Спленэктомия при хроническом идиопатическом миелофиброзе (ХИМФ) - показания, эффективность
Добавил пользователь Skiper Обновлено: 31.01.2026
ГБОУ ВПО «Первый МГМУ им. И.М. Сеченова» Минздрава России, Москва
ФГБУ "Гематологический научный центр" Минздрава России, Москва
Гематологический научный центр Минздрава России, Москва
Научно-клиническое отделение гематологической хирургии Гематологического научного центра Минздрава РФ, Москва
Гематологический научный центр Минздрава РФ, Москва
ФГБУ «Гематологический научный центр» Минздрава России, Москва, Россия
Клинические особенности эссенциальной тромбоцитемии и первичного миелофиброза в зависимости от молекулярных характеристик заболевания
Журнал: Терапевтический архив. 2017;89(7): 4‑9
Аннотация Целью представляемой работы являлась оценка клинических особенностей, риска развития тромботических осложнений (ТО) у больных эссенциальной тромбоцитемией (ЭТ) и первичным миелофиброзом (ПМФ) в зависимости от молекулярной характеристики заболевания. Проведен анализ клинических данных, лабораторных показателей у 50 больных ЭТ и 50 больных ПМФ, наблюдавшихся в отделении стандартизации методов лечения ФГБУ ГНЦ МЗ РФ с февраля 2015 г. по сентябрь 2016 г. Выявлено, что больные ЭТ и ПМФ имеют высокий риск развития ТО. Риск развития ТО у пациентов с ЭТ выше (24% во всей группе), чем у больных ПМФ (14% в обследованной группе). При ЭТ имеется высокий риск тромбозов в случае выявления мутаций генов JAK2 и CALR в сравнении с «тройными негативными» наблюдениями. Больные ПМФ с мутацией V617F гена JAK2 имеют высокий риск развития ТО по сравнению с пациентами — носителями мутации CALR и в «тройных негативных» случаях. Не выявлено достоверной ассоциации ТО с высоким тромбоцитозом. У больных ПМФ не выявлено негативного прогностического значения такого фактора, как возраст.
ИМ — инфаркт миокарда
ИП — истинная полицитемия
ОНМК — острое нарушение мозгового кровообращения
ПМФ — первичный миелофиброз
ТО — тромботические осложнения
ХМПЗ — хронические миелопролиферативные заболевания
ЭТ — эссенциальная тромбоцитемия
Хронические миелопролиферативные заболевания (ХМПЗ) представляют собой клональные заболевания, возникающие на уровне стволовой кроветворной клетки. ХМПЗ характеризуются пролиферацией одной или более клеточной линии миелопоэза в костном мозге с признаками сохранной терминальной дифференцировки и сопровождаются изменением показателей периферической крови. К группе классических Ph-негативных ХМПЗ относят три нозологических варианта: истинная полицитемия, эссенциальная тромбоцитемия (ЭТ), первичный миелофиброз (ПМФ) [1, 2].
Этиология ХМПЗ до сих пор не установлена. Ведущей гипотезой является многоэтапность возникновения заболевания, при которой предрасположенность к болезни реализуется под воздействием внешних факторов, повреждающих геном нормальной клетки и приводящих к ее злокачественной трансформации.
Открытие мутации V617 °F в гене JAK2 в 2005 г. явилось значительным шагом в понимании биологических особенностей Ph-негативных ХМПЗ. Практически у всех пациентов с истинной полицитемией (ИП) выявляется мутация гена JAK2: в 96% случаев мутация V617 °F (14-й экзон), в 2% наблюдений мутация в 12-м экзоне [2, 3]. Мутация JAK2V617 °F выявляется при ЭТ в 55% наблюдений и имеется в 45—68% случаев при ПМФ, тогда как мутация в 12-м экзоне гена JAK2 практически не встречается при ЭТ и ПМФ [4—6].
Помимо мутации гена JAK2 у больных ХМПЗ выявляют мутации в других генах. Мутации гена MPL встречаются в 4% наблюдений при ЭТ, в 8% — при ПМФ и крайне редко при И.П. Причем наиболее частые мутации MPLW515L/K наблюдаются в 10-м экзоне [7—10]. Мутация MPLS505N выявляется как при ЭТ, так и при наследственной тромбоцитемии [9, 10]. Данные мутации не являются строго специфичными для ХМПЗ.
В 2013 г. появились данные о диагностической значимости соматических мутаций в 9-м экзоне гена CALR, кодирующего белок кальретикулин. Выявлены 36 разных видов мутаций в этом гене, которые приводят к образованию дефектного белка. В исследованиях in vitro клетки, экспрессирующие мутированный ген, обладали способностью независимого от цитокинов роста в культуре, что, вероятно, связано с активацией белка STAT5. У пациентов без мутаций в генах JAK2 и MPL мутации в гене CALR выявлены в 67% случаев при ЭТ и 88% при ПМФ. Другие авторы также выявили крайне высокую частоту мутаций гена CALR у пациентов с ХМПЗ (в 70—84% случаев в отсутствие мутации гена JAK2). При этом мутации CALR обнаружены в 8% случаев при миелодиспластическом синдроме и в единичных наблюдениях при других миелоидных неоплазиях. Важно, что ни в одном случае заболеваний немиелоидной природы мутации в данном гене не выявлены [11, 12].
Мутации в генах JAK2, MPL, CALR имеют большое диагностическое значение. Их выявление свидетельствует о клональном характере заболевания и помогает в дифференциальной диагностике ИП, ЭТ, ПМФ от ряда других миелоидных неоплазий, а также вторичных эритроцитозов и тромбоцитозов. Наряду с этим активно изучается роль данных мутаций в прогнозе ХМПЗ [13, 14].
Основными причинами, существенно снижающими качество жизни и угрожающими жизни пациентов с ХМПЗ, являются тромботические и геморрагические осложнения. Совокупная частота тромбозов при ИП составляет 3,8 на 100 пациенто-лет, при ЭТ — от 2 до 4 на 100 пациенто-лет, при ПМФ — 2,23 на 100 пациенто-лет [15]. Возраст и тромботические осложнений (ТО) в анамнезе — основные факторы риска развития Т.О. Высокий тромбоцитоз (≥1000 или 1500·10 9 /л) является фактором риска геморрагических осложнений, но не ТО [16]. Геморрагические осложнения, спонтанные или спровоцированные даже малыми хирургическими вмешательствами, варьируют от незначительных (носовые, десневые кровотечения, экхимозы) до непосредственно угрожающих жизни кровотечений (желудочно-кишечные и другие полостные кровотечения).
Целью данной работы является оценка клинических особенностей, риска развития ТО у больных ЭТ и ПМФ в зависимости от молекулярной характеристики заболевания.
В рамках научно-исследовательской работы «Мутации в генах JAK2, MPL, CALR, SF3B1 при Ph-негативных миелопролиферативных заболеваниях (неоплазиях) и миелодиспластических/миелопролиферативных заболеваниях» сформирована группа пациентов с ЭТ и ПМФ, наблюдавшихся в отделении стандартизации методов лечения ФГБУ ГНЦ МЗ РФ с февраля 2015 г. по сентябрь 2016 г. Всем больным проведено комплексное обследование, включающее как исследования, необходимые для диагностики заболевания, так и дополнительные, позволяющие определить тактику ведения пациента. Во всех случаях диагноз установлен в соответствии с критериями ВОЗ 2008 г.
Эссенциальная тромбоцитемия. В исследование включены 50 больных с впервые установленным диагнозом ЭТ. В исследованной группе 9 мужчин и 41 женщина. Соотношение мужчин и женщин равно 1: 4,5. Возраст больных составил от 19 до 75 лет (медиана — Ме 36 лет). Группа больных старше 60 лет составила 6% (3 пациента).
Всем пациентам выполнено исследование костномозгового кроветворения (трепанобиопсия гребня подвздошной кости с гистологическим исследованием трепанобиоптата). В 47 (94%) случаях костный мозг был нормоклеточным. Гиперклеточность костного мозга выявлена в 3 (6%) наблюдениях. Гипоклеточный костный мозг не выявлен ни в одном случае.
