тревожности, что не подтверждается опытом.
Третья стадия заболевания характеризуется медленным появлением неврологических признаков, таких как пирамидные знаки. Эпилептические припадки на данной стадии развиваются у 2/3-3/4 больных. Нередки предшествующие изменения ЭЭГ, включающие ритмичную тета-активность в лобно-центральной области, пароксизмальные проявления (пики или комплексы «пик-волна»), часто локализованные в задних отделах, вспышки медленных комплексов «пик-волна» (особенно во время сна) и прогрессирующее замедление и деградация фонового ритма (Niedermeyer et al., 1986, Glaze et al, 1987).
На МРТ выявляется уменьшение объема мозга, преимущественно за счет белого вещества, уменьшение объема хвостатого ядра и среднего мозга и нормальное строение извилин (Reiss et al., 1993).

г) Разновидности синдрома Ретта. Фенотип девочек с синдромом Ретта отличается большим разнообразием и варьирует от врожденных форм с практически полным отсутствием психического развития до легких форм, при которых может сохраняться способность к передвижению и даже речь (Huppke et al., 2003). Выделяют «скрытые формы», при которых проявления заболевания типичны, но выражены слабо; тяжелые врожденные формы, при которых практически полностью отсутствует психическое развитие; умеренные формы с регрессом в позднем детском возрасте; скрытые формы с широким диапазоном проявлений и варианты заболевания с сохранением речи (Hagberg и Skjeldal, 1994). В случае сохранения речи девочки могут обладать словарным запасом от 20 до нескольких сотен слов, а некоторые говорят длинными (обычно в виде эхолалий) предложениями (Zappella et al., 1998, 2001).
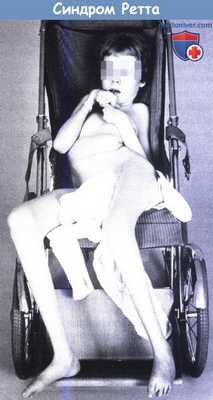
Синдром Ретта у 15-летней девочки.
Характерные стереотипные движения кистей, выраженный сколиоз и атрофия нижних конечностей.
д) Диагностика. Диагноз синдрома Ретта до сих пор устанавливается на основании анамнеза и клинических проявлений, а обнаружение мутации гена МЕСР2 во многих случаях служит подтверждением (Huppke и Gartner, 2005).
Диагностические критерии синдрома Ретта приведены в таблице ниже. У многих девочек с синдромом Ретта на ранних стадиях заболевания отмечаются аутистические проявления без четко выраженных неврологических отклонений. Поэтому в возрасте до трех лет часто ставится диагноз аутизма. Диагноз синдрома Ретта учитывается во всех случаях выявления симптомов аутизма у девочек в очень раннем детском возрасте. В исследовании Witt-Engerstrom и Gillberg (1987) девочек с синдромом Ретта в 80% случаев изначально предполагался аутизм или проявления аутизма, но на основе имеющихся данных о распространенности было установлено, что в 1/3-1/2 случаев выявления симптомов аутизма в течение первых лет жизни в итоге определялся симптом Ретта.
В редких случаях синдром Ретта характеризуется необычно медленным течением. В таких случаях диагноз «чистого аутизма» с серьезной/умеренной умственной отсталостью может не меняться в течение многих лет. Частая встречаемость атипичных форм (Goutieres и Aicardi 1985), отсутствие надежного маркера во многих случаях должны предостеречь от окончательного диагноза. Возможность выявления синдрома Ретта и задержки умственного развития у сибсов позволяет предположить наличие широкого спектра проявлений, но для подтверждения диагноза необходим определенный тест (Huppke et al., 2003). Hagberg и Skjeldal (1994) представили предварительные диагностические критерии для разновидностей синдрома Ретта.
е) Дифференциальная диагностика синдрома Ретта. В основном необходима дифференциальная диагностика с синдромом аутизма. Нейронный восковидный липофусциноз новорожденных с движениями рук, имитирующими вязание, описанный Santavuori, может иметь сходство с синдромом Ретта, но редко встречается за пределами Финляндии (Santavuori et al., 1973; Santavuori 1988). Дефицит орнитинтранскарбомилазы также ошибочно может приниматься за синдром Ретта. Синдром Ангельмана может иметь сходство с синдромом Ретта в связи с судорожной атаксией, наблюдаемой в обоих случаях помимо проявлений аутизма, задержки умственного развития и припадков. Исследование хромосом в сложных случаях позволяет дифференцировать заболевания.
Среди случаев с пограничными симптомами синдрома Ретта и аутизма (Gillberg, 1989), включая «скрытые формы» и формы заболевания с сохранением речи (Hagberg и Rasmussen, 1986), часто отмечаются многие классические проявления синдрома Ретта, но они не соответствуют всем критериям; обычно они также соответствуют большинству или всем критериям аутистического расстройства (или детского аутизма). У некоторых девочек классические симптомы аутизма проявляются только после длительного преморбидного периода (или 1 стадии) заболевания. Ретт-подобные симптомы встречаются также в сочетании с другими неврологическими расстройствами, такими как синдром Мебиуса (Gillberg и Steffenburg, 1989) и мукополисахаридоз.
ж) Лечение. Лечение синдрома Ретта неэффективно. При попытке применения бромокриптина и налоксона положительных результатов получено не было. Пациентам необходима физиотерапия и внимательное отношение к деталям обыденной жизни.
Стимуляторы показаны девочкам с хорошей реакцией на лечение в течение раннего периода отмены препарата. Важной частью терапии синдрома Ретта является ортопедическое лечение для предупреждения развития или уменьшения выраженности сколиоза.
