Синдромы Прадера-Вилли и Ангельмана. Характеристика
Добавил пользователь Alex Обновлено: 28.01.2026
Синдром Прадера-Вилли: причины, диагностика, лечение
Этиология и встречаемость синдрома Прадера-Вилли. Синдром Прадера-Вилли (MIM №176270) - панэтническое заболевание, вызываемое снижением экспрессии генов в хромосоме 15qll-ql3 в хромосоме отцовского происхождения. Утрата отцовских генов может возникать разными способами; приблизительно 70% пациентов имеют делецию 15qll-ql3, 25% имеют однородительскую материнскую дисомию, менее чем 5% имеют мутации в пределах управляющего элемента импринтинга и менее чем 1% имеют хромосомную аномалию. Синдром Прадера-Вилли встречается с частотой 1 на 10 000-15 000 живых новорожденных.
Патогенез синдрома Прадера-Вилли
Многие гены в регионе 15q11-q13 экспрессируются различно, в зависимости от того, наследуются они от отца или от матери. Другими словами, многие гены, экспрессирующиеся в отцовском 15qll-ql3, не экспрессируются в материнском регионе, и наоборот. Этот феномен дифференциальной экспрессии генов в зависимости от того, унаследованы они от отца или матери, известен как импринтинг.
Правильная экспрессия импринтированных генов требует переключения импринтинга при проходе через половые клетки; т.е. отцовский импринтинг переключается на материнский при проходе через материнские половые клетки, а материнский — на отцовский, в его половых клетках.
Переключение импринтинга при проходе через половые клетки регулируется управляющим элементом импринтинга и проявляется эпигенетическими изменениями в метилировании ДНК и хроматина, регулирующих экспрессию генов.
Делеция 15q11-q13 в мужском мейозе приводит к рождению детей с синдромом Прадера-Вилли, поскольку дети, сформировавшиеся из сперматозоида, несущего делецию, теряют гены, активные только в сегменте 15qll-ql3 отцовского происхождения. Механизм, лежащий в основе такой повторной делеции, — неправильная рекомбинация между короткими тандемными последовательностями повторов, ограничивающими область делеции. Реже наследование делеции, захватывающей этот регион, происходит, если пациент унаследует несбалансированный кариотип от родителя, имеющего сбалансированную транслокацию.
Невозможность переключить материнский импринтинг на отцовский в мужском мейозе также вызывает синдром Прадера-Вилли, поскольку дети, сформировавшиеся из сперматозоида с материнским импринтингом региона 15qll-ql3, не могут экспрессировать гены, активные только при отцовском импринтинге 15q11-q13. Неспособность к импринтингу вызывается мутациями в управляющем элементе импринтинга.
Синдром Прадера-Вилли также возникает при материнской однородительской дисомии, поскольку ребенок имеет две материнских хромосомы 15 и не имеет отцовской. Считают, что материнская однородительская дисомия развивается вторично в качестве «компенсации» трисомии, т.е. утрате отцовской хромосомы 15 у концеп-туса с трисомией по 15 хромосоме, вызванной материнским нерасхождением.
Несмотря на наблюдения, указывающие, что утрата отцовского импринтированного региона 15q11-q13 вызывает синдром Прадера-Вилли, и, несмотря на идентификацию множества импринтированных генов в этом регионе, точная причина заболевания все еще неизвестна. До сих пор не показано, что синдром Прадера-Вилли вызван мутациями в каком-либо конкретном гене.

Фенотип и развитие синдрома Прадера-Вилли
В самом раннем детстве синдром Прадера-Вилли характеризуется выраженной гипотонией, трудностями вскармливания и гипогонадизмом с крипторхизмом. Тонус мышц со временем улучшается, хотя взрослые остаются слегка гипотоничными. Гипогонадизм гипоталамического происхождения с возрастом не улучшается и обычно вызывает позднее и неполное половое созревание, а также бесплодие. Трудности с кормлением обычно проходят к концу первого года жизни, и между 1 и 6 годами жизни у пациентов развивается выраженная гиперфагия и поведение «с поиском пищи» (поиск пищи, создание припрятанных запасов).
Такое поведение и низкий метаболизм вызывают выраженное ожирение. Ожирение — основная причина смерти, в основном, вследствие сердечно-легочной патологии и ИНСД (тип II). Если удается эффективно лечить ожирение, длительность жизни может быть практически нормальной.
У большинства детей с синдром Прадера-Вилли моторное и речевое развитие задержано, имеется негрубая задержка умственного развития (IQ от 60 до 80) и выраженная неспособность к обучению. Отмечают также поведенческие проблемы, включая истерики, навязчивые состояния и плохую адаптацию к изменениям в обычной жизни. Эти поведенческие проблемы переходят во взрослую жизнь. Приблизительно у 5-10% пациентов в молодости развиваются психозы.
Другие аномалии, связываемые с синдром Прадера-Вилли, включают низкий рост, сколиоз, остеопороз и дисморфии. Дисморфические признаки представлены сужением битемпорального размера лба, миндалевидными глазами, треугольным ртом и небольшими кистями и стопами. Кроме того, множество пациентов имеют гипопигментацию волос, глаз и кожи.
Особенности фенотипических проявлений синдрома Прадера-Вилли:
• Возраст начала: раннее детство
• Трудности вскармливания
• Повышенный аппетит и ожирение
• Гипотония мышц
• Когнитивные расстройства
• Бесплодие
• Дисморфии
Лечение синдрома Прадера-Вилли
Хотя синдром Прадера-Вилли часто может быть заподозрен на основе анамнеза и данных клинического осмотра, окончательный диагноз устанавливают по отсутствию отцовского импринтированного участка 15q11-q13. Потерю импринтинга обнаруживают при ДНК-анализе, показывающем, что импринтированные гены имеют только материнский тип метилирования. Если исследование ДНК подтверждает диагноз, для генетического консультирования необходимо кариотипирование и FISH-анализ участка 15q11-q13, чтобы определять, не возник ли синдром вследствие унаследованной хромосомной транслокации.
В настоящее время медикаментозное лечение гиперфагии неэффективно; основой помощи при ожирении остаются диета и физические нагрузки. Применение гормона роста может нормализовать рост и улучшить соотношение жира и мышц. Заместительная терапия половыми гормонами улучшает вторичные половые признаки, но часто усиливает поведенческие проблемы у мужчин и увеличивает риск острых нарушений мозгового кровообращения у женщин. Наиболее эффективное лечение поведенческих нарушений в настоящее время — прием ингибиторов серотонина. Взрослые пациенты обычно лучше себя чувствуют в специальном жилом и рабочем окружении.
Риски наследования синдрома Прадера-Вилли
Риск повторения синдрома у детей обусловливается молекулярной причиной заболевания. Для дефектов импринтинга риск может достигать 50%, тогда как при делеции 15q11-q13 и материнской однородительской дисомии риск повторения менее 1%. Риск повторения, если один из родителей несет сбалансированную транслокацию, зависит от природы транслокации, но может достигать 25%; в отличие от этого, все пациенты с синдромом Прадера-Вилли с несбалансированной транслокацией, описанные до настоящего времени, имели новую хромосомную перестройку.
Пример синдрома Прадера-Вилли. Ж.Т., второй ребенок в неродственном браке родителей, родился на сроке 38 нед гестации после неосложненной беременности и родов. Вскоре после рождения родители и медсестра обратили внимание на мышечную гипотонию и плохой аппетит. Родители Ж.Т. и старшая сестра здоровы; в семейном анамнезе нервно-мышечных и генетических болезней, аномалий развития или нарушений питания нет. В медицинской карте нет записей о судорогах, приступах гипоксии, инфекции, пороке сердца, отклонениях глюкозы или электролитов крови.
При осмотре не обнаружено респираторных нарушений или дисморфических признаков, за исключением гипопластичной мошонки и крипторхизма; масса тела и рост соответствовали гестационному сроку; у ребенка выявлены выраженная мышечная гипотония, сонливость, слабый крик, снижение рефлексов, слабое сосание. В обследование включены анализы на врожденные инфекции и врожденный гипотиреоз, МРТ мозга, определение аммиака крови, аминокислот и органических кислот плазмы и мочи, кариотипирование методом in situ гибридизацией (FISH) области делеции локуса синдрома Прадера-Вилли (хромосома 15qll-ql3).
Результаты тестов оказались нормальными, за исключением FISH, показавшего делецию хромосомы 15q11-q13. Генетик объяснил родителям, что у ребенка синдром Прадера-Вилли. После длительного обсуждения и раздумий родители решили, что они не в состоянии ухаживать за больным ребенком и отказались от него.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
Синдром Прадера-Вилли ( Синдром гипотонии-ожирения )
Синдром Прадера-Вилли – это редкое генетическое заболевание, характеризующееся грубыми конституциональными нарушениями, когнитивными и психическими расстройствами. Клиническая картина разнообразна, основные симптомы включают ожирение, задержку роста и умственную отсталость. Часто встречается снижение мышечного тонуса, репродуктивная дисфункция. Окончательный диагноз устанавливается на основании молекулярно-генетического исследования. Специфическое лечение не разработано. Осуществляется симптоматическая терапия по основным компонентам синдрома: назначение гипокалорийной диеты и гормональных средств, индивидуальные занятия с дефектологом и т. д.
МКБ-10

Общие сведения
Синдром Прадера-Вилли (синдром гипотонии-ожирения) является одной из наиболее выраженных форм генетически обусловленного ожирения. Заболевание впервые было описано в 1956 году швейцарскими педиатрами А. Прадером и Х. Вилли. Несмотря на генетическую природу, болезнь носит спорадический характер. По разным статистическим данным, распространенность синдрома составляет 1:15 000 – 1:25 000 новорожденных. Какие-либо значимые гендерные различия отсутствуют.

Причины
Патология развивается в результате мутации 15 хромосомы (сегмента q11.2-q13). Прямой передачи заболевания по наследству не происходит. Хромосомная аномалия возникает в момент оплодотворения яйцеклетки, т. е. обмена родительских генетических материалов. В 65-75% случаев мутация обусловлена дефектом отцовской 15 хромосомы, а в 25-35% – наследованием обеих 15 хромосом от матери. Факторы риска, провоцирующие клинические проявления хромосомной мутации, неизвестны.
Патогенез
Патологические механизмы остаются малоисследованными. Известно, что при этой болезни наблюдается выраженный дисбаланс между процессами липолиза и синтеза жиров в подкожно-жировой клетчатке со сдвигом в сторону последнего. Предполагается, что ведущую роль в ожирении и задержке роста у детей с синдромом Прадера-Вилли играет эндокринная дисрегуляция.
Дисфункция ядер гипоталамуса приводит к снижению выработки многих гормонов, таких как соматотропный гормон, гонадотропины, тиреотропный гормон и пр. Падение концентрации гормона роста и половых гормонов, особенно в детском возрасте, способствует накоплению жировых депо. Характерно повышение уровня пептидного гормона грелина, который является эндогенным стимулятором аппетита.
В генезе нейропсихических расстройств рассматривается роль низкого уровня нейротрофического фактора головного мозга, участвующего в развитии и дифференцировке клеток центральной нервной системы и их функциональной активности. Гипопигментация кожи и волос объясняется подавленной функцией тирозиназы в волосяных фолликулах и меланоцитах.
Симптомы
Клинические проявления начинают манифестировать уже в период внутриутробного развития. Отмечается малая подвижность плода, неправильное предлежание, недоношенность при рождении. Возникает выраженная мышечная гипотония. Значительно ослаблены сосательный и глотательный рефлексы. Это затрудняет кормление ребенка и ведет к недостаточному возрастному набору массы тела. В некоторых случаях необходимо питание через зонд.
Несколько позже присоединяется наиболее характерный симптом – полифагия (патологически повышенный аппетит), вследствие которой ребенок довольно быстро начинает прибавлять в весе, достигая ожирения, вплоть до морбидного. Отложение жира преимущественно происходит в области туловища и проксимальных отделах конечностей.
Выражены нейропсихические нарушения. Речь замедлена, интеллектуальные способности (память, концентрация внимания, последовательная обработка информации) значительно отстают от возрастной нормы. В подростковом периоде нередко наблюдаются обсессивно-компульсивные расстройства, резкие перепады настроения, агрессивное поведение. Из-за недостаточной продукции слюны зубы быстро поражаются кариесом.
Гипогонадизм у мальчиков проявляется гипоплазией мошонки, микропенисом, крипторхизмом, у девочек – недоразвитием половых губ, поздним наступлением менструаций или их полным отсутствием. Возможны нарушения координации, мышечные судороги, косоглазие. Из других конституциональных изменений можно отметить низкий рост, акромикрию (уменьшенный размер кистей и стоп). Типичны гипопигментация кожи, светлые волосы.
Осложнения
Преобладающее число осложнений синдрома Прадера-Вилли связано с морбидным ожирением. Избыток жировой массы способствует раннему развитию инсулинорезистентности, метаболического синдрома и сахарного диабета 2 типа. Нередко встречается неалкогольная жировая болезнь печени (жировой гепатоз). Значительное скопление жира в области шеи обуславливает сужение просвета дыхательных путей.
Вследствие этого более чем у половины пациентов (55-60%) наблюдается синдром обструктивного апноэ сна, который в свою очередь, резко увеличивает риск артериальной гипертензии, инсульта, жизнеугрожающих аритмий. Ожирение также вызывает альвеолярную гиповентиляцию и чрезмерную нагрузку на правые отделы сердца, в результате чего возникает правожелудочковая сердечная недостаточность.
Из-за сниженной минеральной плотности костной ткани любая травма может привести к переломам. Практически все больные страдают первичным бесплодием. Отмечаются частые вирусные инфекции верхних дыхательных путей, бронхиты и пневмонии. Существуют данные о том, что при синдроме ПВ повышается вероятность развития лейкемии и других онкологических заболеваний.
Диагностика
Больных, страдающих синдромом Прадера-Вилли, курируют врачи-педиатры и генетики. При общем осмотре обращают внимание на ослабление мышечного тонуса и сухожильных рефлексов, конституциональные изменения – ожирение, низкий рост. Дополнительное обследование включает следующие исследования:
- Анализы крови. В биохимическом анализе нередко обнаруживается повышение концентрации глюкозы и печеночных трансаминаз (АЛТ, АСТ). Отмечается снижение уровня гонадотропинов (ФСГ, ЛГ), половых гормонов (тестостерона, эстрогенов), соматотропного гормона.
- Денситометрия. При проведении двойной энергетической рентгеновской абсорбциометрии определяются признаки остеопении или остеопороза – показатели плотности костей ниже среднего значения пиковой костной массы более чем на 2,5 SD.
- Определение наличия СОАС. Поскольку обструктивное апноэ представляет угрозу для здоровья и жизни, все пациенты с подозрением на синдром Прадера-Вилли проходят кардиореспираторный мониторинг и полисомнографическое исследование, при которых обнаруживаются высокий индекс дыхательных расстройств и индекс десатурации.
- Генетическое исследование. Выявление микроделеции 15q11-13 с помощью полимеразной цепной реакции, кариотипирования или флуоресцентной гибридизации – основной верифицирующий тест, позволяющий достоверно поставить диагноз.
Дифференциальный диагноз проводится с заболеваниями, которые сопровождаются выраженной мышечной гипотонией и задержкой нейропсихического развития – синдромом Опица-Фриаса, миопатиями, спинальной амиотрофией. Кроме того, синдром ПВ дифференцируется с другими наследственно обусловленными формами ожирения (адипозогенитальная дистрофия, синдром Лоуренса-Муна).
Лечение синдрома Прадера-Вилли
Консервативная терапия
Пациенты подлежат госпитализации в педиатрическое отделение. Эффективные методы этиотропной терапии не разработаны, все лечебные мероприятия носят симптоматический характер. Для борьбы с гипотонией назначаются сеансы массажа и физиотерапевтические методы воздействия. Рекомендуются занятия с логопедом, дефектологом, психотерапевтом. Другие виды лечения синдрома Прадера-Вилли:
- Диета. Основное внимание уделяется изменениям в питании. Необходимо ограничить продукты с высоким содержанием насыщенных жиров и легкоусвояемых углеводов. Общий суточный калораж должен составлять 1000-1200 ккал. Лекарственные препараты, подавляющие аппетит, не используются, так как показали низкую эффективность у больных синдромом ПВ.
- Заместительная гормональная терапия. Рекомендуется подкожное введение рекомбинантного соматотропного гормона даже в раннем детском возрасте еще до наступления ожирения. Для восстановления репродуктивной функции применяются аналоги гонадотропин-рилизинг гормона (гозерелин).
- СИПАП-терапия. Для лечения синдрома обструктивного апноэ наиболее успешным методом является использование специального устройства для автоматической интраназальной вентиляции легких, создающего постоянное положительное давление в верхних дыхательных путях.
- Антиостеопоротическое лечение. При низких показателях плотности костей во избежание патологических переломов назначаются витамин Д (холекальциферол), препараты кальция, бисфосфонаты (золедроновая кислота).
Хирургическое лечение
При наличии определенных показаний (удлиненное мягкое небо, гипертрофия миндалин) для устранения СОАС выполняется хирургическая коррекция – увулопалатофарингопластика, которая заключается в иссечении части мягкого неба, тонзиллэктомии, формировании швов, подтягивающих заднюю стенку глотки. Вероятность рецидива после операции составляет около 50%.
Если не удается добиться снижения массы тела консервативными методами, прибегают к бариатрической хирургии – бандажированию желудка, желудочному шунтированию. Сохранение крипторхизма к концу 1-го года жизни служит показанием к оперативному устранению патологии. Проводится орхипексия – прикрепление яичка к мошонке с помощью швов.
Экспериментальное лечение
Ведутся разработки новых лекарственных средств для терапии синдрома ПВ. Имеются обнадеживающие результаты клинических исследований применения агониста рецепторов окситоцина – карбетоцина. Предлагается воздействовать на кишечную микробиоту больных детей пробиотическими препаратами. В экспериментальных работах на лабораторных животных продемонстрировало лечебный эффект вещество UNC0642, активирующее гены на необходимом участке 15 хромосомы.
Прогноз и профилактика
Продолжительность жизни пациентов, страдающих синдромом ПВ, при своевременной диагностике и адекватном лечении достигает 60-70 лет. В отсутствие превентивных мер смерть может наступить в возрасте 4-5 лет от сердечно-легочной недостаточности. В 50% случаев причиной летального исхода становится обструктивное апноэ сна и вызванные им сердечно-сосудистые катастрофы.
Реже больные погибают от тяжелой респираторной инфекции. Единственным способом предотвращения возникновения заболевания является пренатальная диагностика и прерывание беременности. Основная роль отводится вторичной профилактике – предупреждению осложнений болезни, например вакцинации от гриппа и пневмококковой инфекции.
1. Синдром Прадера-Вилли у детей: новое в этиологии, патогенезе и лечении/ Казанцева Л.З., Новиков П.В., Семячкина А.Н., Николаева Е.А., Курбатов М.Б., Добрыкина Э.В.// Российский вестник перинатологии и педиатрии – 1999 - №4.
2. Синдром Прадера-Вилли в Беларуси: генетическая структура и фенотипическая характеристика/ Хурс О.И., Политыко А.Д., Румянцева Н.В. и др.//Известия Национальной Академии наук Беларуси - 2010 - № 1.
3. Clinical report-health supervision for Children with Prader-Willi Syndrome, the Committee on Genetics/ McCandless S.E.// Pediatrics – 2011 - №1.
4. Prader-Willi syndrome: clinical and genetic fi ndings/ Butler M.G., Thompson T.// Endocrinologist – 2000 - №10.
Синдром Ангельмана ( Синдром марионетки , Синдром Петрушки , Синдром счастливой куклы )
Синдром Ангельмана – генетическое заболевание, характеризующееся наличием неврологической симптоматики, задержкой психического развития. Проявляется интеллектуальным отставанием, слабой сформированностью речи, навыков сидения и ходьбы, хаотичными движениями, гиперактивностью, симптоматической эпилепсией, беспричинным весельем и смехом, сколиозом, своеобразной походкой. Пациенты имеют особую внешность: рот крупный, зубы расположены редко, подбородок выдается вперед. Диагноз устанавливается на основании клинических данных, результатов генетического анализа. Специфическое лечение отсутствует, проводится симптоматическая терапия, оказывается психологическая и педагогическая помощь.
Синдром назван по фамилии британского педиатра Г. Ангельмана. В 1965 году он первым описал симптомы заболевания и назвал его «синдромом счастливой марионетки», поскольку пациенты напоминали ему героя картины «Мальчик-марионетка». В те годы методы генетических исследований были еще не разработаны, установить причину патологии было невозможно. В 1987 году исследователи определили этиологию болезни и переименовали ее в синдром Ангельмана. Сейчас этот термин является официальным, но можно встретить синонимичные названия – «синдром марионетки», «синдром Петрушки», «синдром счастливой куклы». Распространенность составляет 1 случай на 10-20 тысяч новорожденных. Заболевание выявляется после первого года жизни (иногда – к 3-7 годам), чаще болеют мальчики.
Факторы развития синдрома Ангельмана продолжают исследоваться. Генетический дефект обнаружен в 15 хромосоме материнского набора, но его характер и способ возникновения могут различаться. Иногда заболевание дебютирует в результате передачи измененной генетической информации от родителя, иногда является последствием спонтанных нарушений в геноме. Хромосомные аномалии удается определить примерно у 85-88% больных. Причиной синдрома может быть:
- Делеция. При данном дефекте часть генетического материала теряется или инактивируется. У 70% пациентов диагностируются обширные делеции области 15q12 хромосомы, в которой локализован активатор гена.