Бессимптомное течение заболевания (отсутствие любых жалоб, связанных с ЭТ) наблюдалось в 65% случаев. В 5% наблюдений пациенты испытывали дискомфорт/боли за грудиной. Основным клиническим проявлением заболевания, послужившим причиной обращения пациента в клинику, были жалобы на головную боль (30%). Головная боль в большинстве случаев носила характер мигрени.
С умеренной спленомегалией (размеры селезенки не превышали 5 см из-под края реберной дуги) заболевание протекало у 9 (18%) больных, в остальных случаях — у 41 (82%) больного спленомегалия не выявлена.
Всем пациентам, включенным в исследование, проводили молекулярно-генетическое исследование (JAK2, MPL, CALR). Мутация V617 °F гена JAK2 выявлена у 26 (52%) больных. В 2 случаях проведена качественная, в 24 — количественная оценка. Аллельная нагрузка составила от 1 до 80%. Мутация W515L гена MPL выявлена в 1 случае. Мутации гена CALR обнаружены у 15 (30%) пациентов. «Тройные негативные» (triple negative) случаи составили 18% (9 больных).
Основным лабораторным параметром, определяющим диагноз ЭТ, являлось количество тромбоцитов в периферической крови. В исследуемой группе больных этот показатель составил от 525·10 9 до 2525·10 9 /л (Ме 842·10 9 /л). Тромбоциты у пациентов с ЭТ и мутацией гена JAK2 составили от 525·10 9 до 1410·10 9 /л (Ме 730·10 9 /л), с мутациями гена CALR — от 691·10 9 до 2525·10 9 /л (Ме 1000·10 9 /л), «тройные негативные» случаи — от 600 до 1600·10 9 /л (Ме 1165·10 9 /л). Лабораторные особенности ЭТ в зависимости от молекулярных характеристик заболевания представлены в табл. 1.

Таблица 1. Клинические и лабораторные особенности ЭТ в зависимости от молекулярной характеристики заболевания Примечание. ОНМК — острое нарушение мозгового кровообращения; ИМ — инфаркт миокарда.
Несмотря на молодой возраст пациентов, у 41 (82%) из них имелись сопутствующие заболевания. Из них у 21 (42%) диагностировано более двух сопутствующих заболеваний. Заболевания сосудов (геморрой, варикозная болезнь) — у 4 больных, патология женской репродуктивной системы (привычное невынашивание беременности, бесплодие, эндометриоз, миома матки, поликистоз яичников, фиброаденома молочных желез) — у 16, системные заболевания соединительной ткани (антифосфолипидный синдром, артрит) — у 3, заболевания сердечно-сосудистой системы (артериальная гипертония, аритмия) — у 11, эндокринной системы (сахарный диабет, узловой зоб, аутоиммунный тиреоидит, аденома гипофиза) — у 14, пищеварительной системы (хронический гастрит, желчнокаменная болезнь, язвенная болезнь желудка) — у 12, мочевыделительной системы (мочекаменная болезнь, хронический цистит, хронический пиелонефрит) — у 4, новообразования (базалиома, рак матки, рак молочной железы, лейомиома желудка) — у 4.
Известно, что основным осложнением заболевания, приводящим к инвалидности, являются тромбозы. ТО в исследуемой группе выявлены у 12 (24%) больных: ОНМК у 1, ИМ — у 6, тромбоз воротной вены у 1, локтевой вены у 1, подключичной вены у 1, артерий сетчатки глаза у 1, брюшного отдела аорты и почечной артерии у 1 (см. табл. 1). В данной группе соотношение мужчин и женщин составило 1:2,7, возраст от 27 до 75 лет (Ме 38 лет). Концентрация тромбоцитов от 542·10 9 до 1275·10 9 /л (Ме 827·10 9 /л). В 8 случаях у пациентов определялась мутация V617 °F гена JAK2 и только в 4 наблюдениях (тромбоз подключичной вены и 3 случая ИМ) — мутации гена CALR. Вероятность Т.О. не зависела от количества тромбоцитов: у больных с JAK2+ и тромбозами — (630—1275)·10 9 /л (Ме 730·10 9 /л); с JAK2+ и без тромбозов (525—1410)·10 9 /л (Ме 733·10 9 /л); с CALR+ и тромбозами — (691—1031)·10 9 /л (Ме 859·10 9 /л); с CALR+ и без тромбозов — (821—2525)·10 9 /л (Ме 1115·10 9 /л).
Пациенты получали терапию в соответствии с Российскими клиническими рекомендациями по диагностике и лечению Ph-негативных МПЗ: гидроксикарбамид — 17 (34%) больных, интерферон-α — 16 (32%), анагрелид — 4 (8%), симптоматическую терапию (антиагреганты) — 13 (26%) пациентов [1, 17].
Первичный миелофиброз. Исследуемая группа включала 50 больных в возрасте от 24 до 83 лет (Ме 47 лет, старше 60 лет 16 пациентов, или 32%). Соотношение мужчин и женщин равно 1:4.
Всем пациентам проведено гистологическое исследование трепанобиоптата костного мозга. Ранняя/префиброзная стадия диагностирована у 20 (40%) больных, у 4 (8%) больных — постполицитемический миелофиброз, у 26 (52%) определялась характерная картина ПМФ.
Клиническая картина ПМФ складывается из синдромов. Основными проявлениями болезни были утомляемость, не связанная с наличием анемии или других сопутствующих заболеваний, — 46, симптомы опухолевой интоксикации (субфебрилитет, снижение массы тела, потливость) — 10, аквагенный кожный зуд и сухость кожи — 9, неврологические проявления (шум в голове, головная боль, тревожность) — 7, пролиферативный синдром (чувство раннего насыщения, боли в левом подреберье) — 2, боль за грудиной — 1 случай. В 4 (8%) случаях тяжесть состояния пациентов определялась тяжелым анемическим синдромом — анемия носила зависимый от трансфузий характер.
Спленомегалия (пальпируемые размеры селезенки более 5 см ниже края реберной дуги) выявлена у 43 (86%) пациентов.
В большинстве наблюдений диагностированы сопутствующие заболевания: сердечно-сосудистой системы (артериальная гипертония) у 13 (26%) больных, варикозная болезнь и атеросклероз сосудов нижних конечностей у 13 (26%). Данную особенность можно объяснить тем, что большую долю больных ПМФ составляют пациенты старшего возраста. Другими сопутствующими заболеваниями были болезни желудочно-кишечного тракта (хронический гастрит, желчнокаменная болезнь, язвенная болезнь желудка) — 6, эндокринной системы (узловой зоб, аутоиммунный тиреоидит) — 9, женской репродуктивной системы (эндометриоз) — 3, системные болезни соединительной ткани (ревматоидный артрит) — 1, мочевыделительной системы (мочекаменная болезнь, кисты почек) — 4, новообразования (рак желудка, рак простаты) — 2.
Мутация V617 °F гена JAK2 выявлена у 26 (52%) больных, гена CALR — у 16 (32%), гена MPL — у 1, «тройные негативные» случаи — у 7 (14%). Аллельная нагрузка JAK2V617 °F составила от 3,27 до 97% (Ме 17,18%).
ТО в исследуемой группе пациентов выявлены у 7 (14%) больных: ОНМК у 2, тромбоз селезеночной и воротной вен у 2, тромбоз воротной, селезеночной, верхней брыжеечной вен у 1, ИМ у 2, последовательные тромбозы воротной вены, брыжеечной вены, селезеночной артерии, внутренней сонной артерии у 1. Группа пациентов с ТО включала 4 мужчин и 3 женщин в возрасте от 46 до 83 лет. Количество тромбоцитов в периферической крови составило от 27·10 9 до 1587·10 9 /л (Ме 783·10 9 /л), количество лейкоцитов — от 6·10 9 до 25·10 9 /л (Ме 10,7·10 9 /л). В 5 наблюдениях выявлена мутация V617 °F гена JAK2. Следует отметить наблюдение с последовательными венозными и артериальными тромбозами воротной вены, брыжеечной вены, селезеночной артерии, внутренней сонной артерии (табл. 2).