Лечение поведенческих/психиатрических отклонений, вызванных синдромом Ретта, требует знания естественного течения заболевания, чтобы такие симптомы как аутизм и ночной смех не интерпретировались ошибочно как специфические психологические отклонения или проблемы общения. Восприятие речи при синдроме Ретта чрезвычайно снижено. Общение осуществляется с помощью зрительного контакта и жестов. Некоторые функции рук могут быть восстановлены при длительной ежедневной тренировке каждой руки в отдельности.
При применении бромокриптина (20 мг/кг в сутки) были достигнуты некоторые положительные результаты (Zappella et al., 1990), но для подтверждения эффективности необходимо проведение двойных слепых пла-цебо-контролируемых исследований. Результаты применения налоксона неоднозначны.
з) Исход. Отдаленные исходы синдрома Ретта известны только частично. Продолжительность жизни относительно увеличена, некоторые пациенты достигают возраста 80 лет и более. Подавляющее большинство (практически все) пациенты имеют чрезвычайно выраженные неврологические и/или умственные отклонения и зависят от других людей практически во всех областях повседневной жизни. В большинстве случаев клиническая картина осложняется эпилепсией, запорами, сколиозом, прогрессирующими двигательными (и вазомоторными) отклонениями. Психиатрические/по-веденческие отклонения могут являться предметом озабоченности в детском и иногда в подростковом возрасте, но обычно они в меньшей степени затрагивают пациентов старшей возрастной группы.
Синдром Ретта: причины, признаки, фенотип
Синдром Ретта: причины, диагностика, лечение
Этиология и встречаемость синдрома Ретта. Синдром Ретта (MIM № 312750) — панэтническое Х-сцепленное доминантное заболевание с распространением среди девочек 1 на 10 000-15 000.
Вызывается мутациями с утратой функции в гене МЕСР2. Описано несколько мальчиков с выраженными нарушениями развития и неврологическими аномалиями с мутациями, вызывающими частичную потерю функции МЕСР2, но обычно у мужчин типичного синдрома Ретта не бывает, кроме случаев кариотипа 47.XXY или соматического мозаицизма.
У нескольких пациентов с атипическим синдромом Ретта найдены мутации в одном, также Х-сцепленном, аллеле гена CDKL5. Белок CDKL5 — киназа треонина и серина, но о его функции мало известно.
Патогенез синдрома Ретта
Ген МЕСР2 кодирует ядерный белок, связанный с метилированием ДНК и переносящий гистоновую деацетилазу в область метилирования ДНК. Точная функция МеСР2 полностью не определена, но существует гипотеза, что он связан с транскрипционным молчанием и эпигенетической регуляцией генов в областях метилированной ДНК. Соответственно дисфункция или утрата МеСР2, наблюдаемая при синдроме Ретта, должна вызывать неправильную активизацию гена.
Мозг у пациентов с синдромом Ретта небольшого размера, с атрофией коры и мозжечка, но без потери нейронов; синдром Ретта, следовательно, не относится к типичным нейродегенеративным заболеваниям. В коре и гиппокампе нейроны пациентов с синдромом Ретта имеют меньшие размеры и более плотно упакованы, чем в норме, и имеют упрощенное ветвление дендритов.
Эти наблюдения указывают, что белок МеСР2 важен для возникновения и поддержки межнейронного взаимодействия, а не для пролиферации предшественников нейронов или их дифференцировки.
Фенотип и развитие синдрома Ретта
Впоследствии они быстро теряют речь и приобретенные двигательные навыки, особенно целенаправленного использования рук. В ходе непрерывного протекания болезни у них развиваются стереотипные движения рук, нерегулярное дыхание, атаксия и судороги.
После краткого периода псевдостабилизации, обычно в дошкольном или ранним школьном возрасте, состояние пациентов вновь ухудшается, появляется выраженная умственная отсталость, прогрессирующая спастичность, ригидность и сколиоз. Больные обычно доживают до взрослого возраста, однако продолжительность жизни уменьшена из-за повышения встречаемости необъяснимой внезапной смерти.
Кроме синдрома Ретта, мутации в гене МЕСР2 вызывают широкий спектр болезней, поражающих как мальчиков, так и девочек. Среди девочек симптоматика колеблется от сильно пораженных пациентов, не способных говорить, поворачиваться, сидеть или ходить, имеющих выраженную эпилепсию, до слабо пораженных пациентов, которые говорят, имеют сохранные двигательные функции, а также сравнительно хорошо сохранившуюся функцию рук.
У мальчиков колебания симптоматики — от внутриутробной гибели, врожденной энцефалопатии до умственной отсталости с различными неврологическими симптомами, или изолированной легкой умственной задержки; классический синдром Ретта описан только у мальчиков с соматическим мозаицизмом по мутации МЕСР2 или с дополнительной Х-хромосомой.
Лечение синдрома Ретта
Заподозренный на основе клинических признаков, диагноз синдрома Ретта обычно подтверждается ДНК-тестированием; тем не менее в настоящее время такое тестирование обнаруживает мутации в гене МЕСР2 только у 80-90% пациентов с типичным синдромом Ретта.
Клинические критерии диагноза для типичного синдрома Ретта включают нормальный пренатальный и перинатальный период, нормальную окружность головы при рождении, сравнительно нормальное развитие до 6-месячного возраста, задержку роста в возрасте между 6 и 48 мес, утрату приобретенных способностей и целенаправленных движений руками к 5-30 мес жизни, и последующее развитие стереотипных движений руками, потерю речевых навыков, выраженную психомоторную отсталость и развитие апраксическои походки и атаксии в возрасте между 12 и 48 мес жизни.