- Однородительская дисомия. ОРД определяется в 2-3% случаев болезни. В хромосомном наборе присутствуют две копии 15 хромосомы отца. Материнской хромосомы нет, ген также отсутствует.
- Дефект запечатления. Суть аномалии заключается в том, что центр запечатления, регулирующий активность локуса UBЕ3A, оказывается нефункциональным, «выключенным». Ген остается структурно целым, но не выполняет своих функций. Распространенность ДЗ – 3-5%.
- МутацияUBE3A. У 5-10% пациентов причиной болезни являются мутационные изменения гена. Они представлены инверсиями, микроделециями, транслокациями и дупликациями.
Основа синдрома Ангельмана – нарушение функций гена UBE3A, расположенного в пятнадцатой материнской хромосоме. Этот ген кодирует производство протеина Е6АР, который представляет собой ферментный компонент сложной реакции деградации белков. Е6АР участвует в процессе образования убиквитина – белка системы протеасом, стимулирующего протеолиз дефектных белковых молекул в нейронах головного мозга. В норме убиквитин маркирует ненужные (неактивные, нефункциональные) белки с целью инициации их уничтожения. Е6АР обеспечивает закрепление убиквитина на поверхности молекулы белка-мишени. Потом протеасомы расщепляют его на пептидные остатки и на аминокислоты. При синдроме Ангельмана убиквитин не закрепляется на дефектных белках, они скапливаются в нервной ткани мозга, нарушается процесс синаптической передачи. Формируются отклонения, задержки в психическом и моторном развитии.
Клинически заболевание проявляется в возрасте от 6 до 12 месяцев. Постепенно нарастает задержка развития, ранее освоенные навыки сохраняются, но приобретение новых происходит медленно. Дети умеют сидеть, ползать, брать предметы и перекладывать их из руки в руку, поддерживать визуальный контакт, гулить и лепетать. Ходьба дается с трудом, нарушено чувство равновесия, наблюдаются частые падения, ушибы о мебель. Выявляется тремор и хаотичные движения конечностями, особенно руками. Речевые расстройства представлены как задержками, так и полным отсутствием экспрессивной речи. Дети либо совсем не говорят, либо используют лепет, простые слоги и слова общим объемом не более 10 единиц. Сохраняется понимание обращенной речи, стремление к общению, использование невербальных средств коммуникации: жестов, мимики, опосредованных знаков.
Основное поведенческое нарушение – гиперактивность. Дети часто веселятся и смеются без объективной причины, двигательно расторможены, неусидчивы, нецеленаправленны. У многих возникает патологическая привязанность к определенной игрушке или предмету быта, при появлении которого настроение сразу повышается, капризность и плач сменяются смехом. Концентрация внимания снижена, переключаемость быстрая и ненаправленная. Имеются трудности обучения, стойкое снижение интеллектуальных функций. Легко закрепляются стереотипии: раскачивание тела, размахивание руками. У 80% пациентов отмечается микроцефалия, недостаточный объем черепной коробки, эпилептическая активность мозга. Редко наблюдается снижение контроля движений языка, которое проявляется трудностями сосания груди или соски, последующим недостатком массы тела.
Характерные особенности внешности детей – косоглазие, сколиотическое искривление позвоночника, увеличение зубов и губ, разряжение зубного ряда, уплощение затылка, выступание вперед подбородка. Язык часто высунут, рот приоткрыт в улыбке. Развивается мышечная дистония, выраженность рефлексов сухожилий повышается, формируя специфичность моторики: пациенты ходят на прямых несгибающихся ногах, плечи приподнимают, руки сгибают в локтях. Своеобразный симптом – тяга к воде. Большинство детей чувствуют себя спокойнее в водной среде, им нравится плескаться в ванной, играть с корабликами в тазу.
Синдром Ангельмана – редкое малоизвестное заболевание. В связи с этим постановка диагноза и оказание медико-психолого-педагогической помощи зачастую проводятся несвоевременно, к 6-8 годам, что обуславливает низкую успешность коррекционных и лечебных мероприятий. Без физиотерапевтического лечения усугубляются нарушения опорно-двигательного аппарата – больные страдают от тяжелых форм сколиоза, самостоятельно передвигаются с трудом. Своеобразие внешности и поведения становится причиной ухудшения и без того затрудненной социальной адаптации. Все перечисленное влечет за собой утяжеление инвалидизации пациентов.
Обследованием детей с подозрением на синдром Ангельмана занимаются врачи-неврологи, психиатры и генетики. Родители предъявляют жалобы на отсутствие речи, двигательные стереотипии, трудности формирования ходьбы и других двигательных навыков, гиперактивность. Проводится дифференциальная диагностика, в ходе которой должны быть исключены более распространенные заболевания, такие как умственная отсталость, расстройства аутистического спектра, деменции, мутизм, несимптоматические формы эпилепсии. Комплексное исследование включает следующие процедуры:
- Общий осмотр. На наличие синдрома часто указывают специфические черты внешности пациентов: высунутый язык, слюнотечение, крупный рот, широкие редкие зубы, выступающая вперед нижняя челюсть, плоская форма затылка. Характерен светлый оттенок кожи, глаз и волос. Походка детей напоминает движения куклы-марионетки из-за усиления сухожильных рефлексов и снижения мышечного тонуса.
- Осмотр психиатром. В 100% случаев синдрома Ангельмана диагностируется выраженная задержка в развитии психики, отсутствие самостоятельной речи или очень скудный словарный запас. Коммуникация осуществляется с помощью мимики, жестов, рисунков. В поведении отмечается гиперактивность, стереотипные движения руками, беспричинный смех.
- Неврологическое обследование. У всех пациентов определяется атаксия и тремор конечностей. 80% больных имеют постнатальную микроцефалию – окружность головы новорожденного меньше 32 см, к 12 месяцам – около 42 см. По данным ЭЭГ выявляется симптоматическая эпилепсия (высокоамплитудные разряды медленных комплексных волн), клинически возможны судорожные припадки. У некоторых детей имеется косоглазие, диффузное снижение мышечного тонуса, усиление сухожильных рефлексов, гиперкинезы.
- Генетическое исследование. Лабораторная диагностика нацелена на выявление мутаций и делеций в гене UBE3A. Последовательно проводится комплекс процедур, включающий флуоресцентную гибридизацию in situ, анализ мутации центра запечатления, метилирование ДНК СА/ПВС региона, диагностику делеции методом микроматричного анализа, поиск мутационных изменений в локусе UBE3A.
Лечение синдрома Ангельмана
Хромосомные нарушения, лежащие в основе синдрома, устранить невозможно. Пациентам назначается симптоматическое лечение, психолого-педагогическая коррекция, реабилитационные мероприятия. Для уменьшения частоты эпилептических припадков используются антиконвульсанты, для нормализации сна – мелатонин. Занятия лечебной физической культурой и сеансы массаж направлены на развитие мелкой моторики и скоординированной походки, устранение сколиоза. Для улучшения коммуникативных навыков детей обучают языку жестов, вовлекают в групповые занятия, организуют сеансы поведенческой терапии, позволяющей освоить правила взаимодействия в обществе.
Продолжается поиск способов эффективного лечения синдрома. Проводится тестовое применение препаратов на генетически модифицированных мышах. Результаты доказывают, что ингибиторы топоизомеразы способны активировать материнский ген UBE3A. На данном этапе выполняются контрольные исследования, определяется безопасность и риски терапии, но информации пока недостаточно для перенесения экспериментов на группы людей.
Выраженность симптомов синдрома Ангельмана может сильно различаться. Пациенты с легкими формами болезни имеют благоприятный прогноз: их речь становится более развернутой, улучшаются навыки самоконтроля при некоторых нарушениях двигательной сферы. При любой степени тяжести раннее начало и регулярное проведение медико-психологической помощи повышает качество жизни больных. Профилактика сводится к генетическому обследованию пар, в семьях которых есть ребенок с данным синдромом. Характер хромосомного дефекта (спорадический или наследственный) позволяет определить риск рождения второго больного ребенка.
2. Генетические механизмы возникновения синдрома Ангельмана и их фенотипические проявления / Отрошко Е.В., Молодожникова Н.В. // XXIV Студенческая международная заочная научно-практическая конференция «Молодежный научный форум: естественные и медицинские науки» - 2015.
Синдромы Прадера-Вилли и Ангельмана. Характеристика
Возможно, наиболее полно изученные примеры роли геномного импринтинга при болезнях человека — синдромы Прадера-Вилли и Ангельмана.
Синдром Прадера-Вилли — сравнительно частый дисморфический синдром, характеризующийся ожирением, чрезмерным и беспорядочным аппетитом, небольшими кистями и стопами, низким ростом, гипогонадизмом и умственной отсталостью. Приблизительно в 70% случаев синдрома наблюдают цитогенетическую делецию, затрагивающую проксимальный отдел длинного плеча хромосомы 15 (15q11-q13), причем только в хромосоме, унаследованной от отца больного.
Таким образом, геном таких пациентов имеет генетическую информацию в области 15q11-q13, происходящую только от матерей. И наоборот, примерно у 70% пациентов с редким синдромом Ангельмана, характеризующегося необычным лицом, низким ростом, выраженным интеллектуальным отставанием, спастикой и судорогами, отмечают делецию приблизительно той же хромосомной области, но теперь в хромосоме, унаследованной от матери; т.е. пациенты с синдромом Ангельмана имеют генетическую информацию в регионе 15q11-q13, происходящую только от отцов.
Эта необычная ситуация удивительным образом доказывает, что родительское происхождение генетического материала в описанных случаях (в хромосоме 15) имеет выраженное влияние на клиническое проявление дефекта.
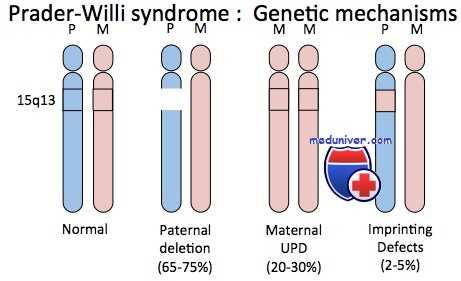
Приблизительно 30% пациентов с синдромом Прадера-Вилли не имеют цитогенетически обнаруживаемых делеций; но у них выявлены две цитогенетически нормальные хромосомы 15, обе унаследованные от матери. Ситуация иллюстрирует однородительскую дисомию — наличие дисомной линии клеток, содержащих две хромосомы или их части, унаследованные от одного родителя. Если оба экземпляра представлены идентичной хромосомой, состояние называют изодисомией; если присутствуют разные гомологи от одного родителя — гетеродисомией.
