Синдром Рувалкаба-Мюре-Смита - клиника, диагностика, лечение
Добавил пользователь Morpheus Обновлено: 30.01.2026
Начиная цикл статей, посвящённый проблемам заболеваний и восстановления кисти, начну с актуальной патологии, а именно-синдрома Зудека. Со времен описания синдрома Зудека (1900) этому заболеванию посвящены многочисленные статьи и монографии (Зудек, Маурер, Лериш, Манастро, Ридер и т.д.), но несмотря на это, причины возникновения его остаются такими же неясными , как и десятки лет тому назад. Но стоит отметить тот факт, что порядка 62-65% случаев развития данного заболевания составляет травма (перелом, последствия гнойных и неврологических заболеваний) которая приходится на лучевую кость предплечья. При этом стоит отметить возрастную группу – это люди старше 45 лет и пожилые. Развитие синдрома Зудека при поражениях нижних конечностей соответственно составляет около 30%, а плечевой кости менее 8%. Также заострить внимание в данной статье хочется на «сезонности» данного заболевания, так как падения на кисти в зимний период всегда были приоритетной травмой.
Посттравматическая рефлекторная дистрофия, дистрофия Зудека, атрофия Зудека - вот те названия, которые часто встречаются в литературе и в статьях, размещенных в интернете. Возникновение заболевания во многом зависит от тяжести травмы, своевременно оказанной травматологической помощи, правильной техники выполнения закрытой ручной репозиции при переломах дистального метаэпифиза лучевой кости, четкого соблюдение правил наложения гипсовой иммобилизации и дальнейшего этапного моделирования гипсовой повязки.


Не стоит забывать и о сопутствующих патологиях пациента, а именно наличии остеопороза, сахарного диабета, неврологических заболеваниях периферической нервной системы, атеросклероза сосудов верхних и нижних конечностей, и т.д. Все указанные предрасположенности могут вызвать синдром Зудека и после небольших травм кисти (ушибы, растяжения, повреждения капсульно-связочного аппарата лучезапястного сустава и т.п.). Но при всем выше перечисленном, одинаковые по характеру и степени повреждения,
даже при одинаковом лечении по неизвестным причинам у одних пациентов приводят, а у других не приводят к дистрофическим состояниям. Во всяком случае, лабильность психики, приводящая к вегетативной дисрегуляции, имеет большое значение. На сегодняшний момент, синдром Зудека – это заболевание всего организма, имеющее периферические симптомы, выступающие на первый план. «Под периферическим синдромом Зудека подразумевается комплекс клинических, рентгенологических и микроскопических признаков дистрофического процесса, поражающего все ткани какого-либо отдела тела человека» (Хенцл).
Восходящие нервные импульсы, исходящие из области повреждения, вызывают возбуждение симпатического нерва и этим сосудосуживающий эффект, за которым следует расширение сосудов. А нарушение равновесия сосудодвигательных нервов вызывает боль, отек и расстройство функции кисти (Лериш).
Для наглядного понимания и осознания протекающих процессов достаточно посмотреть на схему «порочного круга».
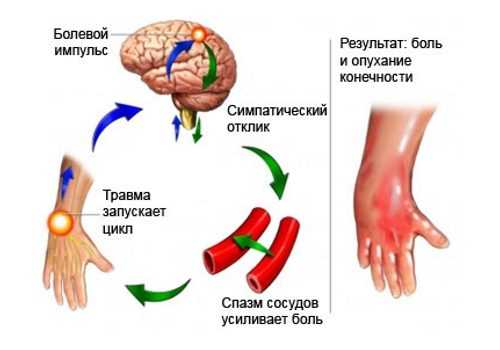
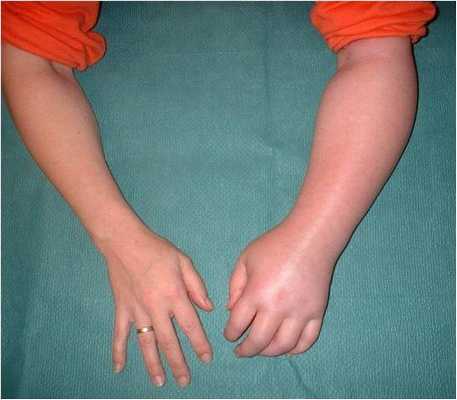
Возбуждение симпатического нерва, вызванное повреждением тканей и особенно нервных окончаний, в нормальных условиях проходит в течение 7-12 дней, и равновесие вазодвигателей восстанавливается. Однако если нормальный ход процесса восстановления конечности по каким-то причинам нарушен (о чем говорилось выше), то трофоневротические расстройства и дистрофическое состояние тканей действует на симпатическую нервную систему негативно, а порой неблагоприятно. А возбуждение последней поддерживает патологическое состояние конечности. При таком патологическом состоянии возбуждение симпатического нерва не только не уменьшается, а только увеличивается, что не создаёт возможности для восстановления равновесия сосудодвигателей. Как следствие данных процессов - «порочный круг» замкнулся в циклический процесс. Разорвать его - основная цель для устранения причины синдрома Зудека, а значит восстановить функции пораженной части тела и вернуть пациента к полноценной жизни.
Вот почему, для достижения этой непростой задачи врач травматолог берет на себя свою зону ответственности (ЛФК, ортезирование, лекарственная терапия, направленная на консолидацию перелома, профилактика контрактуры пальцев, восстановление микроциркуляции в сосудах и тканях и т.п.), а врач невролог, восстанавливает и приводит в «порядок» периферическую нервную систему (симпатическая, вегетативная, периферическая нервная система и т.п.)
Итак, рассказав о всем, что вы могли прочесть, в конечном итоге причиной болезни (синдром Зудека) является повреждение тканей. Маурер в своих монографиях, определяет следующие факторы как причины заболевания:
- Любая травма костей, сосудов и мягких тканей - ушибы, растяжения, переломы, вывихи;
- Неспецифические и специфические гнойные воспаления костей, суставов и мягких тканей;
- Повреждение и воспаление нервов;
- Ожоги, отморожения и электротравма;
- Тромбозы вен и расстройства кровообращения;
- Заболевая кожи (атрофический акродерматит, склеродермия);
- Центральные причины: опухоли мозга, спинная сухотка, сирингомиелия, полимиелит.