К настоящему времени эффективного лечения синдрома Ретта нет, и помощь сосредоточена на уходе и симптоматическом лечении. Медицинская помощь включает антиконвульсанты при судорогах, прием ингибиторов серотонина, карбидопы или леводопы при ригидности и мелатонина для улучшения сна. Семьи часто нуждаются в социальной поддержке, во взаимодействии с аналогичными семьями через группы взаимопомощи, а в некоторых случаях и в профессиональном консультировании.
Риски наследования синдрома Ретта
Приблизительно 99% случаев синдрома Ретта спорадические; большинство мутаций МЕСР2 возникают вновь, хотя в редких случаях они могут наследоваться от здоровой или мало пораженной матери со смещенной инактивацией Х-хромосомы. По крайней мере, 70% новых мутаций возникают в половых клетках отцов.
Если пара имеет больного ребенка, но мутация в гене МЕСР2 у родителей не выявлена, риск для будущих детей низкий, хотя и выше, чем в общей популяции, из-за возможности необнаруженного полового мозаицизма. Если же мать несет мутацию гена МЕСР2, каждый ребенок, независимо от пола, имеет 50% риск унаследовать мутацию.
Тем не менее недостаточная корреляция между генотипом и фенотипом у пациентов с мутациями в гене МЕСР2 обычно не позволяет давать прогнозы, разовьется ли у женского плода с мутацией МЕСР2 классический синдром Ретта или другая патология. Аналогично, идентификация мутации МЕСР2 у плода мужского пола также не позволяет предсказать внутриутробную гибель, развитие врожденной энцефалопатии или другой патологии.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Специалист приемной комиссии свяжется с Вами
в ближайшее время в рабочие часы с Пн-Вс с 9:00-21:00 МСК
Перезвоните мне
Ваш персональный менеджер: Валерия
Ответственная и отзывчивая! 😊
Ожидайте
Специалист свяжется с Вами сразу в рабочее время, ежедневно с 10:00 - 19:00 МСК
Бесплатные занятия с логопедом
Бесплатный курс ИКТ для детей
Когда детям ставят диагноз «психическое расстройство», это всегда плохо, но еще вопрос, что пугает больше: страшное, но знакомое определение (вроде шизофрении или аутизма) или же незнакомые понятия – такие, например, как синдром Ретта. Мало кто из неспециалистов имеет четкое представление о том, что это такое, а ведь лечение (или хотя бы облегчение страданий) маленького пациента, страдающего психическим расстройством, возможно только при полном понимании проблемы.
Характеристика болезни
Современная наука довольно мало знает о синдроме Ретта, поскольку довольно долго он считался обыкновенным слабоумием и не изучался как нечто отдельное. Впервые мысль о том, что некоторые необычные для классического слабоумия признаки позволяют выделить особое заболевание, пришла в голову австрийскому педиатру Андреасу Ретту в 1954 году. Более десяти лет он пытался самостоятельно найти подтверждение собственной теории, что ему и удалось, однако мировое медицинское сообщество широко признало существование открытого синдрома только в 80-х годах прошлого века.
Серьезные исследования в этой отрасли и вовсе начались только на стыке тысячелетий, что не позволяет специалистам располагать достаточным багажом необходимых знаний.
Синдром Ретта в наши дни описывается как генетически обусловленная прогрессирующая деградация центральной нервной системы, что влияет на рост и работу головного мозга и опорно-двигательного аппарата. Дети с таким синдромом – необучаемые, они даже не могут самостоятельно передвигаться. Заболевание относится к редким, встречается приблизительно один раз на 10-15 тысяч младенцев, причем абсолютное большинство группы риска – девочки, а вот случаев развития синдрома у мальчиков за все время зафиксировано лишь несколько.
Из-за непродолжительности изучения болезни никто не может точно сказать, сколько лет живут такие дети, но ученые пришли к выводу, что мальчики, страдая таким диагнозом в тысячи раз реже девочек, при его наличии не выживают.
Явные признаки заболевания заметны на самых ранних этапах жизни – груднички уже демонстрируют некоторые симптомы, которые со временем усугубляются, и к четырем годам всякое развитие организма полностью останавливается.
В нынешнее время недуг считается неизлечимым, но можно облегчить страдания ребенка, а тем временем ученые со всего мира продолжают попытки найти действенные способы победить синдром Ретта.
Причины возникновения
Точные причины возникновения синдрома Ретта до сих пор остаются предметом дискуссий, поэтому можно привести в пример несколько наиболее распространенных теорий. Все они сейчас проверяются путем проведения различных исследований, и хорошо еще, что медики хотя бы установили отличие от слабоумия и теперь движутся в правильном направлении.
Чаще всего специалисты указывают на генное происхождение болезни: они говорят, что недуг вызван мутацией генов. Данное отклонение развивается до масштабов патологии еще на этапе вынашивания плода, поэтому последствия видны уже на ранних этапах жизни.
Указывается, что повышенное количество кровных связей в родословной наверняка влияет на вероятность развития синдрома Ретта, причем процент таких связей высок лишь по сравнению с нормой – 2,5% вместо 0,5%. В пользу такого фактора, как причины возникновения патологии, говорят и ярко выраженные очаги заболеваемости – обычно это небольшие, достаточно изолированные от мира деревушки, где половые связи между членами одной семьи в прошлом вполне вероятны.
Другая группа исследователей указывает на нарушения в Х-хромосоме – ломкость одного из участков ее короткого плеча.
Доказано, что иногда такая аномалия действительно наблюдается, но ученые пока не смогли обнаружить точный участок, отвечающий за развитие патологии, равно как и установить определенную закономерность, позволяющую утверждать стопроцентную взаимосвязь причины и следствия.