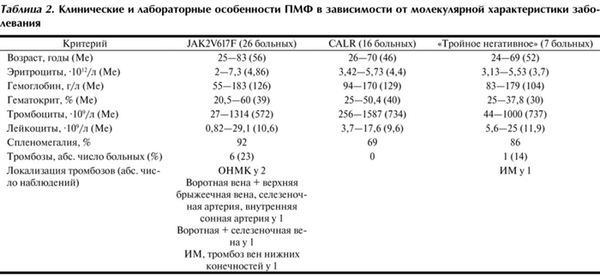
Таблица 2. Клинические и лабораторные особенности ПМФ в зависимости от молекулярной характеристики заболевания
Лечение больных ПМФ проводилось в соответствии с Российскими рекомендациями по диагностике и лечению Ph-негативных ХМПЗ. В 16% случаев проводилась симптоматическая терапия, лечение ограничивалось коррекцией проявлений синдрома, определявшего тяжесть состояния пациента. В 84% наблюдений пациенты получали специфическую терапию. Выбор препарата определялся стадией заболевания, возрастом пациента на момент диагностики ПМФ. В 16 (32%) наблюдениях назначен интерферон-α, в 26 (52%) случаях проводилась циторедуктивная терапия гидроксимочевиной.
Цель данной работы состояла в оценке клинических особенностей, риска развития ТО у больных ЭТ и ПМФ в зависимости от молекулярной характеристики заболевания. Выборка больных включала 50 пациентов с ЭТ и 50 пациентов с ПМФ. В группе больных ЭТ мутация V617 °F гена JAK2 выявлена в 52% наблюдений, гена CALR — в 30%, гена MPL — только в 1 наблюдении, «тройные негативные» наблюдения — в 18%. В группе больных ПМФ мутация V617 °F гена JAK2 выявлена в 52% наблюдений, гена CALR — в 32%, гена MPL — также только в 1 наблюдении, «тройные негативные» случаи составили 14%. Полученные нами данные соответствуют результатам других популяционных исследований по распространенности диагностических мутаций у больных ХМПЗ.
При ЭТ более высокое содержание тромбоцитов наблюдалось в «тройных негативных» наблюдениях: (600—1600)·10 9 /л (Ме 1165·10 9 /л) и при выявлении мутации гена CALR (691—2525)·10 9 /л (Ме 1000·10 9 /л) по сравнению со случаями выявления мутации JAK2V617 °F (525—1410)·10 9 /л (Ме 730·10 9 /л). Риск развития ТО не зависел от количества тромбоцитов. Несмотря на относительно невысокий тромбоцитоз в группе больных ЭТ с мутацией JAK2V617 °F частота тромбозов составила 31% (8 случаев из 26), в то время как у больных ЭТ с мутацией CALR частота тромбозов — 27% (4 случая из 15), а в «тройных негативных» случаях ТО не выявлено. В исследуемой группе не выявлено корреляции риска развития ТО и возраста. Возраст больных ЭТ с ТО составил от 24 до 57 лет (Ме 35 лет), без ТО — 23—62 года (Ме 41 год). Артериальные тромбозы выявлены в 9 случаях (ИМ в 6, ОНМК в 1, артерия сетчатки глаза — в 1, брюшной отдел аорты и почечной артерии — в 1), венозные тромбозы развились в 3 наблюдениях (воротная вена у 1, локтевая вена у 1, подключичная вена у 1). В двух случаях венозных тромбозов провоцирующим фактором служила установка центрального/периферического венозного катетера.
В 2012 г. разработана международная прогностическая шкала риска развития тромбозов при ЭТ — ВОЗ-ЭТ (IPSET-thrombosis), в которой оценивается возраст старше 60 лет, наличие тромбозов в анамнезе, наличие факторов риска развития сердечно-сосудистых заболеваний и выявление мутации V617 °F гена JAK2. Каждому признаку присвоен балл и согласно сумме баллов пациент распределяется в группу низкого, промежуточного или высокого риска развития Т.О. Таким образом, согласно данным литературы, молекулярные особенности заболевания играют роль в развитии ТО при ЭТ: мутация V617FJAK2 определяет высокий риск развития тромбозов [18, 19]. Мы не получили аналогичных результатов о значимости мутации гена JAK2 как фактора риска развития ТО, возможно, из-за небольшой выборки больных.
Заключение
Риск развития ТО у больных ЭТ (24% во всей группе) и ПМФ (14% в исследованной группе) высокий. Больные ПМФ имеют высокий риск развития ТО независимо от возраста. У пациентов с ЭТ риск тромбозов возрастает при выявлении мутаций генов JAK2 и CALR по сравнению с таковым при «тройных негативных» наблюдениях. Пациенты с ПМФ с мутациями V617 °F гена JAK2 имеют высокий риск развития ТО по сравнению с пациентами — носителями мутации CALR или в «тройных негативных» случаях. Не выявлено достоверной ассоциации ТО с высоким тромбоцитозом. Очевидно, что патогенез тромбозов сложен и нуждается в дополнительном детальном изу-чении.
Спленэктомия при хроническом идиопатическом миелофиброзе (ХИМФ) - показания, эффективность
Течение хронического идиопатического миелофиброза (ХИМФ) - прогноз
Распространенное мнение о доброкачественности течения хронического идиопатического миелофиброза (ХИМФ) не относится ко всем больным и не подтверждается статистическими даннными: по результатам анализа итальянских авторов, медиана выживаемости варьирует от 1,4 до 9,1 года.
Частота бластной трансформации достигает 30 % и более. По данным американских авторов, общая средняя продолжительность жизни больных составляет всего 5 лет.
Тем не менее у значительной части больных течение заболевания вполне доброкачественное, и этой группе пациентов свойственны свои клинико-гема-тологические, патогистологические и цитогенетиче-ские характеристики.
В прогностической модели Dupriez, основанной на анализе течения заболевания и медианы выживаемости у 200 больных ХИМФ, факторами плохого прогноза признаны прежде всего анемия (Hb < 100 г/л), лейкоцитоз более 30 • 10 9 /л или лейкопения ниже 3 • 109/л. У больных, не имеющих этих признаков, медиана выживаемости составила 93 мес, а у больных, имеющих хотя бы 1 или 2 таких признака, — 26 мес.
Используя простые клинические и гематологические параметры болезни, F. Cervantes разделил больных на 2 группы — с короткой (20,6 мес) и длительной (98 мес) медианой выживаемости.
При анализе естественного течения заболевания G. Barosi высказал несколько интересных положений прогностического значения:
а) по степени выраженности миелофиброза на момент постановки диагноза можно прогнозировать время развития анемического синдрома: при его резко выраженном варианте анемия развивается в течение 2 лет;
б) большое прогностическое значение имеет содержание CD34-позитивных клеток в периферической крови. Число этих клеток коррелирует с продолжительностью заболевания и вероятностью эволюции в острый лейкоз: при их абсолютном числе 300 • 106/л эта перспектива является неизбежной;
в) автор выделяет отдельную группу хорошего прогноза.

Параметры заболевания в этой группе следующие: более молодой возраст, стабильное течение, небольшая степень миелофиброза или только миелопролиферация, нормальные показатели красной крови и числа лейкоцитов, отсутствие или небольшой сдвиг лейкоцитарной формулы влево, нормальное или повышенное число тромбоцитов. Этот вариант заболевания автор обозначает как «тлеющий» или «замороженный» миелофиброз.
Даже при этой доброкачественной форме осложнения возможны, и особенно это относится к тромбозам в системе чревных артерий.