Приблизительно 3-5% пациентов с синдромом Ангельмана также имеют однородительскую дисомию, только с двумя неповрежденными хромосомами 15 отцовского происхождения. Эти пациенты служат дополнительным подтверждением того, что синдромы Прадера-Вилли и Ангельмана — результат потери соответственно отцовского или материнского вклада генов участка 15q11-q13.
Кроме хромосомной делеции и однородительской дисомии, несколько пациентов с синдромами Прадера-Вилли и Ангельмана, вероятно, имеют дефект в самом центре импринтинга. В результате не происходит переключения от женского к мужскому импринтингу в сперматогенезе или от мужского к женскому в овогенезе.

Оплодотворение сперматозоидом, несущим аномально персистирующий женский импринтинг, приведет к рождению ребенка с синдромом Прадера-Вилли; оплодотворение яйцеклетки, имеющей несвойственный ей мужской импринтинг, закончится рождением ребенка с синдромом Ангельмана.
Наконец, мутации в материнской копии одного гена — убиквитин-протеин лигазы Е6-АР, как оказалось, вызывают синдром Ангельмана. Ген убиквитин-протеин лигазы Е6-АР расположен в области 15q11-q13 и в норме импринтирован, экспрессируется только материнский аллель в центральной нервной системе (ЦНС).
Полагают, что крупные материнские делеции области 15q11-q13 и отцовские однородительские дисомии хромосомы 15, наблюдаемые при синдроме Ангельмана, служат причиной заболевания, так как приводят к утрате материнской копии критически важного импринтированого гена. Мутации для одного импринтированного гена при синдроме Прадера-Вилли пока еще не обнаружены.
Синдром Прадера-Вилли (СПВ) - этиология, клиника, диагностика
Синдром Прадера-Вилли (СПВ) характеризуется перееданием, избыточным весом и рядом других соматических и поведенческих проявлений. Заболевание встречается с частотой около 1 на 7000 детей, доживших до одного года (Akefeldt et al., 1991). Распространенность синдрома среди мальчиков и девочек примерно одинакова.
а) Патогенез. Синдром Прадера-Вилли (СПВ) вызван отсутствием на отцовской ДНК хромосомного локуса 15q11. Возможна делеция отцовского гена или дисомия по материнской линии. В ходе одного из исследований было выявлено, что отцы предварительно контактировали с бензолом (Akefeldt et al., 1995). Примерно в половине случаев синдром связан с интерстициальной хромосомной делецией 15q11—13 (Ledbetter et al., 1982; Mattei et al., 1983; Caldwell и Taylor, 1988). Практически во всех случаях делеция имеет отцовское происхождение (Butler et al., 1986; Knoll et al., 1989). Приблизительно в 40% случаев, без цитогенетических делеций, делеции выявляются при исследовании ДНК, а в 60% случаев отмечается дисомия всей или части 15-й хромосомы матери (Mascari et al., 1992). СПВ и синдром Ангельмана являются классическими образцами импринтинга, то есть различного экспрессирования гена в зависимости от пола родителя, передавшего ген.
Ген синдрома Прадера-Вилли экспрессируется только при наследовании от отца (Wevrick et al., 1994). В случаях, когда не выявлено аномалий 15-й хромосомы, необходимо повторное клиническое обследование для выявления другого заболевания, такого как синдром Смита-Магениса (Greenberg et al., 1991).
Пациенты с делецией имеют более типичные проявления, чем при ее отсутствии (Mattei et al., 1983), хотя атипичные формы заболевания отмечались и в случаях подтвержденной делеции 15q (Pauli et al., 1983; Schwartz et al., 1985; Miike et al., 1988). В рамках исследования было проведено сравнение 21 ребенка с делецией и 18 детей с нормальным хромосомным набором с высокой разрешающей способностью (Butler et al., 1986). Различия между двумя группами были выражены минимально. Чуть больше половины пациентов с делецией (11 из 21) ходили к 24 месяцам (по сравнению с двумя третями пациентов без делеции). У детей с делецией отмечались более светлые волосы, чем у детей без нее, и у всех были голубые глаза, в то время как только 13 из 18 детей без делеции были голубоглазыми (Lee et al., 1994).
б) Диагностика. Клинический диагноз синдрома Прадера-Вилли (СПВ) подтверждается типичными изменениями ДНК, выявляемыми при анализе на ген синдрома Прадера-Вилли на 15-й хромосоме FISH-методом.
В период новорожденности данное заболевание следует заподозрить в случае низкой массы тела при рождении, заметной гипотонии, нарушения глотания, требующего кормления через зонд, и дисморфизма. В более старшем возрасте признаками заболевания могут быть ожирение и аномалии лица. Следует проводить дифференциальную диагностику СПВ и нейромышечных заболеваний и гипотонии, связанных с заболеваниями мозга. Необходимо рассматривать врожденную мышечную дистрофию, миотоническую дистрофию новорожденных и врожденные миопатии. Тем не менее, при СПВ на первый план выступает дисморфизм, а также нарушения сосания и глотания без значимых нарушений дыхания. В более старшем возрасте может вставать вопрос о других причинах гипотонии мозгового происхождения. Результаты биопсии мышц (которой следует избегать), уровень креатинкиназы крови и показатели электромиограммы (ЭМГ) не выходят за пределы нормы.
Синдром Коэна (Norio et al., 1984) может имитировать синдром Прадера-Вилли и передается аутосомно-рецессивным путем. Особую диагностическую значимость имеют аномалии глаз (хориоретинальная дистрофия, атрофия зрительного нерва). Признаком данного заболевания являются аномалии зубов.