Существует и третья (менее известная) теория, согласно которой синдром Ретта может быть следствием метаболических аномалий – нарушенного обмена веществ, вызванного митохондриальной дисфункцией.
Отмечается, что у всех больных данным заболеванием отмечено повышенное содержание в крови двух кислот – пировиноградной и молочной, очевидны также патологии лимфоцитов и миоцитов. Впрочем, это пока только наблюдение, и специалисты сейчас не могут сказать, является ли это причиной возникновения синдрома Ретта или его следствием.
Признаки и синдромы
Существует ряд признаков, по которым можно определить, что у ребенка синдром Ретта – они используются врачами, но обратить на них внимание могут и родители ребенка, чье поведение кажется подозрительным.
Хотя принято считать, что малыши уже рождаются больными, в первые месяцы жизни это нельзя заметить практически никак. Новорожденный выглядит полностью здоровым, из каких-либо болезненных признаков можно выделить разве что незначительную вялость мышечной ткани, пониженную температуру тела и общую бледность, но эти симптомы могут быть малозаметными, а само их наличие может указывать как на синдром Ретта, так и на обычную легкую простуду.
Поскольку синдром Ретта – это замедление и последующая остановка развития, тревожные симптомы можно заметить уже в возрасте 4-6 месяцев, когда малыш начинает отставать от сверстников в таких важных показателях развития, как умение ползать и самостоятельно переворачиваться на спину. Опять же – такое отставание может быть вызвано многими факторами, но оно всегда требует усиленного внимания медиков. Постепенно проявляется замедленный рост головы (по сравнению с остальными частями тела).
Существуют и такие признаки, которые характерны исключительно для данного недуга – или же не являются типичными именно в таком сочетании при других заболеваниях:
Особые манипуляции руками. Деградация при синдроме Ретта довольно глубока – страдающая ею девочка постепенно теряет способность держать в руках предметы даже в том случае, если раньше уверенно это делала. Однако движения рук полностью не исчезают – они становятся необычными.
Для данного недуга характерными являются повторяющиеся движения, напоминающие мытье рук, а также похлопывания в ладоши и по телу, перебирание пальцами.
Отсутствие интереса к познанию. Здоровый малыш очень любопытен и пытается познавать мир любыми доступными способами, тогда как девочка, больная синдромом Ретта, отличается безразличием к окружающему миру и выраженной умственной отсталостью. Иногда ребенок успевает начать различать взрослых и даже говорить, но эти успехи вскоре исчезают.
Очень заметная непропорциональность головы (по отношению к остальным частям тела).
Судорожные припадки, часто сопровождающиеся заламыванием рук – с резким переходом от отрешенности и безэмоциональности к громкому крику; возможны также эпилептические припадки.
Стремительно прогрессирующий сколиоз. Поскольку деградация нервной системы крайне отрицательно действует на тонус мышц, осанка больной девочки очень быстро портится и приобретает явно патологические формы.
Хронология развития
У каждой маленькой пациентки синдром Ретта развивается индивидуально, но все же он позволяет специалистам выделить определенные этапы его развития:
Первая стадия. Начинается с первых признаков, отмечаемых в возрасте около 4 месяцев, и длится до 1-2 года, имеет вид постепенной стагнации развития. Становится заметно, что голова и конечности растут медленнее, чем организм в целом, мышцы также отличаются неестественной расслабленностью. Ребенок сначала не очень активно интересуется окружающей обстановкой, а затем не реагирует даже на попытку увлечь его интересной игрой.
Вторая стадия. Временные рамки – возраст от 1 до 2 лет. Если ребенок уже начал учиться ходить и немного говорить, эти навыки понемногу утрачиваются, зато появляются характерные движения, о которых упоминалось выше. Именно на этом этапе обычно окончательно диагностируют синдром Ретта, поэтому большинство характерных признаков недуга относится именно ко второй стадии. Больной ребенок проявляет беспричинное беспокойство и плохо спит, возникают проблемы с дыханием. Вероятны периодические приступы отчаянного крика, возможны эпилептические припадки. На данном этапе лечение, направленное на устранение симптомов, не дает никакого эффекта.
Из-за отказа восприятия окружающего мира больным синдромом Ретта нередко ошибочно ставят другие диагнозы – обычно аутизм или энцефалит.
Третья стадия. Длится приблизительно до десяти лет, отличается более стабильным состоянием, есть даже минимальные признаки улучшения – девочка теперь лучше спит, меньше кричит и в целом кажется куда более спокойной, иногда даже возможен эмоциональный контакт с родителями. Однако двигательная активность снижается еще больше и сменяется онемением, которое разбавляется только частыми судорогами. Умственная отсталость оценивается как глубокая.
Четвертая стадия. После десяти лет отмечается практически полное разрушение навыков двигательной активности – пациент обычно оказывается полностью или почти полностью обездвиженным. Невозможность самостоятельно передвигаться дополнительно усугубляется крайними формами сколиоза и плохим кровоснабжением конечностей. В то же время сокращается количество припадков, больной в целом может поддерживать эмоциональный контакт со взрослыми. Нет каких-либо существенных отклонений в половом созревании. Нет также какой-либо привязки к продолжительности жизни – есть обрывочные сведения, что в таком состоянии человек может прожить десятки лет.
Особая проблема заключается в том, что синдром Ретта очень часто путают с аутизмом – это вызвано как некоторым подобием симптомов на ранних этапах развития, так и не слишком большой известностью рассматриваемого нами сейчас недуга. Исследователи проблемы обращают внимание на то, что первые признаки аутизма наблюдаются уже в первые недели жизни, тогда как больные синдромом Ретта в первые месяцы выглядят полностью здоровыми.