Прогноз определяет и патоморфологическая картина хронического идиопатического миелофиброза (ХИМФ). У больных с префиброзной или ранней фиброзной стадией медиана выживаемости составила 130 мес, тогда как у больных с выраженным миелофиброзом и остеомиелосклерозом — 82 мес. К параметрам плохого прогностического значения отнесены и пожилой возраст на момент диагностики, степень анемии, а также число лейкоцитов и тромбоцитов.
Прогностическое значение, безусловно, имеют и данные цитогенетического исследования.
Вопросы прогноза дальнейшего течения заболевания на данное время имеют чисто практическое значение при отборе больных на трансплантацию костного мозга, ставшую реальностью.
Ниже приведена суммация показателей плохого прогноза, учитывающая накопленный по этому вопросу опыт, доступный для любого практического врача:
— анемия (Hb— пожилой возраст (старше 64 лет);
— симптомы гиперкатаболизма (потеря массы тела, чрезмерная слабость, ночные поты, субфеб-рильная температура тела);
— число лейкоцитов > 30•10 9 /л;
— число лейкоцитов < 4•10 9 /л;
— циркуляция бластов в периферической крови (> 1 %);
— цитогенетические нарушения (+8, 12р-).
По этому принципу автор разделил больных на 3 группы:
• группа низкого риска — отсутствие плохих прогностических признаков; медиана выживаемости в этой группе может превышать 10 лет;
• группа высокого риска (присутствие 2 и более факторов плохого прогноза независимо от возраста). В этой группе средняя выживаемость не превышает 2 лет;
• группа промежуточного риска.
Отбору на столь ответственную меру лечения, какой является трансплантация костного мозга, подлежит группа больных высокого риска.
Редактор: Искандер Милевски. Дата обновления публикации: 18.3.2021
В практике терапии осложнений хронического идиопатического миелофиброза (ХИМФ) находит применение и спленэктомия (СЭ).
Общими показаниями к ее назначению являются:
• гемолитическая анемия с периодом полураспада меченных 51Cr эритроцитов менее 11 дней (в норме 30—32 дня);
• тромбоцитопения менее 100•10 9 /л;
• массивная спленомегалия и вызываемые ею компрессионные осложнения;
• осложнение портальной гипертонией. Противопоказания к спленэктомии (комплексные данные):
• число тромбоцитов более 500•10 9 /л;
• нарушения гемостаза по типу ДВС-синдрома и тромбоцитопения потребления;
• значительное увеличение печени;
• сцинтитопографическая картина с преимущественной локализацией кроветворения в селезенке;
• нарушения функционального состояния сердца, печени, почек.
Диагноз гемолитической анемии при хроническом идиопатическом миелофиброзе основан на данных радиологического исследования продолжительности жизни эритроцитов, меченных 51 Cr.
В некоторых случаях патогенез тромбоцитопении при хроническом идиопатическом миелофиброзе (ХИМФ) понять сложно, поскольку, хотя соответствующие радиологические методы исследования продукции и деструкции тромбоцитов существуют, в практику они не внедрены. В связи с этим рекомендуется тщательное исследование трепаната костного мозга. При обнаружении уменьшения числа мегакариоцитов, преобладания мелких по величине и дефектных по структуре форм можно ожидать нарушения образования тромбоцитов с худшим прогнозом результатов спленэктомии.
При гиперплазии мегакариоцитарного ростка и преобладании крупных форм мегакариоцитов более вероятна связь тромбоцитопении с повышенным гемолизом в увеличенной селезенке. В данном случае можно ожидать лучшего прогноза спленэктомии в отношении тромбоцитопении, хотя исключения, конечно, возможны. Необходимо исключить и такую причину тромбоцитопении и анемии, как миелодиспластический синдром, при котором спленэктомия не показана и чревата ускоренным развитием острого лейкоза.
Исходно большие размеры печени также надо рассматривать как частичное противопоказание к спленэктомии, поскольку после операции у 25 % больных наблюдается ее прогрессирующий рост, и хотя ее функциональное состояние при этом долго не нарушается, смена спленомегалии на значительную гепатомегалию не может рассматриваться как несущественный фактор.
В оценке прогноза спленэктомии и отборе больных на операцию определенное значение имеют данные сцинтитопографического исследования гемопоэза с помощью 99ТсМ. Картины массивной редукции гемопоэза в плоских и трубчатых костях ставят под сомнение пользу назначения спленэктомии, поскольку селезенка оказывается почти единственным источником гемопоэза. И хотя подобная картина не является абсолютным противопоказанием к спленэктомии, прогнозировать результаты проблематично, но описаны случаи усиления гемопоэтической функции костного мозга после спленэктомии.

Степень выраженности миелофиброза и остеомиелосклероза не имеет решающего значения при отборе больных на спленэктомию, но она оказывает влияние на прогноз ее результатов. Плохое прогностическое значение имеет и гипоклеточность костного мозга.
Высокая исходная нормобластемия, осколки ядер мегакариоцитов, макро- и одновременно микроцитарный характер анемии, резко выраженный пойки-лоцитоз свидетельствуют о глубоких качественных нарушениях гепомоэза, которые не могут быть устранены спленэктомией.
Назначению спленэктомии предшествуют попытки добиться эффекта с помощью консервативной терапии анемического и тромбоцитопенического синдромов с помощью преднизолона. При ее неэффективности назначается спленэктомия.
При внутрипеченочной портальной гипертонии желательно сочетать спленэктомию с наложением спленоренального шунта , но, по нашим данным, эффективна и одна спленэктомия.
Удаление большой селезенки представляет значительные технические трудности, однако практикуемые иногда попытки ее сокращения перед операцией с помощью гамма-терапии не оправданы в связи с развитием спаечных процессов, затрудняющих операцию, и высокой частотой цитопенических осложнений.
Переносимость операции зависит от исходного состояния гемостаза и эффективности коррекции выявленных нарушений, квалификации хирурга, службы интенсивной терапии. Риск хирургических вмешательств с 1970 г. существенно уменьшился, но операционная смертность и послеоперационные осложнения все еще остаются большой проблемой.
По данным анализа 203 спленэктомированных больных, операцию не перенесли 9 %, а частота послеоперационных осложнений составила 31 %. В другом наблюдении (30 спленэктомированных больных) смертность составила 6,7 %, а послеоперационные осложнение — 37 %. Авторы, сопоставив частоту осложнений при раннем и позднем назначении спленэктомии (13 и 64 % соответственно), высказались в пользу более ранней спленэктомии, при первом появлении цитопенических осложнений заболевания.
К послеоперационным осложнениям относятся кровотечения, геморрагический и некротический панкреатит, поддиафрагмальный абсцесс, ателектазы легких, тромботические и инфекционные осложнения. Иногда из-за кровотечения возникает необходимость в релапаратомии. Причиной кровотечений является количественная и качественная неполноценность тромбоцитов и нарушение свертывания крови. Развитию тромбозов сосудов способствуют послеоперационный тромбоцитоз, нормобластемия, повышение уровня тканевого тромбопластина, свойственное массивным операциям, и в некоторых случаях тромбогеморрагический синдром.
Отсюда необходимость квалифицированного гемостазиологического контроля как до, так и во время, и после операции, а также соответствующая подготовка больных. Возникающий после операции тромбоцитоз подлежит быстрому циторедуктивному лечению; назначение аспирина с профилактической целью нежелательно из-за угрозы кровотечений.
Следует отметить улучшение качества жизни у перенесших данную операцию. Это объясняется устранением симптомов, вызванных большой селезенкой, уменьшением клеточного гиперкатаболизма, нормализацией или улучшением показателей красной крови, существенной прибавкой массы тела.
Вопрос об удлинении продолжительности жизни больных под влиянием спленэктомии остается открытым. Отдельные больные имеют уникальную продолжительность жизни после спленэктомии, равную 10—11 годам. Расчет средней продолжительности жизни на большом клиническом материале не показал ее увеличения, но не было и сокращения, даже с учетом больных, у которых после спленэктомии развился острый лейкоз.