Возможен широкий разброс клинических проявлений отдельных случаев, включая слабовыраженные формы. У многих детей могут отсутствовать выраженные трудности в обучении. Таким образом, диагноз синдрома Прадера-Вилли стоит рассматривать во всех случаях «чрезвычайно толстых и голодных» детей с проявлениями поведенческого фенотипа.
в) Клинические проявления. Клинические проявления синдрома очень характерны, особенно в период новорожденности; при осмотре в более позднем возрасте большую роль при постановке диагноза играет типичный неонатальный анамнез. Новорожденные с синдромом Прадера-Вилли обычно рождаются в срок, но масса тела не соответствует гестационному возрасту. 30% детей рождаются в ягодичном предлежании. С рождения у пациентов отмечается тяжелая мышечная гипотония, поэтому часто предполагается наличие мышечного заболевания. Нарушения глотания требуют питания через зонд в течение 2-5 недель. Дисморфические проявления соответствуют кариотипу, определение которого требуется практически во всех случаях (Stephenson, 1980). В дальнейшем клиническая картина меняется. Наиболее характерными признаками являются гиперфагия и ожирение, развивающиеся у большинства детей к концу второго года жизни.
Для поздней стадии заболевания характерно выраженное ожирение, стойкая гипотония и часто слабость проксимальных отделов и относительно слабо выраженная задержка умственного развития со средним коэффициентом IQ 65 (Butler et al., 1986), хотя до 10% взрослых пациентов могут иметь нормальный коэффициент IQ (Clarke et al, 1989; Greenswag, 1987). К частым проявлениям относятся также гипогонадизм, крипторхизм, низкорослость и маленький размер лица, носа, ушей, кистей и стоп (Holm et al., 1993). Часто встречается характерный внешний вид с маленькой «круглой» головой и миндалевидными глазами.
Взрослые с синдромом Прадера-Вилли характеризуются низким ростом, избыточной массой тела, когнитивными нарушениями и эмоциональной лабильностью. У многих пациентов отмечается кифоз, сколиоз и остеопороз. Слабо развиты двигательные навыки, повышен аппетит (Greenswag, 1987; Lee, 2002) и имеется характерный поведенческий фенотип, включающий дерматилломанию и другие варианты поведения со склонностью к повторяющимся действиям (Akefeldt et al., 1991).
В атипичных случаях возможна задержка умственного развития тяжелой степени (3%) или нормальные когнитивные способности. Иногда у пациентов может не быть ожирения, а в редких случаях отмечалась кахексия (Miike et al, 1988).
Синдром Прадера-Вилли (СПВ)
Если ребенок начинает толстеть на втором или третьем году жизни, родители часто обращаются за помощью. В это время ребенок может быть необычно послушным или спокойным, но быть подверженным эпизодам раздражительности и чрезвычайно тяжелым и деструктивным приступам гнева. Гнев обычно немедленно сменяется «исходной» пониженной активностью и угрызениями совести. Результаты недавних исследований позволяют предположить, что обсессивно-компульсивное поведение иногда чрезвычайно напоминает расстройства аутистического спектра, но в ходе контрольных исследований не удалось выявить превышающую ожидаемую частоту расстройств аутистического спектра, принимая во внимание степень общей неспособности к обучению (Descheemaeker et al., 2006).
Большинство детей с синдромом Прадера-Вилли обладает странной привычкой ковырять кожу и наносить себе раны, синяки и царапины. Большая часть детей с синдромом Прадера-Вилли страдает булимией во время обострения. Они готовы на что угодно для получения доступа к еде в холодильнике, морозилке или кладовке. Большинство родителей вынуждено применять замки для ограничения доступа детей к еде. Даже в ходе научных исследований была наглядно продемонстрирована неспособность данных пациентов прекратить есть (в экспериментальных условиях).
г) Лечение синдрома Прадера-Вилли. Следует информировать родителей о природе состояния их детей. Во всех случаях необходимы беседы и печатные пособия. Врачи, диетологи, психологи (и сами родители) часто винят родителей в выраженном ожирении детей. Сопутствующие нарушения поведения часто приводят к направлению пациентов в детскую психиатрическую службу. Без подтвержденного диагноза существует большой риск, что консультирование будет сконцентрировано на воспитании как основе имеющихся отклонений. Положительное психологическое влияние на родителей оказывает информация о практически идентичных поведенческих проблемах у других детей с синдромом Прадера-Вилли. Важным моментом является обращение родителей в национальную или региональную группу поддержки больных с синдромом Прадера-Вилли.
Последние 10 лет в качестве компонента полной программы лечения детей и взрослых с синдромом Прадера-Вилли начато применение гормона роста. Применение данного препарата приводит к достижению более высокого роста и некоторому улучшению умственных способностей. Тем не менее, присутствует предположение о возможности развития значимых (и иногда опасных) побочных эффектов у некоторых пациентов, что означает необходимость назначения терапии гормоном роста только врачами-специалистами, работающими в данной области.
Читайте также:
- Лучевая диагностика эпидермоидной кисты позвоночника и спинного мозга
- Отравление противосудорожными лекарствами и их побочные эффекты
- Вакуум-экстракция межпозвоночного диска. Внутридисковая радиочастотная терапия.
- Танатофорная дисплазия. Диагностика и прогноз танатоформной дисплазии
- Псориатический артрит: причины, симптомы и лечение