Аутисты манипулируют окружающими предметами, их движения не выглядят какими-то чрезвычайно неловкими, а вот страдающие синдромом Ретта дети очень скованны в движениях, хуже дышат, отличаются маленькой головой и склонностью к эпилептическим припадкам.
Важно правильно отличить синдром Ретта от аутизма, чтобы более эффективно бороться с его проявлениями и не усугубить состояние маленькой пациентки еще больше.
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами.
Синдром Ретта – это генетическое заболевание, сопровождающееся тяжелыми психоневрологическими симптомами. Диагностика его затруднительна: оно практически никогда не обнаруживается внутриутробно, а после рождения проявляется не ранее, чем через 6 месяцев. Своему носителю оно грозит глубоким слабоумием, двигательными ограничениями, дезадаптацией в социуме.
ИСТОКИ ЗАБОЛЕВАНИЯ
В масштабном формате о расстройстве заговорили в 1983 году благодаря шведскому ученому Бенгту Хагбергу. В это время он со своей группой изучал 35 подобных между собой случаев в 3 разных странах: в Португалии, Франции и Швеции.
Однако Хагберт не является первооткрывателем синдрома. Впервые его обнаружил педиатр Андреас Ретт, имя которого носит заболевание. Он наблюдал за двумя девочками, имеющими одинаковые симптомы. Их он заметил в очереди на прием. Они сидели на коленях у матерей, а те держали их за руки. Девочки раскачивались как маятники, а затем внезапно обе начали совершать стереотипные движения руками. Дети застыли в одном положении, отстраненные от окружающего мира. Взгляд был направлен в одну точку. Поражала их синхронность в движениях и поведении.
В своих письменных архивах врач отыскал подобные истории болезни, а затем отправился в Европу, чтобы разыскать и там таких же пациентов. В 1966 он сделал первые публикации своих исследований, которые, однако, не вызвали особого интереса.
Зафиксированную им болезнь Ретт назвал синдромом атрофии мозга. Сначала ее считали проявлением аутизма или шизофрении, и только лишь в 1983 году вывели в отдельную нозологическую единицу.
В настоящее время синдром относят к категории довольно редких генетических заболеваний. Он встречается с частотой случаев 1 на 15000. Причиной его называют мутацию гена МЕСР2. Этот ген отвечает за синтез определенного белка, влияющего на развитие мозга. В норме этот белок, спустя некоторое время после рождения, должен подавляться другими генами, чтобы обеспечить нормальное развитие мозга.
Если же ген МЕСР2 мутирован, то белок инактивируется не полностью, что вызывает аномальное мозговое созревание, и провоцирует развитие синдрома Ретта.
Обычно мутирующий ген располагается в Х хромосоме, потому заболеванием страдают преимущественно девочки.
ПОЧЕМУ МАЛЬЧИКИ НЕ БОЛЕЮТ
Учитывая, что мутирующий ген несет в себе Х-хромосома, то девочки в плане заболевания находятся в более «выигрышной» позиции. У них присутствует две Х-хромосомы. Поэтому если одна из них «бракованная», то вторая функционирует нормально. Это дает девочке хоть малый шанс на нормальное существование.
У мальчика Х-хромосома одна. Если она имеет мутационный ген, значит, выпадает из работы полностью, и ее нечем заменить. Такие малыши мужского пола, как правило, погибают еще внутриутробно, так и не родившись. Поэтому синдром Ретта у мальчиков встречается крайне редко.
Но, несмотря на такую особенность заболевания, очень редко, но все-таки мальчики с подобным синдромом выживают. Это может быть связано с тем, что не все гены в Х-хромосоме подвергаются мутации. Из-за этого заболевание развивается не столь остро.
Другая причина – наличие у мальчика синдрома Клайнфельтера. При этом наблюдается полисомия половых хромосом, то есть их набор составляет ХХУ. И, если одна Х-хромосома имеет патологический ген, то вторая может регулировать синтез белка и дарить мальчику возможность жизни. Получается такая же картина, как и у девочки.
КАК РАЗВИВАЕТСЯ ЗАБОЛЕВАНИЕ
Синдром Ретта у детей – довольно коварное заболевание. При рождении оно практически не проявляет себя. Первые его симптомы появляются в период от 6 мес. до полутора лет. Однако некоторые, еле заметные признаки, в первом полугодии все-таки имеются. Но они настолько ничтожны, что не привлекают внимания.
Вот что говорит мама одной из девочек с синдромом по поводу первого полугодия ее жизни. Она придала значение этим мелочам только по прошествии 1 года и 7 месяцев с рождения ее дочери, когда проявления стали уже явными. Из предвестников болезни она отметила, что ее малышка начала держать голову в 3 месяца, а не в 2, как это положено. В 6 месяцев она еще не могла сидеть, а ходить начала только в 1 год и 4 месяца. Психологически развивалась нормально, и говорить начала рано, но это были не стандартные слова «мама», «папа», а «зайчик», «мишка» и др.
В 1 год и 7 мес. она перестала узнавать родителей и, казалось, не нуждалась в них. Весь день проводила за одним однообразным занятием: кидала мяч или катала коляску. Часами ходила по кругу, пока ее не останавливали или она запиналась. Такое стереотипное поведение носит название полевого, когда действие затягивает больного, и он не может ничего сделать.
В четыре года к симптомам присоединились эпилептоидные припадки. Однако по достижении школьного возраста девочка находилась на домашнем обучении, и делала некоторые успехи.