Как видно с факторами, перечисленными выше, трудно поспорить, а надо принять и понять, что зачастую даже с виду «здоровый пациент» может иметь подобный синдром, причина может крыться далеко не в травме.
Многочисленные исследования последних 10-15 лет утверждают, что синдром Зудека чаще всего наблюдается у пожилых пациентов с гиперфункцией щитовидной железы, у пациентов с лабильной вегетативной нервной системой, у дистоников, у больных с лабильной психикой и страдающих сахарным диабетом. Наблюдались случаи, что лечение синдрома Зудека не давало эффекта у пациентов, которые после травмы имели неприятности семейного, служебного характера или же непосредственно связанные с лечением. Хенцел в своих статьях и монографиях писал о том, что «несомненно, прав утверждая, что во время лечения следует принимать во внимание как психическое и психологическое состояние, так и социальное положение пациента». Многие, получив травму или заболев недугом, в первую очередь боятся потерять работу, и все их мысли настроены не на выздоровление, а на скорейший выход на работу.
Хочется отметить, что у детей синдром Зудека наблюдается крайне редко, единичные случаи, которые обусловлены врожденными патологиями вышеуказанных систем организма, или же тяжелейшими полифокальными травмами (переломы с обморожением или сдавлением или ожогами и т.п.).
Переходя к описанию стадий синдрома Зудека, еще раз в первую очередь хочу напомнить, что он и наступает после переломов , сопровождающихся сильными болями. Поэтому некоторые авторы придерживаются мнения, согласно которому синдром Зудека является последствием предварительного неправильного лечения.
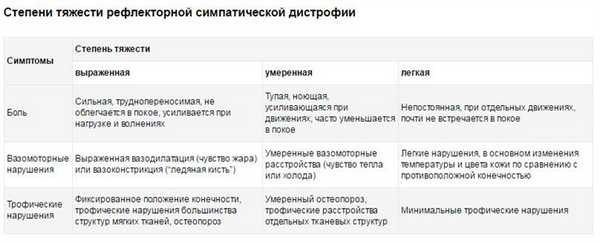
Характеристики, на которые нужно обращать пристальное внимание, при осмотре и опросе пациента для оценки тяжести рефлекторной симпатической дистрофии с целью этиотропного лечения и восстановления, приведены в таблице.
- Острая стадия (Зудек I),
- Дистрофия ( Зудек II),
- Атрофия (Зудек III),
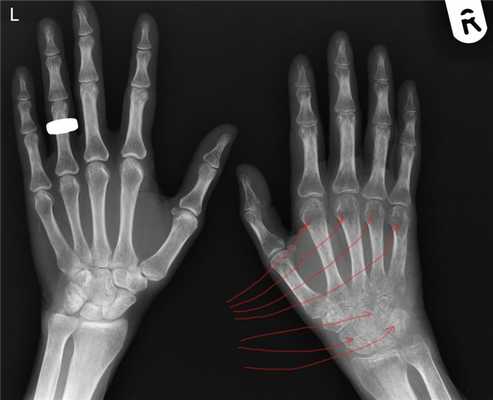
Стадия Зудек I (острая). Характеризуется отсутствием клинических симптомов улучшения (уменьшение боли, гиперемии кожи, уменьшением припухлости, сморщивания кожи т.д.), которые в нормальных условиях появляются уже в конце первой недели после повреждения. Отёк не только не уменьшается, но даже увеличивается, кожа гиперемирована, боли усиливаются, присоединяются неукротимые, не котирующиеся препаратами из группы НПВС - ночные боли. Пациент не может уснуть, как следствие истощение нервной системы на фоне прогрессирующей бессонницы. Боли не уменьшаются ни при иммобилизации (гипсовая, с использованием ортеза при растяжениях и ушибах), ни при поднятом положении конечности. Прикосновение к этому отделу конечности, особенно надавливании на мышцы, вызывает нестерпимую боль. К концу второй недели красный цвет кожи может переходить в синюшный, отмечается атрофия мускулатуры (пальцы «не слушаются, не сгибаются - сильная боль и .т.п.). Активные движения в суставах кисти резко ограничены, болезненны. На четвертой недели рентгенограмма костей кисти с захватом лучезапястного сустава показывает неравномерное затенение (на рентгене как просветление). Если имеется рентгенограмма обеих кистей и предплечья, то признаки остеопороза наиболее рано выявляются в дистальных эпифизах пястный костей.
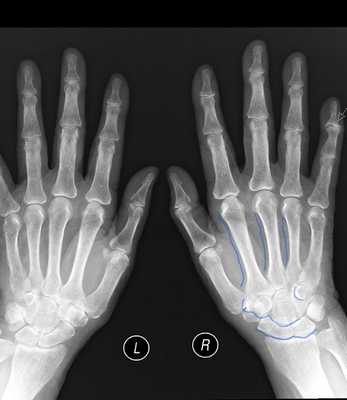
Стадия Зудек II (дистрофия). Боли в кисти держатся и не проходят. Суставы неподвижны, попытки пассивного (самостоятельно здоровой рукой двигать пальцы больной кисти) движения вызывают резкое усиление постоянно нестерпимой боли. Кожа цианотичная, на ощупь холодная, часто отмечается сильное потоотделение (ладонь больной кисти мокрая от пота). На рентгенограмме неравномерное затенение выражено не так ясно, как в 1-й стадии. Рисунок принимает облачный характер, балочки губчатой кости исчезают, корковый слой суживается. Контуры кости выявляются так ясно, как будто бы они очернены карандашом.
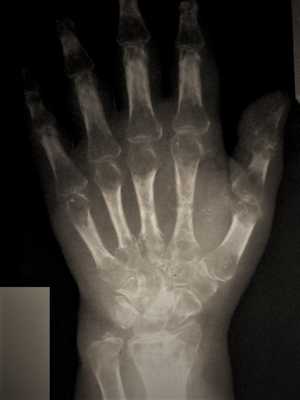
Стадия Зудек III (атрофия). В отдельных, далеко запущенных случаях и при крайне позднем обращении за квалифицированным этиотропным лечением, несмотря на предпринятые меры, патологический процесс не улучшается и в течение полутора-двух лет, от момента заболевания или травмы, доходит до конечной стадии. Кисть атрофична, холодная, суставы неподвижны. Ногти четко показывают трофические расстройства. Боли полностью прекращаются. Дифференцированные скользящие ткани кисти замещаются соединительной тканью (кисть как в панцире-перчатке, кожа в складки на тыле при защипывании не собирается). В этой стадии на рентгенограмме выявляется бедность костей известью, и помимо этого хорошо видны балки губчатой кости (крайне выраженная картина «тигрового» остеопороза).
Прогноз и лечение болезни Зудека.
В течение первой стадии процесса, при условии своевременного направления на лечение или самостоятельного обращения пациента, процесс может остановиться, при условии, если это произойдет в течение первых четырёх недель с момента травмы или заболевания. Возможно полное восстановление при комплексном подходе в терапии : врача- травматолога, врача невролога, врача-физиотерапевта , врача ЛФК и сопутствующих специалистов при наличие дополнительной патологии, которая усугубляет протекание синдрома Зудека. При второй стадии также имеется возможность восстановления, но при условии комплексного подхода указанных выше специалистов (как при первой стадии). К сожалению, третья стадия заболевания является стадией необратимых изменений, и комплексный подход в терапии будет направлен в первую очередь на поддержание функционально значимых для повседневной жизни двигательных функций кисти (самообслуживание, удержание ложки, ручки, сохранение и по возможности восстановление движений большого, указательного и среднего пальца).
Лечение (лекарственная терапия, ФТЛ, ЛФК) меняется соответственно стадиям. В первой стадии требуется соблюдение строго постельного режима! Пациент нуждается как в физическом, так и в психическом покое. Поврежденная конечность освобождается от всяких повязок, в том числе и от циркулярной гипсовой. Ей придается поднятое положение, в кровати с использованием подушек, валиков. Это так называемое - лечебное положение (лечение положением) конечности. Лекарственная терапия включает в себя сосудосуживающие, болеутоляющие (вплоть до сильнодействующих), препараты воздействующие на вегетативную нервную систему, транквилизаторы, антидепрессанты, поливитамины, гормональные препараты. Активные самостоятельные движения, занятия лечебной физкультурой ( строго с инструктором ЛФК), могут быть начаты только после купирования, исчезновения отека и болей! Уменьшение гипергидроза (повышенной потливости ладоней кисти) является благоприятным признаком, поэтому на данном этапе в терапию восстановления, при отсутствии противопоказаний, включаются физиотерапевтические процедуры, массаж и т.п. Оставлять на произвол судьбы не следует даже пациентов в конечной стадии заболевания. Продолжительное квалифицированное, этиотропное функциональное лечение все же может восстановить хотя бы минимальную функцию конечности.

Врач травматолог-ортопед по направлению ортопедической реабилитации, ЛФК и СМ Александр Борисович Тарасов
Синдром Рувалкаба-Мюре-Смита - клиника, диагностика, лечение
Синонимы: синдром Райли—Смита, синдром Баньяна—Зонана, синдром Баньяна—Райли—Рувалкаба.
Определение. Генетическое заболевание, проявляющееся 5 основными симптомами: макроцефалия, умственная отсталость, полипоз кишечника, лентиго полового члена и множественные опухоли (подкожные липомы и сосудистые мальформации).
Историческая справка. В 1960 г. американские врачи Харрис Райли и Уильям Смит описали наследственное заболевание у матери и ее четверых детей из семи, проявляющееся макроцефалией, псевдопапиллоэдемой и несколькими гемангиомами. Ими было указано, что данное заболевание ранее не описывалось.
В 1971 г. Джордж Баньян описал один случай с макроцефалией, несколькими липомами и гемангиомами. В 1976 г. Джонатан Зонана с соавторами описали пациентов с макроцефалией, липомами и гемангиомами. В 1980 г. R. Ruvalkaba, S. Myhre и D. Smith описали двух больных с макроцефалией, полипозом кишечника и лентиго на половом члене.
Этиология и патогенез. Синдром наследуется аутосомно-доминантно и относится к дисплазиям. Отмечаются мутации гена PTEN (phosphatase and tensin homolog — гомолог фосфатазы и тензина). Ген расположен в локусе 10q23.31.
Возраст и пол. Клинические признаки могут быть обнаружены при рождении. Преимущественно синдром отмечается у пациентов мужского пола.
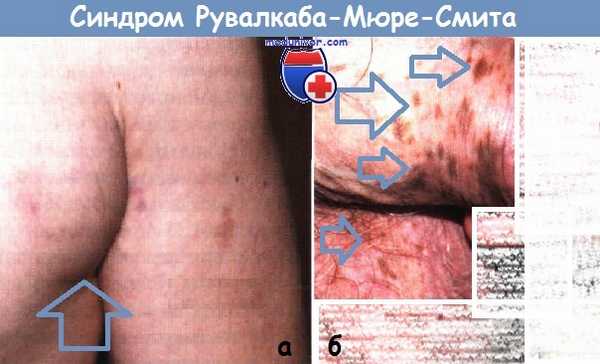
а - Подкожная липома в правой подмышечной области у того же пациента.
Липома имеет размеры 54x43x116 мм. Впервые заметил данное образование в начальных классах школы.
б - Лентигиноз полового члена и мошонки у пациента 19 лет. Очаги появились в 18 лет.
Поражение кожи и подкожной клетчатки. Определяются лентиго тела и головки полового члена, возможно увеличение полового члена, а также подкожные липомы. Липомы у детей встречаются крайне редко и их сочетание с макроцефалией позволяет заподозрить данный синдром.
Обнаруживаются, как правило, множественные подкожные липомы. Кроме того, выявляются множественные кожные сосудистые мальформации, которые могут быть врожденными, но чаще возникают после 3 лет. Они представлены подвижными кожными или подкожными узлами диаметром от 0,5 до 2 см, обычно коричневато-фиолетовой окраски, иногда болезненными.
Узлы чаще располагаются в области брюшной стенки, кистей, стоп и бедер. При гистологическом исследовании обнаруживаются мальформации смешанного типа (капиллярного, венозного и, возможно, лимфатического). Спонтанная инволюция этих образований не наблюдается. Иногда отмечаются внутричерепные сосудистые мальформации. Иногда встречается лимфедема нижних конечностей.
Другие поражения. Дети рождаются с избыточными массой тела и ростом, но затем отмечается постнатальная задержка роста. С рождения наблюдают макроцефалию, у некоторых — псевдоотек соска зрительного нерва. В дальнейшем выявляются полипоз тонкого и толстого кишечника, гамартомы внутренних органов, серозных полостей и костей, тиреоидит Хашимото, отставание в умственном развитии.
Клиническое наблюдение о сочетании лентигиноза полового члена и мошонки с липомой, сосудистой мальформацией и поражением внутренних органов представлено ниже.
Вследствие родовой травмы в возрасте 10 дней у пациента было диагностировано перинатальноепоражение головного мозга, гидроцефальный синдром. Наблюдался у педиатра. Длительно проводилась дегидратационная терапия, дважды получал стационарное лечение.
В 15 лет диагностирован диффузно-узловой зоб II степени, эутиреоз. Постоянно получал препараты йода. В 18 лет внезапно появились множественные пигментные элементы на головке и стволе полового члена, атакже на мошонке. В 19лет поступил на стационарное лечение в госпиталь. Выявлены множественные узлы в обеих долях щитовидной железы. Не исключалась возможность наличия рака, в связи с чем была выполнена тиреоидэктомия.
Назначена заместительная терапия L-тироксином в дозе 100 мг и кальций Д3-никомед 1 табл. утром. В период данной госпитализации были диагностированы лентигиноз полового члена и мошонки, липома правой подмышечной области, сосудистая мальформация в области брюшной стенки слева и очаговый фиброз в S2—S6 правого легкого без нарушения функции дыхания.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Смита-Лемли-Опица
Синдром Смита-Лемли-Опица — это редкое наследственное заболевание, которое связано с генетическим дефектом обмена холестерина. Возникает при мутации гена DHCR7 в 11-й хромосоме, в локусе 11q12-q13. Патология проявляется низкорослостью, умственной отсталостью, многочисленными костными аномалиями и врожденными пороками внутренних органов. Диагностика синдрома предполагает биохимические анализы, генетическое тестирование, инструментальные способы визуализации соответственно превалирующей симптоматике. Лечение поддерживающее: нейрореабилитационные мероприятия, помощь пластических хирургов, кардиохирургов, челюстно-лицевых хирургов.
МКБ-10
Общие сведения
Синдром назван в честь американских педиатров Д. Смита, Л. Лемли и генетика Дж. Опица, которые в 1964 г. описали его клинические признаки. Генетические механизмы развития заболевания были установлены в 1998 г. Частота встречаемости синдрома составляет в среднем 1:29000, чаще болеют европейцы (1:22000). При этом носительство мутантного гена широко распространено: до 1,2% у светлокожего населения и до 0,8% у темнокожих людей. Состояние характеризуется тяжелым течением, частой инвалидизацией, высокой смертностью, что обуславливает его актуальность в современной генетике.
Причины
Синдром Смита-Лемли-Опица связан с аномалией (миссенс- и нонсенс-мутацией) гена DHCR7, который располагается на длинном плече 11 хромосомы (11q12-q13). Он кодирует фермент 7-дегидрохолестерин-редуктазу, участвующий в заключительном этапе синтеза холестерина. Заболевание имеет аутосомно-рецессивный тип наследования. Риск возникновения синдрома у ребенка, оба родителя которого являются носителями поврежденного гена, составляет 25%.
Патогенез
Основным механизмом, который провоцирует появление патогномоничных фенотипических признаков синдрома, считается холестериновый дефицит. При этом заболевании конечная молекула не образуется, вместо нее в организме накапливаются липопротеины низкой плотности, другие промежуточные метаболиты. Эти вещества в избыточных количествах обладают токсическим влиянием на клетки.
Неврологические нарушения вызваны недостаточным образованием липидов, которые необходимы для миелинизации нервных волокон, а специфический сигнальный белок sonic hedgehog провоцирует нарушения внутриутробного формирования мозговой ткани. Дефицит холестерина коррелирует с недостатком гормонов (тестостерона, эстрогенов), что объясняет нарушения формирования наружных половых органов у мальчиков.
Симптомы
Для болезни Смита-Лемли-Опица специфичны микроцефалия, волчья пасть, микрогнатия. Реже встречается синдром Пьера-Робена в сочетании с выступающими резцами, что создает затруднения при жевании пищи, проведении анестезиологического пособия. У 95% детей наблюдается Y-образная синдактилия 2-3 пальцев ног, в половине случаев отмечается постаксиальная синдактилия. Также характерны вывих тазобедренных суставов, укорочение конечностей.
При генетическом дефекте DHCR7 типично отставание в росте, физическом развитии. С самого рождения младенцы плохо набирают вес, длина их тела ниже нормы. По мере взросления ребенка задержка роста становится более заметной, во взрослом возрасте параметры тела находятся ниже 3-го перцентиля. Нарушается половое созревание: у мальчиков выявляются гипоспадия, крипторхизм, у девочек — гипертрофия клитора.
У большинства пациентов определяется выраженная неврологическая симптоматика. Она проявляется нарушениями сна (70% больных): частыми ночными пробуждениями, неспособностью уснуть, отсутствием четкого режима дня. Психические симптомы включают повышенную агрессивность, в том числе аутоагрессию, склонность к навязчивому поведению, расстройства аутистического спектра. Большинство детей, страдающих синдромом Смита-Лемли-Опица, умственно отсталые.
Осложнения
36-38% случаев синдрома осложняется врожденными сердечными пороками. Аномалии желудочно-кишечного тракта встречаются у 25-29% больных. Они включают гастроэзофагеальный рефлюкс, болезнь Гиршпрунга, мальротацию кишечника. Распространены пороки мочеполовой системы: кистозная дисплазия почек (29%), дистопия почек (19%), гидронефроз (16%) и обструкция лоханочно-мочеточникового соединения (13%). Около 12-18% пациентов страдают от снижения зрения.
Большинству больных устанавливается инвалидность из-за выраженной умственной отсталости, отставания темпов физического развития. Синдром Смита-Лемли-Опица ассоциирован с высоким уровнем смертности как в перинатальном периоде, так и после рождения ребенка вследствие грубых врожденных аномалий. Хотя многие пациенты доживают до зрелого возраста, им требуется постоянный уход и медицинское наблюдение.
Диагностика
При физикальном осмотре ребенка удается выявить патогномоничные изменения фенотипа, что вместе с признаками задержки роста и психомоторного развития позволяет поставить предварительный диагноз. Для подтверждения синдрома Смита-Лемли-Опица назначается обследование у генетика, ряд диагностических мероприятий:
- Биохимический анализ. В плазме крови и тканях организма проверяется содержание липидов. При этом генетическом заболевании наблюдается повышение уровня 7-дегидорохолестерина одновременно со снижением количества холестерина.
- Генетическое исследование. Для обнаружения специфической мутации выполняется секвенирование экзона. При выявлении аномалии DHCR7 диагноз синдрома Смита-Лемли-Опица подтверждается со 100% точностью. Такая же диагностика показана родственникам больного.
- Пренатальная диагностика. Парам с отягощенной наследственностью рекомендуется исследование во время беременности путем измерения концентрации 7-дегидрохолестерола, генетического анализа амниотической жидкости или ворсин хориона.
- Инструментальные методы. Пациентам проводится комплексное обследование сердечно-сосудистой (эхокардиография, ЭКГ), мочеполовой (экскреторная урография, УЗИ органов малого таза), нервной системы (ЭЭГ, нейросонография, КТ или МРТ головного мозга).
Лечение синдрома Смита-Лемли-Опица
Консервативная терапия
При наличии синдрома Смита-Лемли-Опица проводится симптоматическая терапия, которая подбирается индивидуально на основании ведущих клинических признаков. Пациентам требуется помощь мультидисциплинарной команды врачей, поскольку у одного больного сочетаются разнообразные поражения внутренних органов, нарушения костно-мышечной системы, нервно-психические расстройства. Выделяют следующие направления лечения генетического синдрома:
- Диетотерапия. Для нормализации липидного профиля плазмы крови применяются пищевые добавки с кристаллическим холестерином, рацион обогащается яичными желтками, животными жирами.
- Реабилитация. Для улучшения моторных функций назначается комплексная программа ЛФК, массаж, кинезитерапия. Чтобы скорректировать нервно-психический дефицит, показаны длительные занятия с коррекционными педагогами, логопедами, психологами.
- Квалифицированный уход. Ввиду умственной отсталости пациенты не могут проживать самостоятельно. Уход за ними осуществляют родители с участием медицинского персонала, также рассматривается вариант пребывания больных в специализированных интернатах.
- Диспансерное наблюдение. Пациенты с синдромом Смита-Лемли-Опица должны регулярно проходить медицинские осмотры у ортопеда-травматолога, невролога, уролога и других специалистов. Частота обследований определяется тяжестью клинических симптомов.
Хирургическое лечение
Для устранения аномалий лицевого черепа, синдактилии, гипоспадии проводятся пластические и челюстно-лицевые вмешательства. Объем и сроки операции подбираются с учетом степени тяжести фенотипических изменений. Коррекция врожденных пороков сердца выполняется по стандартным протоколам в зависимости от характера аномалии, наличия расстройств кровообращения. Сложные пороки требуют вмешательства кардиохирургов уже на первом году жизни ребенка.
Прогноз и профилактика
При своевременной диагностике синдрома и раннем начале комплексной терапии прогноз относительно благоприятный. Врачам удается ускорить психомоторное развитие, устранить аномалии строения скелета, скорректировать соматические патологии. Однако низкий рост, умственная отсталость, проблемы с фертильностью остаются на всю жизнь. Профилактика предполагает генетическое консультирование носителей мутировавшего гена перед планированием беременности.
1. Федеральные клинические рекомендации по диагностике и лечению синдрома Смита-Лемли-Опица/ Л.П. Назаренко. — 2015.
2. Врожденные пороки развития. Синдром Смита-Лемли-Опица/ О.А. Милованова, Н.В. Чернышева, А.И. Чубарова, Л.В. Калинина// Неврологический журнал. — 2013. — №3.
3. Случай синдрома Смита-Лемли-Опица/ Л.И. Ширяева, А.М. Поздняков, Е.А. Соляник// Научно-медицинский вестник Центрального Черноземья. — 2006. — №24.
4. Синдром Смита-Лемли-Опица у детей/ А.Н. Семячкина, П.В. Новиков, М.И. Яблонская, М.Б. Кубатов// Российский вестник перинатологии и педиатрии. — 2006. — №3.
Синдром Смит-Магенис
Синдром Смит-Магенис — это редкое генетическое заболевание, которое возникает при микроповреждении короткого плеча 17-й хромосомы. Патология проявляется множественным врожденными пороками развития, среди которых преобладают аномалии лицевого скелета, а также интеллектуальными нарушениями, разнообразными поведенческими особенностями. Для диагностики синдрома Смит-Магенис назначается молекулярно-генетическое тестирование, неврологический обследование, методы инструментальной визуализации. Больным требуется помощь логопедов, реабилитологов, коррекционных педагогов, по показаниям проводится лечение соматических осложнений.
Синдром назван в честь американских врачей ‒ клинициста Энн Смит и цитогенетика Эллен Магенис, которые в 1980 г. впервые описали характерный симптомокомплекс у группы детей. Ранее считалось, что заболевание встречается с частотой 1:25000, но с развитием молекулярно-генетических методов диагностики подтвердилась более высокая распространенность — 1:15000. Чаще всего патология наблюдается у детей с задержкой психического развития — 1 случай на 569 пациентов.
Заболевание возникает вследствие спонтанной хромосомной аномалии — делеции 17 хромосомы в локусе 17p11.2, которая предполагает потерю до 4 млн. нуклеотидных пар. До 25% клинических случаев синдрома Смит-Магенис составляют атипичные микроделеции, которые выражаются в утрате меньшего участка короткого плеча хромосомы. Болезнь носит спорадический характер, пока ученым не удалось установить типичные закономерности и провоцирующие факторы.
Развитие характерной для синдрома симптоматики прежде всего связывают с нарушением гена RAI1, который является индуктором ретиноловой кислоты. Ученые обсуждают его участие в дифференцировке тканей, что объясняет формирование множественных врожденных пороков. Также RAI1 предположительно используется при дифференцировке нейронов. Некоторые психические проявления синдрома Смит-Магенис вызваны дефектом синтеза и деградации меланина.
Дети имеют типичные особенности внешности, которые обнаруживаются уже в младенческом возрасте. Обычно у них широкая квадратная форма головы, выпуклый лоб, глубоко посаженные глаза с монголоидным разрезом. Также для синдрома характерно недоразвитие средней зоны лица, короткий нос с вывернутыми ноздрями, шатровидная верхняя губа при относительно маленькой нижней челюсти. У детей непропорционально короткие руки и ноги, с раннего возраста отмечается задержка роста.
Специфическим диагностическим признаком синдрома Смит-Магенис являются нарушения сна. На первом году жизни наблюдается патологическая сонливость, такие дети много спят днем, их приходится будить для очередного кормления. Со временем нарушаются циркадные ритмы, преобладает поздний отход ко сну с частыми ночными пробуждениям. С возрастом пациент все больше спит днем, не может заснуть ночью.
Психоневрологические изменения включают умственную отсталость умеренной степени тяжести, возникающую практически у всех больных. Характерна задержка психоречевого развития, затруднения при произношении слов, дефицит внимания с гиперактивностью. До 30% пациентов имеют неврологические нарушения: расстройства координации движений, периферическую нейропатию, судорожные приступы.
Болезнь Смит-Магенис сопровождается специфическим поведенческим профилем. Дети страдают стереотипным поведением, проявляют дезадаптивные черты личности — вспышки гнева, непослушание, постоянное требование внимания к себе со стороны взрослых. Некоторые больные склонны к самоповреждению: кусают, щиплют или бьют себя, вводят инородные предметы в физиологические отверстия тела.
Большинство детей имеют мышечную гипотонию с рождения, вследствие чего возникают трудности вскармливания, нарушения глотания, отказ от пищи. В 80% случаев синдром Смит-Магенис ассоциирован с офтальмологическими проблемами: дисплазией радужки, микрокорнеа, пятнами Брушфильда. Более 60% больных страдают хроническими отитами, кондуктивной или нейросенсорной тугоухостью. У половины пациентов диагностируется гиперхолестеринемия.
Пороки сердца встречаются в 25% случаев синдрома. Наиболее распространены дефекты межпредсердной и межжелудочковой перегородок, стеноз трикуспидального клапана, тетрада Фалло. До 15% детей страдают от аномалий мочевыводящих путей, которые включают унилатеральную агенезию почек, удвоение мочеточников, увеличение линейных размеров органов.
Обследование осуществляет мультидисциплинарная команда специалистов с участием педиатра, детского невролога, психиатра, генетика. Синдром Смит-Магенис имеет патогномоничные фенотипические и поведенческие признаки, при выявлении которых устанавливается предварительный диагноз. Для верификации патологии используются следующие методы:
- Генетический анализ. Для обнаружения мутации применяется FISH гибридизация со специфическими зондами, которая определяет патологию с точностью 96-100%. Если тест не показал синдром Смит-Магенис при наличии типичных клинических признаков, выполняются дополнительные молекулярные анализы.
- Пренатальная диагностика. При отягощенном семейном анамнезе или настораживающих результатах УЗИ для исключения синдрома возможно исследование путем амниоцентеза во втором триместре беременности. Учитывая спонтанный характер мутации, такая диагностика производится редко.
- Неврологический осмотр. Пациенту требуется полное обследование с оценкой рефлексов, мышечного тонуса, проб на координацию движений. Обязательно проводятся тесты на уровень интеллекта, степень развития когнитивных функций. Зачастую осмотр дополняется консультацией психиатра.
- Инструментальная визуализация. Методы исследования назначаются дифференцировано. Может потребоваться офтальмоскопия, аудиометрия, исследование вызванных слуховых потенциалов. Для выявления соматических нарушений, типичных для клинической картины синдрома, рекомендованы эхокардиография, УЗИ брюшной полости и забрюшинного пространства.
Лечение синдрома Смит-Магенис
Показано ежегодное комплексное обследование состояния здоровья для контроля динамики нарушений. Больному подбирается индивидуальная программа симптоматической терапии. Медикаментозные средства назначаются редко, в основном для коррекции бессонницы, а психотропные препараты не используются из-за риска усугубления психических проблем. Лечение включает следующие направления:
- Логопедическая помощь. В раннем возрасте занятия с логопедом необходимы для улучшения состояния артикуляционного аппарата и структур полости рта, что облегчает кормление, создает условия для развития речи. В дальнейшем требуется продолжительная коррекция для усовершенствования вербальных навыков.
- Эрготерапия. Частые проблемы с мышечным тонусом и координацией движений требуют участия в программе лечения реабилитологов, которые помогают восстановить моторные функции, развить мелкую моторику, усовершенствовать зрительную и слуховую перцепцию.
- Коррекционные программы. Дети с болезнью Смит-Магенис хорошо учатся в небольших тихих коллективах до 5-7 человек, где каждый получает достаточное внимание и поддержку от преподавателя. Учебные программы подбираются с учетом степени интеллектуальных нарушений.
При осложненном течении синдрома схема лечения расширяется. Неврологические расстройства требуют применения физиотерапии, упражнений ЛФК, противоэпилептических препаратов. Для нормализации зрения некоторым необходима оптическая коррекция, исправление косоглазия. Хирургическое лечение пороков сердца осуществляется в соответствии с выявленной аномалией.
При своевременном начале комплексной терапии синдрома Смит-Магенис удается значительно развить моторные и речевые навыки пациента, адаптировать его к жизни в социуме. Прогноз относительно благоприятный при пожизненной реабилитации, социально-психологической поддержке. Специфические меры профилактики синдрома не разработаны, что обусловлено его спорадическим характером.
1. Smith-Magenis Syndrome / Smith, A. C., & Gropman, A. L. // Cassidy and Allanson's Management of Genetic Syndromes. — 2021.
2. Синдром Смит-Магенис: клиникогенетическая характеристика и молекулярно-цитогенетическая диагностика/ О.М. Хурс, Н.В. Румянцева, Л.В. Исакович, О.Л. Зобикова// Педиатрия. Восточная Европа. — 2015. — №1.
Синдром Кронкхайта-Канада
Синдром Кронкхайта-Канада — редкий вариант диффузного полипоза органов ЖКТ, сочетающегося с лентигинозом, ониходистрофией, алопецией. Проявляется диареей, нелокализованными болями в брюшной полости, гиперпигментацией, выпадением волос, разрушением ногтей, атрофическим глосситом, ангулярным хейлитом, истощением. Диагностируется с помощью данных копрограммы, эзофагогастродуоденоскопии, колоноскопии, рентгенографии желудка, кишечника. Для лечения применяют кортикостероиды, производные кромоглициевой кислоты, блокаторы H2-гистаминорецепторов, антибиотики, ингибиторы протонной помпы. По показаниям проводят резекцию тонкого или толстого кишечника, стомирование.
Причины синдрома Кронкхайта-Канада
Этиология заболевания не сегодняшний день не установлена. Ранее считалось, что расстройство наследуется по аутосомно-доминантному типу, однако убедительных доказательств семейной предрасположенности не получено. Кроме того, у больных отсутствуют мутации гена PTEN, характерные для других врожденных полипозов пищеварительного тракта (болезни Рувалкаба-Мири-Смита, синдрома Коудена и др.). В качестве одной из рабочих теорий происхождения патологии рассматривается развитие аутоиммунной реакции, что косвенно подтверждается выявлением в сыворотке антинуклеарных антител, гиперреактивностью тучных клеток, положительными результатами назначения кортикостероидов и мембраностабилизаторов.
Из-за неясности этиологии механизм развития заболевания изучен недостаточно. Пусковые моменты, провоцирующие возникновение диффузного полипоза, неизвестны. Патоморфологические изменения в слизистой кишечника проявляются расширением и кистозной трансформацией желез с образованием ретенционных кист, признаками поверхностного воспаления, резко выраженной отечностью стромы, умеренным утолщением и воспалительной инфильтрацией мышечного слоя. По мере прогрессирования патологии образовавшиеся полипы подвергаются множественным поверхностным некрозам. У большинства пациентов полипозные образования выявляются на слизистой желудка и толстого кишечника, реже поражается тонкий кишечник и пищевод.
Ведущую роль в патогенезе синдрома Кронкхайта-Канада играет расстройство секреторной, пищеварительной и всасывающей функций пораженных отделов ЖКТ в сочетании с нарушениями моторики кишечной стенки. Результатом хронической диареи и недостаточности пристеночного пищеварения становится интенсивная потеря белка и микроэлементов, приводящая к развитию гипопротеинемии, гипокалиемии, гипокальциемии, гипомагниемии с соответствующей клинической симптоматикой. Нарушения белково-минерального обмена, вероятнее всего, служат основной причиной кожных проявлений болезни.
Симптомы синдрома Кронкхайта-Канада
Клиническая картина заболевания нарастает постепенно. Первым признаком болезни становится диспепсический синдром, который проявляется обильным жидким стулом с примесями слизи до 5-7 раз в сутки, болезненностью в животе без четкой локализации. Спустя несколько месяцев после начала желудочно-кишечных расстройств возникают характерные кожные симптомы – у пациентов выявляются гиперпигментированные пятна до 10 см в диаметре, расположенные на кистях рук, лице, шее, бедрах, голенях, тыльной поверхности стоп, изредка на слизистых оболочках. У некоторых больных на коже появляется депигментация (витилиго).
Постоянным симптомом болезни Кронкхайта-Канада является истончение и ломкость ногтей, в тяжелых случаях происходит полное отслоение ногтевой пластины. На поздних стадиях синдрома присоединяются отеки (в основном на нижних конечностях), судороги, наблюдается диффузное выпадение волос. Характерны изменения со стороны слизистой ротовой полости: язык становится опухшим, болезненным, на нем образуются красные пятна, изъязвления, в уголках рта формируются пузырьки, эрозии, гнойно-кровянистые корки. Пациенты стремительно теряют массу тела вплоть до развития кахексии.