Сравнительно высокая продолжительность жизни после операции — 55 мес у 65 больных, перенесших спленэктомию, отмеченная итальянскими авторами, может найти объяснение в отборе на операцию больных сравнительно молодого возраста (средний возраст 55 лет).
Противоречивость мнений по этому вопросу, а также по влиянию спленэктомии на частоту последующего развития острого лейкоза A. Tefferi справедливо объясняет целым рядом факторов, прежде всего неоднородным отбором больных на спленэктомию. Это легко понять, если принять во внимание, что есть сторонники раннего назначения спленэктомии и такие специалисты, которые назначают спленэктомию на поздних стадиях заболевания, когда все консервативные методы лечения больного исчерпаны. Результаты и последствия заведомо будут разными.
Среди 203 перенесших операцию больных A. Tefferi острый лейкоз развился у 16 % больных, а среднее время жизни после спленэктомии до развития острого лейкоза составило 27 мес. По мнению автора, эти данные выглядят благополучно по сравнению с исходами у 321 больного, по данным литературы. Хотя частота развития острого лейкоза среди этих больных составила 11— 27 %, средний срок периода до развития острого лейкоза равнялся всего 13 мес.
В итальянской группе имела место высокая пропорция острого лейкоза у больных после спленэктомии — 43 %, а у не-оперированных — 22 %. Суммарные данные по Италии (462 перенесших спленэктомию больных): частота острого лейкоза у перенесших спленэктомию — 26 %, а у неоперированных — 12 %. По мнению авторов, спленэктомия является независимым фактором риска развития острого лейкоза.
Какими бы субъективными не выглядели различные точки зрения по этому вопросу, проблема промоторного действия спленэктомии на развитие острого лейкоза существует. Между тем сама операция не может считаться первопричиной острого лейкоза; она может выявить скрытую тенденцию к этому исходу, которая до спленэктомии не была должным образом оценена. При анализе своих наблюдений post factum было обращено внимание на очень большую продолжительность заболевания у большинства наших больных, наличие больных с лейкопенией, или лейкоцитозом, или всем набором симптомов миелодисплазии, при которой возможность развития острого лейкоза в ближайшие сроки может легко прогнозироваться.
Из этого следует необходимость улучшения отбора больных на спленэктомию. Если прогностическое значение высокого содержания CD34-позитивных клеток в периферической крови подтвердится, появится еще один критерий, исключающий назначение спленэктомии.
Таким образом, спленэктомия как метод лечения осложнений хронического идиопатического миелофиброза не потеряла своего позитивного значения, обладает достаточной эффективностью, особенно при портальной гипертонии, компрессионном синдроме и гемолитической анемии, способна значительно улучшить качество жизни больных, хотя и не продлевает существенно ее продолжительность.
Клиника хронического идиопатического миелофиброза (ХИМФ) - спленомегалия, портальная гипертензия, анемия, асцит
Картина крови при хроническом идиопатическом миелофиброзе (ХИМФ) отличается большим разнообразием. Показатели красной крови на момент постановки диагноза чаще нормальны или несколько снижены, но у 15—25 % больных уже имеется анемия. Повышение показателей красной крови, обычно клинически бессимптомное, отмечается у 5—6 % больных.
У большинства больных наблюдается нейтрофильный лейкоцитоз (10—15•10 9 /л), очень характерен выраженный палочкоядерный сдвиг и присутствие единичных мета- и миелоцитов.
У 10—15 % больных наблюдается более выраженный лейкоцитоз (>20•10 9 /л) и большая степень левого сдвига лейкоцитарной формулы крови, циркуляция единичных бластов. Число тромбоцитов нормально у 30—40 % больных и увеличено примерно у 15 %. Отмечается повышение уровня лактатдегидрогеназы, коррелирующее с числом лейкоцитов. Морфологические изменения эритроцитов — частая находка при хроническом идиопатическом миелофиброзе (ХИМФ). Клинические симптомы хронического идиопатического миелофиброза можно разделить на:
1) ассоциированные со значительным увеличением селезенки;
2) обусловленные усиленным клеточным катаболизмом;
3) обусловленные недостаточностью костного мозга.
Под последними имеются в виду анемический и тромбоцитопенический синдромы, хотя в действительности в их развитии участвуют разные патогенетические механизмы.
В зависимости от давности хронического идиопатического миелофиброза (ХИМФ) размеры селезенки широко варьируют. Со временем она достигает гигантских размеров, занимает всю левую и часть правой половины живота, отличается повышенной плотностью и бугристостью. Субъективные расстройства, вызываемые большой селезенкой, — это чувство тяжести, ощущение сдавления (малой вместимости) желудка и кишечника (неустойчивый стул), периодические острые боли, вызываемые инфарктом селезенки и периспленитом.
Классической причиной увеличения селезенки оказывается миелоидная метаплазия селезенки (ММС), но следующей возможной причиной спленомегалии является осложнение портальной гипертонией, а также увеличение депонирующей и секвестрирующей функций селезенки, т. е. рабочая гипертрофия органа.

Гепатомегалия и портальная гипертензия при хроническом идиопатическом миелофиброзе
Более чем у половины больных при установлении диагноза определяют гепатомегалию. Изолированное увеличение печени изредка возможно, как и преобладание гепатомегалии над спленомегалией. Значительное увеличение печени наблюдается обычно у больных, перенесших спленэктомию (СЭ), а частота прогрессирующей гепатомегалии после СЭ составляет 26—22 %.
Функциональные нарушения печени редки, чаще наблюдаются в терминальной стадии заболевания.
Развитие синдрома портальной гипертонии сопровождается значительным увеличением селезенки, не обусловленным ее участием в кроветворении, варикозным расширением вен пищевода, а затем периферическими отеками и асцитом. По данным Silverstein, портальная гипертония осложняет течение хронического идиопатического миелофиброза в 6—8 %, из них в 70 % она является результатом гиперкинетического тока крови и в 30 % — внутрипеченочных блоков. Их причины — миелоидная метаплазия, которая локализуется в синусоидах печени, вызываемый ею реактивный фиброз, а в отдельных случаях — постнекротический цирроз печени, обусловленный перенесенным гепатитом.
Среди вариантов портальной гипертонии, частота которой другими авторами оценивается в 10—15 %, выделяют пресинусоидальный тромботический блок, синусоидальную обструкцию, вызываемую миелоидной метаплазией печени в сочетании с гиперкинетическим током крови, постсинусоидальный, очевидно, тромботический блок, аналогичный синдрому Бадда — Киари.
Считается, что при функциональном внутрипеченочном портальном блоке анатомических изменений в печени нет, но есть и такая точка зрения, что для развития портальной гипертонии одного гиперкинетического тока крови недостаточно и структурные нарушения в печени должны присутствовать.
Уровень блока току крови в портальной системе в настоящее время определяется неинвазивным методом — ультразвуковой допплерографией.
При подпеченочной портальной гипертензии (ПГ) (тромбоз селезеночной или воротной вены) имеются большие размеры селезенки и отсутствие или незначительность увеличения печени. При пункции селезенки кровь поступает в шприц под большим давлением, в пунктате селезенки элементов миелоидной метаплазии обычно мало, за исключением случаев с большой продолжительностью заболевания и смешанным генезом спленомегалии.
При осложнении тромбозом надпеченочных вен клиническая картина выглядит значительно более драматично: болевой синдром имеет различную выраженность, но может и отсутствовать; формируется массивный, резистентный к лечению асцит, нередко желтуха, признаки печеночной недостаточности (клинические и лабораторные) тяжелое общее истощение; возможны периодические кровотечения из расширенных вен пищевода и желудка (кровавая рвота и мелена). Печень обычно значительно увеличена, тогда как селезенка — умеренно. Течение синдрома Бадда — Киари может быть острым, подострым и хроническим.
Неизвестно почему, но у всех шести наблюдаемых нами больных это осложнение возникло на раннем этапе, до постановки гематологического диагноза, и у женщин молодого возраста. Изменения в анализах крови были не столь однозначными, чтобы на их основании поставить гематологический диагноз и уточнить, какой именно. С подобной ситуацией сталкиваются и другие авторы. Расширение возможностей углубленного обследования больных с помощью мегакариоцитарной и эритроидной культур и цитогенетического анализа позволило прийти к заключению, что у 2/3 больных этот синдром является осложнением ХМПЗ.
Склонность к тромбозам в системе воротной, надпеченочных и чревных вен в целом отмечена преимущественно у больных эссенциальной тромбоцитемией (ЭТ) с моноклональным ге-мопоэзом, у носителей гена PRV-1, у больных со спонтанным ростом эритроидной культуры. Пока неизвестно, в какой степени эти патогенетические особенности распространяются на больных хроническим идиопатическим миелофиброзом (ХИМФ).
В клиническом отношении значение проблемы портальной гипертензии у больных хроническим идиопатическим миелофиброзом весьма существенно. Ее своевременная диагностика позволяет принять решение в пользу назначения спленэк-томии у больных с под- и внутрипеченочной портальной гипертензией и назначить адекватное консервативное или хирургическое (наложение шунтов) лечение при осложнении тромбозом надпеченочных вен.
Чаще всего большие размеры селезенки врачи относят за счет основного заболевания и проводят довольно агрессивную терапию с целью ее сокращения, что в случаях осложнения портальной гипертензией обречено на неудачу и может привести к серьезным проблемам.
В одном нашем наблюдении больную лечили по месту жительства миелосаном в течение 6 мес с целью сокращения размеров селезенки. В анализах крови наблюдались только небольшой тромбоцитоз и нейтрофилез. Заболевание расценивалось как сублейкемический миелоз. Селезенка была увеличена до уровня пупка. К концу этого лечения развился тяжелый геморрагический тромбоцитопенический синдром, в течение 3 мес больная находилась в критическом состоянии. Еще через 3 мес она была подвергнута спленэктомии. На операции выявлен цирроз печени без миелоидной метаплазии селезенки (ММС). Диагноз пересмотрен в пользу эссенциальной тромбоцитемии (ЭТ).
В течение последующих 5 лет больная находилась в хорошем состоянии и принимала гидроксимочевину в небольшой дозе, которая контролировала тромбоцитоз. Затем внезапно развился тромбоз в системе мезентериальных сосудов, распознанный с опозданием. Во время операции удалена значительная часть тонкого кишечника.
Случай демонстрирует часто имеющую место неточность гематологического диагноза в группе ХМПЗ, нераспознанную внутрипеченочную портальную гипертензию, в данном случае, видимо, обусловленную сопутствующим заболеванием, неадекватную цитостатическую терапию, осложнившуюся гипоплазией кроветворения и геморрагическим синдромом, а также развитие тромбоза мезентериальных сосудов при контролируемом тромбоцитозе.
Причиной развития асцита может оказаться не только портальная гипертензия, но и имплантация очагов кроветворения на брюшине и сальнике. В таких случаях в асцитической жидкости обнаруживают мегакариоциты и гранулоциты. Плевральный и абдоминальный выпот часто носит геморрагический характер. Эта и другие атипичные локализации миелоидной метаплазии: в лимфатических узлах со сдавлением спинного мозга, тонком кишечнике, средостении, почках, легких, других висцеральных органах — относятся к числу раритетов. Имеются данные об увеличении периферических лимфатических узлов у 32 % больных, но, по нашим наблюдениям, это значительно более редкий феномен.
К симптомам, обусловленным клеточным гиперкатаболизмом, относятся потеря массы тела и повышение температуры тела, гиперурикемия. Она может быть бессимптомной или протекать с признаками подагрической полиартралгии, подагры, мочекаменной болезни, осложняться хроническим пиелонефритом, обтурацией мочеточников, хронической почечной недостаточностью. У отдельных больных интенсивность камнеобразования в почках необычайно велика. Развитию урикемии способствует проведение массивной цитостатической терапии .
Хотя повышение температуры тела может быть результатом клеточного гиперкатаболизма, это справедливо по отношению к умеренному субфебрилитету, а значительные подъемы температуры тела обычно обусловлены инфекцией, особенно мочевыводящих путей, или латентно протекающим МДС-синдромом, который может проявиться как типичный, развернутый острый лейкоз через ряд месяцев и даже лет.

В случаях, протекающих с количественной и качественной патологией тромбоцитов, возможны сосудистые осложнения: тромботические микроциркуляторные расстройства, тромбозы артерий и вен, геморрагический синдром, ДВС-синдром.
Внутренние кровотечения обычно обусловлены разрывом вен пищевода при осложнении портальной гипертонией. Присущая этому заболеванию, как и другим ХМПЗ, качественная дефектность тромбоцитов объясняет появление экхимозов на коже при сравнительно умеренной тромбоцитопении. Несостоятельность гемостаза особенно четко проявляет себя при спленэктомии.
Анемический синдром нередко выходит на передний план, особенно в поздних стадиях заболевания. Его причины разнообразны. Среди них могут быть:
• недостаточное образование эритроцитов;
• гиперволемия;
• усиление депонирования и секвестрации клеток крови в увеличенной селезенке (гиперспле-низм);
• аутоиммунный гемолиз эритроцитов;
• ускоренный гемолиз эритроцитов в результате синдрома пароксизмальной ночной гемоглобинурии или ферментных дефектов (дефицит Г-6-ФДГ и др.);
• дефицит железа и фолиевой кислоты.
Количественная недостаточность эритропоэза определяется замещением кроветворного костного мозга миелофиброзом и остеомиелосклерозом с возможным присутствием жировой ткани. Компенсаторный эритропоэз в трубчатых костях со временем также редуцируется, а компенсаторные возможности селезеночного эритропоэза ограничены его частой неэффективностью и одновременным усилением депонирования и деструкции клеток крови в большой селезенке.
Гемодилюционная анемия является результатом гиперволемии, обусловленной спленомегалией. Она хорошо переносится больными и является по существу только лабораторным феноменом.
Дефекты мембраны эритроцитов, сходные с наблюдаемыми при пароксизмальной ночной гемоглобинурии (ПНГ), описаны при хроническом идиопатическом миелофиброзе многими авторами. Их последствием является синдром гемолитической анемии. Повышенному гемолизу эритроцитов способствует и усиление перекисного окисления липидов клеточных мембран эритроцитов.
Дефицит фолиевой кислоты, приводящий к появлению макроцитарной анемии с кольцами Кебота, базофильной пунктацией эритроцитов, тельцами Жолли, наблюдается в поздней стадии заболевания. Его объясняют повышенным расходом фолиевой кислоты при усиленном гемопоэзе.
К количественной недостаточности эритропоэза приводит и его подавление при прогрессирующей гиперплазии лейкоцитарного ростка, хронической и острой. Возможно развитие сидеробластной анемии без- и с малопроцентной бластемией, которая является предстадией острого лейкоза. Описаны случаи парциальной красноклеточной аплазии, завершившиеся острым лейкозом. Отметим, что обычно к анемии приводит сочетание нескольких причинных факторов. Удельный вес каждого из них подлежит уточнению, что особенно необходимо при решении вопроса о назначении спленэктомии. Это же относится и к тромбоцитопеническому синдрому. Причинами его развития являются:
• усиление депонирования и деструкции тромбоцитов в увеличенной селезенке (и печени);
• вторичный аутоиммунный гемолиз тромбоцитов;
• нарушение образования тромбоцитов в результате редукции числа мегакариоцитов или их качественной дефектности;
• сочетание этих процессов;
• ДВС-синдром (тромбоцитопения потребления).
Среди известных клинических проявлений заболевания могут иметь место и аутоимунные симптомы, такие как дерматиты и кожные васкулиты, опосредованные активацией Т-лимфоцитов.
При рентгенографическом исследовании костного скелета обнаруживаются признаки уплотнения структуры плоских костей, особенно позвонков, иногда эбурнеация трубчатых костей с сужением их просвета, иногда определяют очаговый остеолиз.
Эволюция хронического идиопатического миелофиброза характеризуется постепенным нарастанием лейкоцитоза при исходно различном числе лейкоцитов: нормальном, сниженном и повышенном. Развитие острого лейкоза наблюдается как в случаях прогрессирующего лейкоцитоза (более 30•109/л), так и лейкопении (3•109/л). Большинство больных не доживает до развития типичного острого лейкоза. Картины рефрактерной анемии, тромбоцитопении, панцитопении или, наоборот, нарастающего лейкоцитоза и левого сдвига лейкоцитарной формулы, выход из-под контроля размеров селезенки, появление упорной асептической лихорадки являются показателями терминальной фазы заболевания.
Терминальное состояние больных может определяться и висцеральными осложнениями: сердечной, печеночной и почечной недостаточностью, к которым имеются соответствующие патофизиологические предпосылки. Жизнь больных может оборвать острое кровотечение из расширенных вен пищевода при осложнении портальной гипертонией. Гематологические и соматические терминальные состояния нередко сочетаются с тяжелой общей дистрофией и компрессионными осложнениями.
Течение заболевания обычно хроническое, медленно прогрессирующее. Длительное время оно может протекать почти полностью бессимптомно, и только случайное исследование анализа крови выявляет небольшие отклонения или осмотр позволяет обнаружить спленомегалию. При доброкачественном, медленно протекающем варианте хронического идиопатического миелофиброза (ХИМФ) наблюдаются «мягкая» степень развития миелофиброза или только клеточная пролиферация, отсутствие анемии, нормальное или незначительно повышенное число лейкоцитов и тромбоцитов, отсутствие циркулирующих бластов и незначительное число CD334-позитивных клеток в периферической крови. У подобных больных прогрессия заболевания может длительное время отсутствовать.
Возможны варианты более злокачественного течения, которые терминологически обозначаются как варианты с ускоренным развитием терминальной фазы. Выделяют и вариант с подострым течением, при котором морфологические изменения костного мозга не отличаются от обычного хронического идиопатического миелофиброза, но при этом нет значительной спленомегалии, и довольно быстро развиваются анемия и другие проявления недостаточности кроветворения.
К атипичным хронически протекающим вариантам хронического идиопатического миелофиброза можно отнести и так называемые гибридные формы, имеющие признаки двух ХМПЗ:
• истинной полицитемии и хронического идиопатического миелофиброза — упорная плетора, но раннее и значительное увеличение селезенки за счет миелоидной метаплазии, лейкоцитозные формы, устойчивость к цитостатической терапии, эволюция в острый лейкоз через длительный период зрелоклеточного лейкоцитоза, частые цитогенетические аномалии;
• хронического идиопатического миелофиброза и эссенциальная тромбоцитемия. С первым заболеванием их сближает выраженный миелофиброз, со вторым — небольшая величина селезенки, значительный тромбоцитоз, отсутствие лейкоэритробластической картины периферической крови, характерной для иМФ.
Атипичными в определенном смысле являются и случаи хронического идиопатического миелофиброза с существенным увеличением печени, а не селезенки, а также осложненные портальной гипертонией, синдромом Бадда — Киари, при которых проявления гематологического заболевания по анализу крови могут быть минимальными, и только культуральные и цитогенетические исследования выявляют их принадлежность к ХМПЗ.
Миелофиброз ( Агногенная миелоидная метаплазия , Сублейкемический миелоз )
Миелофиброз – это хроническое гематологическое заболевание, характеризующееся опухолевой пролиферацией гемопоэтических стволовых клеток и фиброзом костного мозга. Основные клинические проявления включают симптомы опухолевой интоксикации и анемического синдрома (прогрессирующую слабость, бледность кожи и слизистых оболочек, потерю веса), а также увеличение селезенки (спленомегалию). Диагноз устанавливается на основании молекулярно-генетических исследований, изучения гистологической картины костного мозга. Лечение проводится с помощью химиотерапевтических препаратов. Хирургические методы лечения подразумевают трансплантацию костного мозга и удаление селезенки.
МКБ-10
Общие сведения
Миелофиброз (агногенная миелоидная метаплазия, сублейкемический миелоз) – злокачественное заболевание, при котором происходит постепенное замещение костного мозга опухолевыми стволовыми клетками и разрастающейся соединительной тканью. Впервые эту патологию описал немецкий врач Г. Хойк в 1879 году. А в 1951 году американским гематологом Уильямом Дамешеком миелофиброз был выделен в самостоятельную нозологическую единицу. При неблагоприятном течении миелофиброз способен трансформироваться в более тяжелую болезнь ‒ острый лейкоз. Распространенность миелофиброза составляет от 0,3 до 0,7 случаев на 100 тыс. населения. Пик заболеваемости приходится на возраст от 50 до 70 лет, но встречаются и молодые пациенты. Чаще страдают мужчины.
Причины миелофиброза
Существует первичный и вторичный сублейкемический миелоз. Точная причина первичного миелофиброза до сих пор не установлена. Наибольшей популярностью среди специалистов в области гематологии пользуется теория влияния генетической мутации. У большинства пациентов выявляются мутации гена тирозинкиназы (JAK2V617F), кальретикулина (CALR), тромбопоэтина (MPL), регулирующих экспрессию белков JAK-STAT сигнального пути. Гены локализуются в локусе хромосомы del3p24.
В качестве этиологического фактора изучается действие большой дозы радиоактивного излучения. Также рассматривается роль персистирующих вирусных инфекций (вируса простого герпеса, Эпштейна-Барра, цитомегаловируса), длительного приема оральных контрацептивов, миелосупрессивных лекарственных препаратов, контакта с различными органическими и неорганическими соединениями (бензолом, мышьяком). Вторичный миелофиброз развивается как исход других хронических миелопролиферативных заболеваний – истинной полицитемии, эссенциальной тромбоцитемии, хронического миелолейкоза.
Патогенез
В результате повышенной экспрессии сигнальных белков в одной из стволовых костномозговых клеток запускается активная пролиферация (опухолевая трансформация). Этот процесс сопровождается вторичным воспалением с выделением цитокинов и факторов роста. Факторы роста фибробластов и эндотелия сосудов индуцируют выработку стромальными клетками костного мозга большого количества коллагена и разрастание соединительной ткани (собственно фиброз). Постепенно нормальная ткань костного мозга замещается опухолью и соединительной тканью.
При массивном поражении опухолью костного мозга клетки крови, не достигнув стадии полного созревания, попадают в системный кровоток. Это приводит к образованию очагов экстрамедуллярного (внекостномозгового) кроветворения, главным образом в печени и селезенке. Распад опухоли ведет к высвобождению мочевой кислоты, которая откладывается в тканях суставов и почечных канальцах.
Симптомы миелофиброза
Длительное время пациент чувствует себя удовлетворительно. Через несколько лет от начала заболевания постепенно появляется опухолевая интоксикация в виде общей слабости, повышения температуры до субфебрильных цифр, потливости, усиливающейся по ночам. У больного снижается аппетит, он стремительно теряет в весе. Присоединяется анемический синдром (бледность кожных покровов, головокружение, учащение сердцебиения). Характерны носовые, десневые кровотечения, геморрагические высыпания на коже. Возникают боли в суставах, кожный зуд, боли в костях.
Пациент ощущает тяжесть и боли в левом подреберье вследствие выраженного увеличения селезенки. На фоне спленомегалии развивается синдром гиперспленизма, который заключается в массивном разрушении клеток крови (в основном эритроцитов) в синусоидах селезенки. В этом случае встречаются признаки гемолиза (желтушность кожи, слизистых оболочек, потемнение мочи).
Редкие симптомы связаны с необычной локализацией очагов экстрамедуллярного кроветворения – в легких (кашель, затруднение дыхания, кровохарканье), желудочно-кишечном тракте (боли в животе, кровавая диарея). При расположении очагов в центральной и периферической нервной системе наблюдаются эпилептические судороги, нарушения чувствительности, слабость движений в конечностях, вплоть до полного паралича.
Осложнения
При миелофиброзе часто образуются тромбы, которые приводят к острому нарушению мозгового кровообращения, инфаркту миокарда, тромбоэмболии легочной артерии. Стойкое снижение уровня лейкоцитов нередко сопряжено с различными инфекциями, приобретающими тяжелое течение. Наиболее неблагоприятным осложнением считается трансформация миелофиброза в миелолейкоз (бластный криз), трудно поддающийся терапии. К нетипичным осложнениям следует отнести патологические переломы из-за деструкции трубчатых костей и портальную гипертензию, причиной которой служит длительная обструкция микротромбами внутрипеченочных вен.
Диагностика
Курацией пациентов с миелофиброзом занимаются врачи-гематологи. При общем осмотре обращает на себя внимание изменение цвета кожных покровов, слизистых (бледность или желтушность), спленомегалия при пальпации и перкуссии селезенки, иногда достигающей гигантских размеров (до лобкового симфиза). Дополнительные методы диагностики включают:
- Общие лабораторные исследования. В начале заболевания в общем анализе крови выявляется увеличение эритроцитов, тромбоцитов, лейкоцитов, со временем сменяющееся на низкие показатели. Часто в периферической крови присутствуют незрелые формы эритроцитов, лейкоцитов (миелоциты, промиелоциты). В биохимическом анализе крови наблюдаются повышенные концентрации лактатдегидрогеназы (ЛДГ), ионизированного кальция. Отмечаются изменения коагулограммы – ускорение свертывания крови, уменьшение активированного частичного тромбопластинового времени, торможение процессов фибринолиза. В анализе мочи обнаруживаются уробилин, гемоглобин, ураты (соли мочевой кислоты).
- Исследование костного мозга. Образец костного мозга получают с помощью трепанобиопсии. Гистологическая картина зависит от фазы заболевания. Для ранней (префибротической фазы) характерны гиперплазия всех ростков кроветворения (гранулоцитарного, мегакариоцитарного, эритроидного) с незрелостью клеток. В позднюю (фибротическую) фазу определяется большое количество коллагеновых и ретикулярных волокон (фиброз), замещающих гемопоэтическую ткань, выраженная клеточная атипия. Высокий уровень бластных клеток (более 20%) свидетельствует о трансформации миелофиброза в острый лейкоз.
- Молекулярно-генетические тесты. Диагностика мутации генов JAK2V617F, CALR, MPL осуществляется методом FISH. Для идентификации аллельной нагрузки мутации проводится полимеразная цепная реакция real-time. Также выполняется HLA-типирование для решения вопроса о возможности трансплантации костного мозга.
- Цитогенетические и цитохимические анализы. При цитогенетическом исследовании (кариотипировании) клеток костного мозга находят аномалии 1, 3, 6 хромосом (транслокация, трисомия, комплексные нарушения). При анализе химического состава (цитохимии) нейтрофилов активность щелочной фосфатазы оказывается в 3 раза выше нормы.
Для достоверной постановки диагноза гематологическим сообществом разработаны специальные критерии. Большие критерии включают повышенную клеточность костного мозга с ретикулярным и коллагеновым фиброзом, наличие мутаций генов JAK2V617F, MPL, CALR. К малым критериям относятся анемия, спленомегалия, лейкоэритробластоз (присутствие в крови незрелых форм лейкоцитов, эритроцитов), а также повышение лактатдегидрогеназы. Диагноз считается подтвержденным, если имеются 2 больших критерия или 1 большой и 3 малых критерия.
Миелофиброз следует дифференцировать в первую очередь с гематологическими заболеваниями, такими как аутоиммунные гемолитические анемии, гемобластозы (лейкозы, лимфомы). Сочетание спленомегалии с симптомами интоксикации (слабостью, субфебрилитетом, ночной потливостью) требует исключения туберкулеза, подострого инфекционного эндокардита.
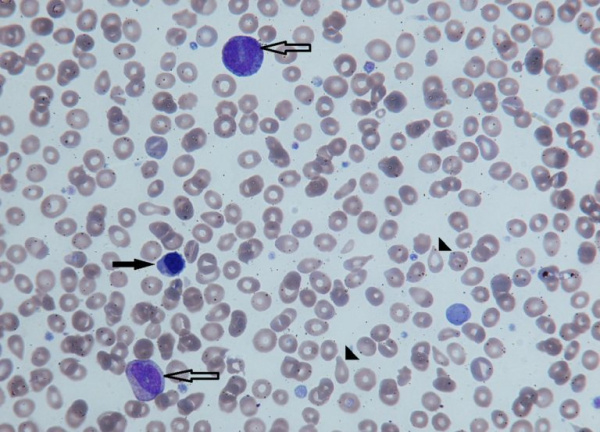
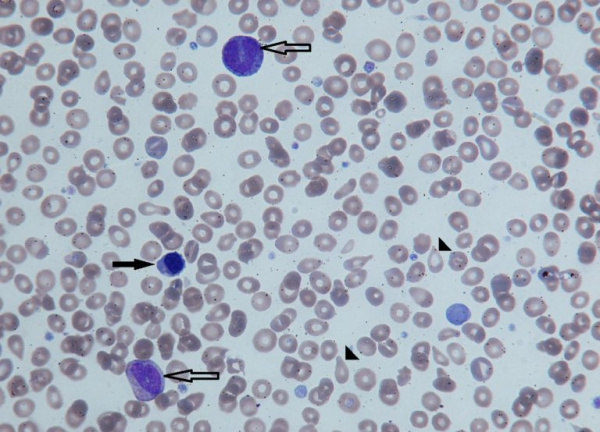
Незрелые формы эритроцитов (черная стрелка) и гранулоцитов (контурная стрелка) в периферической крови
Лечение миелофиброза
После постановки диагноза пациент должен быть госпитализирован в гематологический стационар. Для принятия решения о выборе тактики лечения необходимо определить степень риска, а именно - вероятность бластной трансформации и ориентировочную продолжительность жизни. С этой целью была создана «Международная шкала оценки риска и прогноза» (DIPSS). Она учитывает возраст пациента, количество форменных элементов крови, выраженность симптомов опухолевой интоксикации. Каждый признак соответствует одному баллу. Различают низкий, первый и второй промежуточный, высокий риски, при которых проводится дифференцированная терапия:
Прогноз и профилактика
Миелофиброз – это тяжелое заболевание с неблагоприятным прогнозом. С момента постановки диагноза средняя продолжительность жизни составляет около 5 лет. При манифестации в более молодом возрасте миелофиброз имеет менее агрессивное течение, что сопряжено с лучшим ответом на терапию и большей выживаемостью больных. Эффективных методов профилактики не разработано ввиду неизвестности этиологического фактора. Предупреждение развития вторичного миелофиброза заключается в своевременной диагностике и лечении патологий, на фоне которых он возникает - истинной полицитемии и эссенциальной тромбоцитемии.
2. Патофизиологические основы лечения сублейкемического миелоза. Патофизиология крови. Экстремальные состояния/ Под ред. А.И. Воробьева и Н.А. Горбуновой - 2004.
3. Критерии диагностики и современные методы лечения первичного миелофиброза/ Абдулкадыров К. М., Шуваев В. А., Мартынкевич И. С.// Вестник гематологии. - 2013 - №9(3).
4. Клинические рекомендации по диагностике и терапии Ph-негативных миелопролиферативных заболеваний (истинная полицитемия, эссенциальная тромбоцитопения, первичный миелофиброз. - 2014.
Читайте также:
- Противопоказания к бронхоспирометрии. Комплексное бронхологическое исследование
- Причины фибрилляции предсердий и их прогноз
- Контрастность рентгеновского снимка. Регулирование контрастности рентгеновского изображения
- Спазм желудка. Признаки и диагностика спазма желудка
- Дифференциальная диагностика патологического прелиминарного периода. Тактика при патологическом прелиминарном периоде.