12–6 лет – это был период ремиссии, когда болезнь практически не беспокоила. Но с 16 лет появились новые, более глубокие проблемы, связанные с костными деформациями и болезнями внутренних органов. Одна нога девочки была короче другой почти на 10 см, что не могло не препятствовать ходьбе. В 20 лет она весила всего 24 кг с ростом 158 см.
Обычно СР протекает в 4 стадии.
Первая стадия, которая, как правило, стартует с 6 месяцев до полутора лет, проявляется нарастанием раздражительности и лабильностью настроения у ребенка. Эпизоды плача и психомоторного возбуждения сменяются все большей пассивностью. Малыш бесцельно передвигается по комнате, пропадает интерес к игрушкам. Но контакт с матерью сохраняется.
Вот как описывает женщина поведение своей дочери на заре заболевания: она кричала целый день без остановки, билась головой о стены, не могла уснуть. Что бы мы ни делали, она не успокаивалась. Это был настоящий ад. Но больше угнетало то, что ни один врач не мог поставить вразумительный диагноз.
Развивается диспропорция головы и конечностей по отношению к телу. Они становятся несоизмеримо маленькими. Замедляется рост, и снижается тонус мышц.
Вторая стадия, длящаяся несколько лет, отличается пестротой симптомов. Сразу обращает на себя внимание снижение интеллектуальных способностей, развивается умственное слабоумие. Происходит регресс практически всех полученных навыков. Речь полностью исчезает или переходит в степень эхолалии – механического повторения услышанного.
Приобретенные двигательные навыки, предметно-ролевое поведение теряются и замещаются двигательными стереотипами. Характерный симптом: многочисленно повторяющиеся движения, напоминающие мытье рук. Кроме этого, ребенок постоянно заламывает или потирает их, размахивает ими, хлопает в ладоши. Сжатие пальцев рук вполне нормально в 4 месяца, но в более позднем возрасте говорит об остановке развития. Малыш утрачивает хватательный рефлекс, не способен производить вращательные движения руками.
Постепенно двигательная активность сходит на нет. Нарушается походка, ребенок ходит, не сгибая коленей.
Третья стадия длится 10 лет и более, характеризуется она развитием стойкого, глубокого слабоумия, вплоть до идиотии. Наблюдается полная потеря способности говорить и понимать обращенную к ребенку речь. Появляется тремор всего тела, отягчающий движения. Усиливаются судорожные припадки.
Четвертая, конечная стадия – это период усугубления ранее проявляемых симптомов. Стойкая утрата умственных способностей, двигательных навыков, развитие мышечных дистрофий, приводящих к полному обездвиживанию.
Продолжительность жизни таких больных в среднем колеблется до 30 лет, хотя известны случаи, когда они доживали и до 50-летнего возраста.
САМЫЕ ЧАСТЫЕ СИМПТОМЫ РАССТРОЙСТВА
Типичные симптомы для синдрома Ретта – мышечные и двигательные нарушения. Мышцы находятся в гипертонусе или же, наоборот, теряют его. В этом случае у ребенка развивается неправильное положение тела, прогрессирует частичные параличи и нарушение координации. Например, девочки скрещивают ноги во время ходьбы.
Синкинезии – патологические сокращения мышц, возникают вслед за произвольным движением: простая улыбка способна вызвать резкий взмах ногой. Такое явление постепенно приводит к повреждению суставов, сухожилий и связок, провоцирует ортопедические нарушения. Последние проявляются во всевозможных деформациях и также очень часто сопровождают таких детей. Среди них выделяют вывих тазобедренного сустава, провоцируемый малой подвижностью.
Статическая деформация стопы чаще развивается из-за нарушенного мышечного тонуса. Распространенной считается патология под названием «конская стопа», связанная со снижением подвижности голеностопного сустава. Ее можно узнать по пятке, которая не достигает земли, стопа при этом смещается кнаружи или вовнутрь. Причина патологии – гипертонус икроножной мышцы.
Сколиоз – боковое искривление позвоночника, который провоцирует массу проблем у таких пациентов: деформации суставов и костей, боли во время ходьбы, в стоячем или сидячем положении, утрата способности передвигаться. Сколиоз грудного отдела вызывает легочную недостаточность. Появляются также проблемы с пищеварением.
У детей с синдромом Ретта наблюдается повышенное слюнотечение. Но это происходит не из-за избытка количества слюны, а потери способности сглатывать ее.
Нарушение питания может развиваться из-за частых приступов тошноты. Она появляется на любые аспекты питания: на определенный продукт, его температуру, на способ приготовления. Так, ребенок способен отрицательно реагировать на пищу, поданную кусочками, или на комочки в блюде.
Постоянная тошнота провоцирует отказ от питания, а значит, потерю в весе.
Плохое сглатывание слюны, которая регулирует кислотность в желудке, и повышенное внутрибрюшное давление вызывают желудочно-пищеводный рефлюкс, то есть забрасывание содержимого желудка в пищевод. Это чревато такими последствиями, как воспаление стенки пищевода, респираторные инфекции.
Малоподвижный образ жизни, неврологические расстройства, неправильное питание провоцируют возникновение запоров у детей с синдромом Ретта. Они носят тяжелый характер, поскольку способны вызывать закупорку кишечника и сильные боли.
Повышенное слюнотечение, тошнота, рефлюкс снижают потребление ребенком пищи и даже развивают на нее негативную реакцию. В результате этого ребенок теряет в весе. Этот процесс стоит строго контролировать, поскольку он чреват истощением.
Другое тяжелое расстройство связано с работой дыхательной системы, развивающееся вплоть до приступов апноэ. Это явление настолько часто среди детей с синдромом, что нередко стает причиной их гибели.