При симптомокомплексе Кронкхайта-Канада существует риск изъязвления полипов с профузными желудочно-кишечными кровотечениями. В случае малой хронической кровопотери возникает железодефицитная анемия с бледностью, снижением трудоспособности, головокружениями, гипотонией, тахикардией. Вследствие нарушения процессов переваривания и всасывания питательных веществ развивается синдром мальабсорбции, что приводит к гипопротеинемии, дистрофии мышечной ткани, в том числе миокардиодистрофии, остеомаляции, дефициту микроэлементов, значительному снижению иммунитета. При полипах большого размера может развиваться механическая кишечная непроходимость. У 13% пациентов полипы малигнизируются, в 8% случаев возникает аденокарцинома толстого кишечника, в 5% — желудка. Основными причинами смерти больных становятся кишечные кровотечения, сердечная недостаточность, сепсис.
Постановка диагноза зачастую затруднена, что обусловлено незначительной распространенностью заболевания и сходством клинической картины с другими болезнями желудочно-кишечного тракта. О возможном наличии синдрома Кронкхайта-Канада свидетельствует сочетание диспепсических проявлений с гиперпигментацией, ониходистрофией, алопецией. Наиболее информативны для постановки диагноза:
- Копрограмма. В лабораторном анализе кала определяются признаки синдрома мальабсорбции: большое количество нейтральных жиров, непереваренных мышечных волокон, зерен крахмала. Дополнительно для обнаружения скрытой крови в кале, свидетельствующей о хроническом кровотечении из ЖКТ), проводится реакция Грегерсена.
- Эзофагогастродуоденоскопия. При эндоскопическом исследовании верхних отделов пищеварительного тракта оценивается состояние слизистой оболочки, выявляются типичные полипозные разрастания эпителия. Для цитологического исследования тканей выполняется биопсия измененных участков.
- Колоноскопия. При наличии у пациента синдрома дистального колита целесообразно исследование толстого кишечника гибким эндоскопом. В ходе осмотра врач обнаруживает диффузный полипоз, отбирает биоптаты для гистологического анализа, по показаниям проводит лечебные мероприятия (остановку кровотечения, резекцию).
- Рентгенография желудка и кишечника. Серия рентгеновских снимков после перорального введения контрастного вещества при заболевании Кронкхайта-Канада позволяет визуализировать полипы в виде округлых дефектов наполнения с четкими контурами. Метод высокоинформативен при размере образований более 5 мм.
В общем анализе крови обнаруживают снижение уровня эритроцитов и гемоглобина, микроцитоз. В биохимическом анализе крови наблюдается гипопротеинемия, уменьшение концентрации электролитов (калия, магния, кальция). Проведение ангиографии кишечных сосудов позволяет дифференцировать полипы с интрамуральными опухолями. При недостаточной информативности других методик назначается КТ брюшной полости. Дифференциальная диагностика осуществляется с истинными и гиперпластическими полипами, синдромом Пейтца-Егерса, язвенной болезнью, опухолями пищеварительного тракта мезенхимального и эндотелиального происхождения, болезнью Менетрие, ювенильным полипозом, синдромом Гарднера, кишечной лимфангиэктазией. Кроме осмотра гастроэнтеролога пациенту рекомендованы консультации онколога, гематолога, генетика, дерматолога.
Лечение синдрома Кронкхайта-Канада
Выбор врачебной тактики зависит от локализации и масштаба распространения диффузного полипоза. При поражении желудка, тонкого кишечника, пищевода рекомендовано консервативное ведение пациента с назначением пробной противовоспалительной терапии и коррекцией возникших расстройств метаболизма. Диета аналогична рациону при других энтеропатиях: рекомендовано ограничение количества жиров (не более 30 г/сут), употребление продуктов, богатых белками, жирорастворимыми витаминами, основными макроэлементами, прием специальных жировых эмульсий, содержащих среднецепочечные триглицериды. При значительном снижении протеинов крови пациентам показано парентеральное питание с введением белково-содержащих растворов (2-3 одномесячных курса инфузий в год). Схема медикаментозного лечения включает:
- Кортикостероиды. На фоне гормональной терапии обычно уменьшается диарея, улучшаются процессы пищеварения. Отмечается стабилизация дерматологических симптомов: замедляется или прекращается выпадение волос, восстанавливается структура ногтей, цвет кожи становится более светлым и однородным.
- Стабилизаторы мембран тучных клеток. Улучшению состояния часто способствует дополнение гормонотерапии производными кромоглициевой кислоты. Угнетение кальций-зависимой дегрануляции тучных клеток тормозит выделение из них гистамина, предотвращает или уменьшает развитие местной воспалительной реакции.
- Н2-гистаминоблокаторы. При преимущественном поражении диффузным полипозом Кронкхайта-Канада слизистой желудка эффективен прием конкурентных ингибиторов гистаминовых рецепторов. Количество полипов заметно уменьшается на фоне снижения базальной и стимулированной секреции соляной кислоты.
По результатам наблюдений, динамика течения синдрома улучшается при назначении ингибиторов протонной помпы, макролидов, β-лактамных пенициллинов, других антибактериальных препаратов для элиминации хеликобактерий. Хирургические методы показаны при локальном поражении кишечника – проводится сегментарная, лапароскопическая резекция тонкой кишки, удаление различных отделов толстой кишки с выполнением при необходимости колостомии, сигмостомии, цекостомии, последующих реконструктивных вмешательств.
Исход заболевания зависит от выраженности экссудативной энтеропатии и наличия осложнений со стороны других органов. Прогноз при расстройстве Кронкхайта-Канада неблагоприятный, около 45% пациентов умирают в течение первых 5 лет после манифестации клинических проявлений. Меры специфической профилактики не разработаны. Для предупреждения развития осложнений необходимо динамическое наблюдение за больными с установленным диагнозом диффузного полипозного синдрома, которое включает регулярное посещение гастроэнтеролога, ежегодное выполнение диагностического эндоскопического исследования.
Читайте также:
- От чего немеет стопа? Причины онемения стопы
- Возможные осложнения нейролептанальгезии (НЛА) при инфаркте миокарда ( оим, ОИМ ).
- Диагностика анапластической астроцитомы по КТ, МРТ, ПЭТ
- Клиника острой печеночной недостаточности.
- Патологическая анатомия бронхиальной астмы. Физиологические формы бронхиальной астмы