Важными патогномоничными признаками синдрома считаются проявления аутизма. Именно из-за них заболевание изначально считали одной из форм этого расстройства, а в настоящее время относят к болезням аутистического спектра.
Аутистические признаки проявляются в отстранении от окружающего мира, в том числе и от родственников. Ребенок замыкается в себе, может не откликаться, когда его зовут. Предпочитает одиночество. Дети боятся чужих людей и непривычных ситуаций.
Лицо такого ребенка становится похожим на каменное. Взгляд блуждающий или устремлен в одну точку. Поведение часто непредсказуемо: случаются приступы неутомимого смеха или плача. Склонны к самоповреждениям: царапают кожу, кусают пальцы, вырывают волосы.
НЕТИПИЧНАЯ КАРТИНА
Наряду с типичной формой заболевания, описанной выше, встречаются и атипичные формы. Они имеют свои особенности, от которых зависит тяжесть заболевания.
- Zapella – форма синдрома с неярко выраженными признаками. Речь частично сохранена, умеренно выражен сколиоз, умственная отсталость средней степени тяжести. Физически развиваются нормально.
- Hanefeld – в клинической картине преобладает раннее развитие судорожных приступов. Часто они случаются даже до появления умственной деградации.
- Rolando – на первый план выходят признаки задержки психомоторного развития. Ребенок теряет возможность передвигаться, нарастает стереотипия движений, его беспокоят дыхательные нарушения.
Синдром Ретта – сложное генетическое заболевание. Прежде всего, его сопровождает полная умственная деградация и психоневрологические нарушения, влекущие за собой многочисленные патологии других систем организма.
К сожалению, в мире еще не существует способа кардинального искоренения болезни, хотя ученые ведут постоянные разработки в этом направлении.
Лечение синдрома сводится к трем основным направлениям. Медикаментозная терапия назначается для купирования судорожных припадков и стимуляции работы головного мозга.
Диетотерапия включает в себя контроль массы тела, употребление в пищу высококалорийных, витаминизированных продуктов.
Однако наибольшее внимание уделяется реабилитационным мероприятиям, направленным на укрепление опорно-двигательного аппарата и поддержание умственного, психомоторного развития.
Важно сохранить комплексный, всесторонний подход к проблеме. Такие дети нуждаются в постоянной поддержке со стороны взрослых и веры в них. Сотрудничество с ними, как с полноценной ячейкой общества, способствует их лучшей адаптации в социуме и более благоприятному развитию.
Синдром Ретта
Синдром Ретта считается редким прогрессирующим генетическим заболеванием нервной системы. Синдром проявляется в раннем и быстром регрессе развития с последующим ухудшением ряда функций организма.
Болезнь носит имя врача – педиатра Андреаса Ретта, впервые описавшего проявления синдрома в 1966 году. На сегодняшний день его распространенность по разным данным составляет 1:10000 – 1: 15000 случаев.

Синдром Ретта - генетическое заболевание
Чтобы человеческий организм развивался, нужна информация: из чего будет построена клетка, как она будет действовать в случае изменения внешней среды, каким образом в конечном итоге положено работать тому или этому органу… Сведения о бесконечном множестве подобных "мелочей", необходимых для правильного формирования невероятно сложного человеческого тела, содержатся в молекулах ДНК. Отдельные фрагменты ДНК называются генами. Они хранят зашифрованную информацию относительно того или иного признака организма. В свою очередь, гены "уложены" в хромосомы, набором которых укомплектована каждая наша клетка.
Отдельного внимания заслуживают половые хромосомы – они отличают женский пол от мужского, а также служат средством передачи информации о строении и функционировании организма от родителей к потомству. В норме женский организм содержит две Х-хромосомы, а мужской – Х и У. Из информации родительских Х и/или У хромосом, заключенных в половых клетках – сперматозоиде и яйцеклетке – зародыш складывает свой собственный генетический код, определяющий его дальнейшее развитие.
Поломка в какой-либо из родительских хромосом (Х или У) может привести к возникновению заболевания у ребенка, даже в случае, если сами родители внешне здоровы.
При синдроме Ретта, как правило, поломка находится в Х-хромосоме, и изменяет ген МеСР2, отвечающий за развитие нервной системы, в том числе головного мозга, что ведет к проявлению болезни у девочек. У мальчиков в случае такого нарушения в единственной Х-хромосоме, не имеющей здорового дублера, болезнь оказывается летальной, такие дети гибнут вскоре после рождения. В редких случаях в мужском организме может иметься "лишняя" Х-хромосома, что позволит избежать гибели и проявиться заболеванию. Крайне редко у детей могут встречаться атипичные формы синдрома Ретта с более "мягкими" проявлениями.
Синдром Ретта у детей. Как заметить?
В большинстве случаев до 6 – 18 месяцев болезнь никак себя не обнаруживает. В раннем возрасте родители порой обращают внимание на вялость ребенка, потливость, слабый мышечный тонус, что относится к неспецифической симптоматике, то есть может наблюдаться при множестве разных состояний.
Как уже было сказано, суть синдрома Ретта – поражение нервной системы. "Сломанный" ген отвечает за формирование связи нервных клеток друг с другом (синаптогенез), а потому и внешние проявления синдрома будут выражены прежде всего в нарушении психоневрологического развития.
Стадии болезни
В своем развитии заболевание проходит 4 последовательные стадии.
I стадия (аутистическая) проявляется после 6-18 месяцев, и сопровождается отставанием в формировании возрастных навыков. К примеру, малыш не сидит, не встает или не ползает, не начинает говорить. Ранее приобретенные навыки (указательный жест, интерес к игрушкам и пр.) пропадают. Появляется "отрешенность" от внешнего мира, "уход в себя", снижается зрительный контакт, часто замедляется рост головы.
Регулярное наблюдение у врача-педиатра, который отслеживая динамику развития ребенка, нередко первым обращает внимание на несоответствие возрастным показателям и может рекомендовать родителям более полное, подробное обследование у специалистов узкого профиля.
Первая стадия болезни длится от нескольких месяцев до года, затем сменяется так называемой "быстрой деструкцией" или "быстрым регрессом".
II стадия ("быстрая деструкция") начинается в 1-4 года и длится от нескольких недель до нескольких месяцев. По мере прогрессирования поражения нервной системы появляется все больше симптомов:
● усиление аутистической отрешенности, снижение интереса к общению;
● трудности поведения: импульсивность, неусидчивость, суетливость:
● утрата ранее приобретенных навыков разговорной речи;
● двигательные расстройства: утрата произвольных (управляемых сознанием) движений рук (захват и удержание предмета и пр.), смена ведущей руки, присоединение стереотипных движений руками (заламывание, хлопанье, постукивание, скручивание, потирание, "моющие" движения, поднесение руки ко рту, битье по подбородку, заведение рук назад).
Все это – свидетельства распада двигательных актов, регулируемых корой головного мозга.
В дальнейшем к вышеописанному присоединяются признаки нарушения работы мозжечка – атаксия походки (расстройство координации движений, ребенок становятся неловкими, нарушается равновесие, при ходьбе дети широко расставляют ноги и шатаются); тремор (мышечные сокращения, "дрожание").
Утрачивается способность менять позу, вставать и садиться.
● нарушение дыхания (во время бодрствования остановка дыхания сменяется интенсивным, учащенным дыханием, зачастую сопровождаемым криком, может наблюдаться форсированное (усиленное) изгнание воздуха и слюны).
● расстройства настроения: беспричинные перепады от веселости до тоски, страхи и тревога, спонтанный плач, раздражительность.
● периферические вазомоторные расстройства (синюшность кистей и стоп, их зябкость и холодность на ощупь).
Наряду с этими признаками во II стадии синдрома Ретта может быть: скрежет зубов, расстройства сна, снижение реакции на боль, задержка роста.
III стадия (псевдостационарная) начинается с 2-10 лет и длиться годами. В это время наблюдается некоторое смягчение определенных симптомов: уходит аутистическая отрешенность, частично восстанавливается способность к общению, появляется понимание речи и жестов, в собственной речи – возникают отдельные слова.
Сглаживаются поведенческие сложности, улучшается общий фон настроения, несколько повышаются показатели внимания.
Параллельно формируются серьезные ортопедические нарушения (деформируются конечности, у ребенка появляется вывих тазобедренного сустава; вследствие прогрессирования неврологической симптоматики и атрофии мышц спины изменяется осанка, что ведет к усугублению расстройств дыхания и пищеварения).
Могут возникать эпилептиформные приступы: от так называемых "абсансов" с кратковременной утратой сознания и остановкой взора, до "больших припадков" с выраженными судорожными проявлениями.
IV (тотальная деменция) длится до десятков лет. Нарастает мышечная скованность, снижается подвижность вплоть до полной утраты навыков ходьбы.
Усугубляющиеся расстройства глотания и жевания могут приводить к отказу от твердой пищи, избирательности рациона, что служит причиной изменения веса (как снижения, так и увеличения), дефицита витаминов и минералов, утяжеления состояния ребенка.
Упомянутые расстройства глотания являются причиной слюнотечения, что в свою очередь способно опосредованно провоцировать гастроэзофагальный рефлюкс (забрасывание содержимого из желудка в пищевод) и запускать последующие нарушения работы желудочно-кишечного тракта.
Интеллектуальное развитие детей с синдромом Ретта на протяжении жизни остается на уровне умственной отсталости, а это значит, что такие дети будут постоянно нуждаться в уходе и заботе со стороны окружающих, а также специальном психолого-педагогическом сопровождении.
Атипичные формы синдрома Ретта
При атипичных формах синдрома Ретта IV стадии болезни обычно не наступает, и способность к ходьбе не утрачивается. Отрешенность от внешнего мира может сохраняться в течение всей жизни. В речи этих детей могут оставаться слова и короткие фразы в виде повторов – эхолалий и эхофразий. Возможно раннее проявление судорожного синдрома.
Постановка диагноза и лечение синдрома Ретта
Ставится исключительно врачами после полного обследования на основании сведений анамнеза, клинических признаков, данных лабораторно-инструментальных исследований, включающих ДНК-тестирование. Некоторые проявления синдрома Ретта схожи с признаками ряда других болезней, потому в вопросе диагностики важно комплексное обследование с консультацией таких врачей, как психиатр, невролог, генетик.
Дифференциальный диагноз проводится с аутизмом, детским церебральным параличом, синдромом Ангельмана, болезнями обмена веществ, умственной отсталостью.
К моменту написания этого материала специфического лечения синдрома Ретта не существует. С целью абилитации активно применяются психокоррекционные занятия, работа с дефектологами и логопедами, семейная психотерапия. При необходимости под строгим врачебным контролем используются медикаментозные средства, направленные на коррекцию тяжелых поведенческих трудностей, судорожных припадков, дыхательных расстройств, нарушений настроения и сна.
Учитывая, что синдрому Ретта часто сопутствует соматическая патология, то есть нарушение работы внутренних органов, важно помнить о необходимости дальнейшего наблюдения не только у психиатра, невролога и генетика, но еще и педиатра, ортопеда, гастроэнтеролога.
Римма Кондратьева, врач-психиатр Адаптационного отделения Центра им. Г.Е. Сухаревой ДЗМ
Читайте также: