Тромбоцитарный сгусток. Ингибиторы факторов свертывания
Добавил пользователь Alex Обновлено: 27.01.2026
Основными функциями гемостаза являются поддержание жидкого состояния крови и быстрое купирование кровоточивости из места повреждения сосудистой стенки. В этом процессе принимают участие клетки крови, эндотелий сосудов и циркулирующие в крови белки – факторы свертывания крови и фибринолиза, активаторы и ингибиторы. Нарушение тонкого баланса взаимодействия всех этих компонентов, в том числе из-за врожденной недостаточности некоторых из них, приводит к развитию различных заболеваний (тромбоэмболических синдромов, диссеминированного внутрисосудистого свертывания, ишемии и инфарктов органов, гемофилии), нередко фатальных. Нарушение гемостаза происходит при таких видах патологии, как атеросклероз, иммунные и онкологические заболевания, акушерские осложнения и многие другие.
Создание первой научной теории свертывания крови в 1872 г. принадлежит русскому ученому Шмидту А.А., позже была дополнена, модифицирована и сформулирована как классическая ферментативная теория свертывания крови, которая в литературе получила название теории Шмидта-Моравица. До разработки современной клеточной теории свертывания крови для понимания механизмов гемостаза использовали «каскадную» модель свертывания крови (Davie E.W., Ratnoff O.D.; Macfarlane R.G., 1964 г.), где процесс свертывания крови подразделяется на первичный и вторичный гемостаз, с выделением «внешнего», «внутреннего» путей активации тромбина и «общего пути» (см. схему свертывания крови). Условным третьим этапом свертывания крови является процесс лизиса кровяного сгустка (фибринового тромба) — фибринолиз.
Итак, согласно классической каскадной модели свертывания крови, активация коагуляционных факторов, приводящая к образованию фибрина, осуществляется двумя путями: внешним (тканевым) и внутренним (тромбоцитарно-сосудистым), в зависимости от характера активирующей поверхности на начальных этапах процесса свертывания крови.
Для внешнего пути такой поверхностью является тканевой фактор (ТФ), который выделяется из поврежденного эндотелия и активирует фактор (ф.) VII при участии ионов кальция.
Внутренний путь рассматривается как процесс, который начинается с активации ф.XII при контакте крови с поврежденной сосудистой стенкой (субэндотелиальными компонентами сосудистой стенки, в частности с коллагеном), и проходит цепь последовательных реакций активации факторов XI, IX и VIII.
Оба пути приводят к активации ф.Х, и с этого момента процесс свертывания крови протекает по общему пути свертывания. Фактор Ха со своим кофактором Va образует протромбиназный ферментативный комплекс, который на поверхности тромбоцитов активирует протромбин с образованием тромбина. Образовавшийся тромбин поступает в ток крови и превращает фибриноген в фибрин-мономеры. Последние спонтанно соединяются, образуя полимеры фибрина.
Несмотря на то, что каскадная модель не совсем верно отражает суть функционирования системы гемостаза, ее до сих пор с успехом используют для интерпретации базовых (скрининговых) коагуляционных тестов, в которых искусственно воспроизведены условия активации ф.X по внешнему пути (протромбиновый тест) или внутреннему пути (активированное частичное тромбопластиновое время, АЧТВ).
Примечательно, что слово « каскад» не совсем верно отражал суть функционирования системы гемостаза, так как в процессе коагуляции имеется множество перекрёстных реакций, а «каскад» подразумевает лишь последовательную цепочку процессов. Цепочки реакций внутреннего, внешнего и общего путей гемостаза in vivo работают не изолированно, а в тесном взаимодействии друг с другом и с тромбоцитами. Как оказалось, взаимосвязь путей свертывания подтверждается способностью комплекса ТФ-ф.VIIa активировать фактор IX, а также способностью факторов XIIa и Ха активировать фактор VII.
Несмотря на сходную структуру мембранных липидов, клетки, несущие тканевой фактор, и активированные тромбоциты экспрессируют рецепторы, которые локализуют на их поверхности различные компоненты свертывающей системы крови.
Именно факт локализации различных коагуляционных факторов на поверхностях субэндотелиальных клеток и тромбоцитов позволил по-новому пересмотреть последовательность включения их в процесс формирования фибринового сгустка.
Классическая теория свертывания крови оставляла слишком много вопросов. Например, с одной стороны, было неясно, какая поверхность в физиологических условиях является активатором. С другой стороны, было не понятно, почему возможность образования протромбиназного комплекса (ф.Xa и ф.Va) по одному пути не компенсирует поломку в другом. А именно, почему активация фактора X внешним путем через комплекс ТФ-ф.VIIa не компенсирует недостаток факторов VIII или IX у больного гемофилией.
Аналогичный вопрос возникает и в отношении пациентов с дефицитом фактора VII, у которых при отсутствии нарушений во внутреннем пути развиваются тяжелые проявления кровоточивости. Если внутренний путь начинается с активации фактора XII, то почему его дефицит не вызывает тенденции к кровоточивости. Почему дефицит факторов внутреннего пути (ф.VIII, ф.IX, ф.XI) приводит к выраженной кровоточивости при нормальной активности факторов внешнего пути, а глубокий дефицит факторов внешнего пути не сопровождается геморрагическим синдромом. В современной теории гемостаза предпринята попытка разрешить данные противоречия.
Клеточная модель свертывания не отрицает реакции взаимодействия и свойства факторов свёртывающей, противосвёртывающей и фибринолитической систем, представленных в каскадной модели свертывания. Она признает наличие внешнего и внутреннего пути свертывания, но существенно их модифицирует.
1 фаза – инициация процесса свертывания крови, которая развивается за счет образования комплекса ТФ-ф.VIIa на поверхности субэндотелиальных клеток в месте повреждения сосудистой стенки и приводит к образованию незначительного стартового количества тромбина.
2 фаза – усиление процесса свертывания крови за счет активации тромбоцитов и целого ряда коагуляционных факторов тромбином, который образуется под влиянием комплекса ТФ-ф.VIIa.
3 фаза – распространение процесса свертывания крови с формированием теназного (ф.VIIIa/ф.IXa) и протромбиназного ф.Vа/ф.Xа/кальций/фактор III тромбоцитов) комплексов на поверхности активированных тромбоцитов. В результате образуется значительное количество тромбина («тромбиновый взрыв»), способного сформировать сгусток фибрина.
Инициация
Повреждение сосудистой стенки и/или внутрисосудистая активация клеток, экспрессирующих тканевой фактор, обеспечивает его контакт с ф.VII крови. Поскольку ТФ – интегральный мембранный белок, комплекс ТФ-ф.VIIa всегда связан с мембранной поверхностью клеток. Это важный момент, который объясняет локализацию коагуляционного каскада в зоне повреждения сосуда, то есть именно в том месте, где он необходим для остановки кровотечения.
Активный комплекс ТФ-ф.VIIa путем ограниченного протеолиза активирует факторы X и IX. При этом образовавшийся фактор IХа мигрирует с поверхности субэндотелиальных клеток, несущих ТФ, и связывается со специфическим рецептором на активированных тромбоцитах, которые находятся в непосредственной близости в зоне повреждения сосуда. Фактор Ха, оставаясь на поверхности субэндотелиальных клеток, вместе со своим кофактором – фактором Va расщепляет протромбин с образованием небольшого количества тромбина (ф.IIа).
Таким образом, в ходе инициации происходит активное взаимодействие эндотелия, тромбоцитов и факторов свёртывания крови. Повреждение (и/или дисфункция) эндотелия сосудов вызывает адгезию и агрегацию тромбоцитов и инициирует свёртывание крови. Активированные тромбоциты способствуют образованию тромбина. В свою очередь тромбин – мощный стимулятор агрегации тромбоцитов.
Усиление
Небольшое количество тромбина, образовавшегося в фазу инициации, активирует тромбоциты, факторы V, VIII и XI. Тромбин способствует высвобождению фактора VIII из комплекса с фактором Виллебранда, в результате образуется ф.VIIIa. Ф.XIa приобретает способность связываться с поверхностью тромбоцитов. Активированные небольшим количеством образовавшегося в фазу инициации тромбина факторы в следующую фазу (фазу распространения) обеспечивают формирование на тромбоцитарной матрице огромного количества тромбина, которое способно перевести фибриноген в фибрин. Таким образом, вышедший из фазы инициации тромбин выступает в качестве мощного усилителя коагуляции.
Распространение
В фазу распространения на поверхности активированных тромбоцитов происходит сборка, концентрация, активация факторов свёртывания и формируется теназный (ф.VIIIa/ф.IXa) комплекс. Теназный комплекс на поверхности тромбоцитов активирует ф.X, связанный со своим кофактором ф.Va, что ведет к образованию протромбиназного (ф.Va/ф.Xa) комплекса. Образовавшийся протромбиназный комплекс, в свою очередь, обеспечивает лавинообразное нарастание уровня тромбина. Тромбин вызывает гидролиз фибриногена до фибрина, а также активирует ф.ХIII, обеспечивающий стабилизацию фибриновых нитей и образование множества ковалентных перекрёстных связей между ними; такие прочные фибриновые волокна способны удерживать тромбоцитарную массу на месте повреждения. Затем происходит ретракция кровяного сгустка. Одновременно активируется фибринолитическая система.
Таким образом, по современным представлениям in vivo процесс свертывания крови является единым и связан с гемостатическими реакциями тромбоцитов. Благодаря их сложному рецепторному аппарату они не только участвуют в активации коагуляционных факторов, но и выполняют функцию регуляции всего процесса свертывания крови. Взаимосвязь тромбоцитов, факторов свертывания крови и сосудистой стенки постоянно уточняется.
Обзор гемостаза (Overview of Hemostasis)
Сосудистые факторы снижают кровопотерю, обусловленную травмой, с помощью механизма локальной вазоконстрикции (немедленная реакция на повреждение) и компрессии поврежденных кровеносных сосудов путем экстравазации крови в окружающие ткани. При повреждении сосудистой стенки происходят адгезия и агрегация тромбоцитов и генерация фибриновых полимеров из фибриногена; взаимодействие тромбоцитов с фибрином формирует тромб.
Тромбоциты
Ток крови, благодаря предотвращению агрегации тромбоцитов и расширению интактных кровеносных сосудов, сохраняется с помощью различных механизмов, к которым относятся высвобождаемые эндотелиальными клетками простациклин и оксид азота. Производство этих медиаторов прекращается при повреждении эндотелия сосудистой стенки. В этих условиях происходят адгезия тромбоцитов к поврежденной интиме сосудистой стенки и формирование тромбоцитарных агрегатов. Начальная адгезия тромбоцитов к волокнам коллагена связана с фактором Виллебранда (ФВ), который был предварительно секретирован и связан стимулированными эндотелиальными клетками. ФВ взаимодействует с рецепторами на поверхности тромбоцитарной мембраны (гликопротеин Ib/IX). Тромбоциты, которые зацепились за стенки сосуда, подвергаются активации. При этом тромбоциты высвобождают медиаторы агрегации, включая аденозин дифосфат (АДФ), из накопительных вакуолей.
Другие биохимические изменения, происходящие в результате активации тромбоцитов, включают
Гидролиз мембранных фосфолипидов
Мобилизацию внутриклеточного кальция
Фосфорилирование внутриклеточных белков
Арахидоновая кислота преобразуется в тромбоксан A2; эта реакция требует участия циклооксигеназы тромбоцитов и необратимо ингибируется аспирином и обратимо многими НПЗП (нестероидными противовоспалительными препаратами).
АДФ, тромбоксан A2 и другие медиаторы провоцируют агрегацию дополнительных тромбоцитов к поврежденному эндотелию и активируют их. Тромбоцитарные рецепторы АДФ включают рецептор P2Y12, который посылает сигналы для подавления аденилатциклазы, уменьшает уровень циклического аденозин монофосфата (цАМФ) и способствует активации рецептора гликопротеина IIb/IIIa (образующегося на поверхности мембраны активированного тромбоцита из гликопротеинов IIb и IIIa). Фибриноген связывается с гликопротеиновым комплексом IIb/IIIa расположенных рядом тромбоцитов и соединяет их друг с другом.
Сборка и активация коагуляционных комплексов и образование тромбина проходят на поверхности тромбоцитов. Тромбин превращает фибриноген в мономеры фибрина, а мономеры фибрина полимеризуются в полимеры фибрина, которые соединяют агрегированные тромбоциты в тромбоцитарно-фибриновые гемостатические пробки.
Плазменные факторы свертывания
Факторы свертывания крови воздействуют друг на друга на поверхности тромбоцитов и эндотелиальных клеток с целью производства тромбина, который превращает фибриноген в фибрин. Фибрин укрепляет тромб за счет формирования гемостатической пробки и ее закрепления.
При внутреннем пути свертывания фактор XII, высокомолекулярный кининоген, прекаликреин и активированный фактор XI (фактор XIa) взаимодействуют для производства активированного фактора IХa из фактора IX. Фактор IХa затем взаимодействует с фактором VIIIа и прокоагулянтными фосфолипидами (присутствуют на поверхности активированных тромбоцитов, эндотелиальных клеток и клеток тканей) для создания комплекса, активирующего Х.
При внешнем пути фактор VIIа и тканевой фактор (ТФ) непосредственно активируют фактор Х (и, возможно, также фактор IX – см. рисунок Пути свертывания крови Метаболические механизмы свертывания крови [Pathways in blood coagulation] и таблицу Компоненты реакции свертывания крови Составляющие реакций свертывания крови [Components of Blood Coagulation Reactions]).
Многие (или большинство) коагуляционных протеинов вырабатываются в эндотелиальных клетках сосудов, включая эндотелиальные клетки, выстилающие синусоиды печени. Некоторые коагуляционные белки также могут вырабатываться другими типами клеток (например, тканевым фактором фибробластами).
Метаболические механизмы свертывания крови
Активация внутреннего или внешнего пути стимулирует общий путь свертывания, что приводит к образованию фибринового сгустка. Общий путь активации состоит из трех шагов:
Протромбиназа вырабатывается на поверхности активированных тромбоцитов, эндотелиальных и других тканевых клеток. Протромбиназа представляет собой комплекс из фермента, фактора Ха и кофактора, фактора Va на поверхности прокоагулянтного фосфолипида.
Протромбиназа расщепляет протромбин до тромбина.
Тромбин стимулирует создание мономеров и полимеров фибрина из фибриногена и активирует растворимый фактор VIII и фактор XI. Тромбин также активирует фактор XIII – фермент, катализирующий образование более сильных ковалентных связей между мономерами фибрина.
Ионы кальция требуются для большинства реакций образования тромбина и, соответственно, кальций-хелатирующие агенты (например, цитрат, этилендиаминтетрауксусная кислота) используются в качестве антикоагулянтов в условиях in vitro. Витамин К-зависимые факторы свертывания крови (факторы II, VII, IX и X) в норме связываются с фосфолипидами с помощью кальциевых мостиков для участия в процессе свертывания крови. Реакции коагуляции не могут протекать должным образом при отсутствии витамина К. Витамин К-зависимые регуляторные белки свертывания включают протеины С, S и Z.
Хотя пути свертывания крови достаточно полезны для понимания механизмов и лабораторной оценки коагуляционных нарушений, в естественных условиях в процессе коагуляции не участвуют фактор XII, прокалликреин или высокомолекулярный кининоген. У людей с наследственным дефицитом этих факторов отсутствуют нарушения гомеостаза. У людей с наследственным дефицитом фактора XI может наблюдаться легкое или умеренное нарушение свертываемости крови. In vitro растворимый фактор XI может быть активирован тромбином. Однако между уровнем плазматического фактора XI и вероятностью или степенью кровотечения устойчивая связь отсутствует. Растворимый фактор IX может активироваться in vitro как фактором XIa, так и фактором VIIa/комплексами тканевого фактора.
В естественных условиях инициирование внешнего пути возникает, когда повреждение кровеносных сосудов приводит кровь к контакту с тканевым фактором на мембранах клеток в пределах и в окружении сосудистой стенки. Контакт с тканевым фактором запускает образование комплекса фактор VIIa/тканевой фактор, который активирует фактор X (и, возможно, фактор IX, внутренний фактор). Фактор IXa в комбинации со своим кофактором, фактором VIII на поверхности фосфолипидной мембраны также генерирует фактор Ха. Активация фактора X комплексами фактора IXa/VIIIa необходима для нормального гемостаза. Необходимость этого процесса для факторов VIII и IX объясняет, почему при гемофилии Гемофилия Гемофилия является общим наследственным нарушением свертываемости крови, вызванным недостатком фактора свертывания крови VIII или IX. Частоту и тяжесть кровотечений определяет степень дефицита. Прочитайте дополнительные сведения типа А (дефицит фактора VIII или фактора IX) или типа В (дефицит фактора IX) возникает кровотечение. Активация фактора X комплексами фактор VIIa/тканевой фактор на внешнем пути свертывания не генерирует достаточное количество тромбина (и фибрина) для предотвращения кровотечения у пациентов с тяжелыми формами гемофилии А или В.
Обзор гемостаза (Overview of Hemostasis)
Регуляторные механизмы уравновешивают процесс формирования тромбов. Нарушения системы гемостаза могут привести к повышенной кровоточивости или тромбозам.
Сосудистые факторы гемостаза
Сосудистые факторы снижают кровопотерю, обусловленную травмой, с помощью механизма локальной вазоконстрикции (немедленная реакция на повреждение) и компрессии поврежденных кровеносных сосудов путем экстравазации крови в окружающие ткани. При повреждении сосудистой стенки происходят адгезия и агрегация тромбоцитов и генерация фибриновых полимеров из фибриногена; взаимодействие тромбоцитов с фибрином формирует тромб.
Тромбоциты
При помощи различных механизмов, к которым относятся высвобождаемые эндотелиальными клетками простациклин и окись азота, сохраняется ток крови, предотвращается образование тромбоцитарного стаза, расширяются интактные кровеносные сосуды. Производство этих медиаторов прекращается при повреждении эндотелия сосудистой стенки. В этих условиях происходят адгезия тромбоцитов к поврежденной интиме сосудистой стенки и формирование тромбоцитарных агрегатов. Начальная адгезия тромбоцитов к волокнам коллагена связана с фактором Виллебранда (ФВ), который был предварительно секретирован и связан стимулированными эндотелиальными клетками. ФВ взаимодействует с рецепторами на поверхности тромбоцитарной мембраны (гликопротеин Ib/IX). Тромбоциты, которые зацепились за стенки сосуда, подвергаются активации. При этом тромбоциты высвобождают медиаторы агрегации, включая аденозиндифосфат (АДФ), из накопительных вакуолей.
Другие биохимические изменения, происходящие в результате активации тромбоцитов, включают
- Гидролиз мембранных фосфолипидов
- Ингибирование аденилатциклазы
- Мобилизацию внутриклеточного кальция
- Фосфорилирование внутриклеточных белков
Арахидоновая кислота преобразуется в тромбоксан A2; эта реакция требует участия циклооксигеназы и необратимо ингибируется аспирином и обратимо многими нестероидными противовоспалительными препаратами.
АДФ, тромбоксан A2 и другие медиаторы провоцируют агрегацию дополнительных тромбоцитов к поврежденному эндотелию и активируют их. Тромбоцитарные рецепторы АДФ включают рецептор P2Y12, который посылает сигналы для подавления аденилатциклазы, уменьшает уровень циклического аденозин монофосфата (цАМФ) и способствует активации рецептора гликопротеина IIb/IIIa (образующегося на поверхности мембраны активированного тромбоцита из гликопротеинов IIb и IIIa). Фибриноген связывается с гликопротеиновым комплексом IIb/IIIa расположенных рядом тромбоцитов и соединяет их друг с другом.
Сборка и активация коагуляционных комплексов и образование тромбина проходят на поверхности тромбоцитов. Тромбин превращает фибриноген в мономеры фибрина. Полимеры фибрина соединяют агрегированные тромбоциты для сохранения тромбоцитарно-фибриновой гемостатической пробки.
Плазменные факторы свертывания
Плазменные факторы свертывания крови воздействуют друг на друга с целью производства тромбина, который превращает фибриноген в фибрин. Фибрин укрепляет тромб за счет формирования гемостатической пробки и ее закрепления.
При внутреннем пути свертывания фактор XII, высокомолекулярный кининоген, прекаликреин и активированный фактор XI (фактор XIa) взаимодействуют для производства активированного фактора IХa из фактора IX. Фактор IХa затем взаимодействует с фактором VIIIа и прокоагулянтными фосфолипидами (присутствуют на поверхности активированных тромбоцитов, эндотелиальных клеток и клеток тканей) для создания комплекса, активирующего Х.
При внешнем пути фактор VIIа и тканевой фактор (ТФ) непосредственно активируют фактор Х (и, возможно, также фактор IX – см. рисунок Пути свертывания крови [Pathways in blood coagulation] и таблицу Компоненты реакции свертывания крови [Components of Blood Coagulation Reactions]).
Метаболические механизмы свертывания крови
Составляющие реакций свертывания крови
Номер или название фактора
Синоним
Назначение
Факторы плазмы
Факторы плазмы
Предшественник тромбина, который превращает фибриноген в фибрин; активирует растворимые факторы V, VIII, XI и XIII; и связывается с тромбомодулином, активируя протеин С
Является витамин К-зависимым
Активируется до фактора Va – кофактора для ферментативного фактора Xa в комплексе фактор-Ха/Va/фосфолипид, который расщепляет протромбин до тромбина
Присутствует в альфа-гранулах тромбоцитов
Активация внутреннего или внешнего пути стимулирует общий путь свертывания, что приводит к образованию фибринового сгустка. Общий путь активации состоит из трех шагов:
- Протроминовая конвертаза вырабатывается на поверхности активированных тромбоцитов, эндотелиальных и других тканевых клеток. Протромбиновая конвертаза представляет собой комплекс из фермента, фактора Ха и кофактора, фактора Va на поверхности прокоагулянтного фосфолипида.
- Протромбиновая конвертаза расщепляет протромбин до тромбина.
- Тромбин стимулирует создание мономеров и полимеров фибрина из фибриногена. Тромбин также активирует фактор XIII – фермент, катализирующий образование более сильных ковалентных связей между мономерами фибрина, а также активирующий растворимый фактор VIII и фактор XI.
Ионы кальция требуются для большинства реакций образования тромбина и, соответственно, кальций-хелатирующие агенты (например, цитрат, этилендиаминтетрауксусная кислота) используются в качестве антикоагулянтов в условиях in vitro. Витамин К-зависимые факторы свертывания крови (факторы II, VII, IX и X) в норме связываются с фосфолипидами с помощью кальциевых мостиков для участия в процессе свертывания крови. Реакции коагуляции не могут протекать должным образом при отсутствии витамина К.
Хотя пути свертывания крови достаточно полезны для понимания механизмов и лабораторной оценки коагуляционных нарушений, в естественных условиях в процессе коагуляции не участвуют фактор XII, прокалликреин или высокомолекулярный кининоген. У людей с наследственным дефицитом этих факторов отсутствуют нарушения гомеостаза. У людей с наследственным дефицитом фактора XI наблюдается легкое или умеренное нарушение свертываемости крови. In vitro растворимый фактор XI может быть активирован тромбином. Между уровнем плазматического фактора XI и вероятностью или степенью кровотечения отсутствует устойчивая связь, хотя растворимый фактор IX может активироваться in vitro как фактором XIa, так и комплексом фактор VIIa/тканевой фактор.
В естественных условиях инициирование внешнего пути возникает, когда повреждение кровеносных сосудов приводит кровь к контакту с тканевым фактором на мембранах клеток в пределах и в окружении сосудистой стенки. Контакт с тканевым фактором запускает образование комплекса фактор VIIa/тканевой фактор, который активирует фактор X (и, возможно, фактор IX, внутренний фактор). Фактор IXa в комбинации со своим кофактором, фактором VIII на поверхности фосфолипидной мембраны также генерирует фактор Ха. Активация фактора X обоими путями необходима для нормального функционирования гемостаза. Необходимость этого процесса для факторов VIII и IX объясняет, почему при гемофилии А (дефицит фактора VIII или фактора IX) возникает кровотечение, несмотря на неповрежденный внешний путь коагуляции, инициированный комплексом фактор VIIa/тканевой фактор.
Регуляция процесса коагуляции
Ряд ингибиторных механизмов предотвращает неконтролируемую активацию реакций коагуляции, которая может привести к локальному тромбозу или диссеминированному внутрисосудистому свертыванию. Эти механизмы включают:
- Инактивация факторов свертывания
- Фибринолиз
- Печеночный клиренс активированных факторов свертывания
Инактивация факторов свертывания
Ингибиторы плазменных протеаз (антитромбин, ингибитор тканевого фактора, α2-макроглобулин, кофактор гепарина II) инактивируют ферменты коагуляции. Антитромбин ингибирует тромбин, фактор Xa, фактор XIa и фактор IXa.
Два витамин К-зависимых протеина, протеин С и свободный протеин S образуют комплекс, который путем протеолиза инактивирует факторы VIIIa и Va. Тромбин, соединяясь с рецептором на эндотелиальных клетках (тромбомодулин [CD141]) активирует протеин С. Активированный протеин С совместно со свободным протеином S и рецепторами протеина С эндотелиальных клеток протеолизует и инактивирует факторы VIIIa и Va.
Кроме присутствующих в норме инактиваторов, существует целый ряд антикоагулянтов, которые потенцируют инактивацию факторов свертывания (см. рисунок Антикоагулянты и сферы их действия [Anticoagulants and their sites of action]).
Гепарин усиливает активность антитромбина. Варфарин является антагонистом витамина К. Он ингибирует восстановление активной формы витамина К и, следовательно, ингибирует образование функциональных форм витамин К-зависимых факторов свертывания II, VII, IX и X (так же, как и протеины С и S). Нефракционированный гепарин (НФГ) и низкомолекулярные гепарины (НМГ) повышают активность антитромбина к инактивации факторов IIa (тромбин) и Xa. НМГ включают эноксапарин, дальтепарин и тинзапарин. Фондапаринукс представляет собой небольшие синтетические молекулы, содержащие основную пентасахаридную часть структуры гепарина; усиливает инактивацию фактора Ха (но не фактора IIа) антитромбином. Парентеральные прямые ингибиторы тромбина включают в себя аргатробан и лепирудин. Более новые пероральные антикоагулянты прямого действия включают ингибитор тромбина (дабигатран) и ингибитор фактора Ха (апиксабан, ривароксабан, эдоксабан). Применение этих препаратов, включая описание рисков, выгоды и нейтрализующих средств, обсуждается в разделах Руководства, касающихся фибрилляции предсердий, тромбоза глубоких вен (ТГВ) и тромбоэмболии легочной артерии (ТЭЛА).
Антикоагулянты и точки приложения действия
НМГ = низкомолекулярный гепарин; ТФ = тканевой фактор; НФГ = нефракционированный гепарин.
Фибринолиз
Отложение фибрина и фибринолиз должны быть сбалансированы для временного поддержания и последующего удаления гемостатического сгустка при восстановлении поврежденной сосудистой стенки. Фибринолитическая система растворяет фибрин при помощи плазмина, являющегося протеолитическим ферментом. Фибринолиз активируется посредством активаторов плазминогена, продуцируемых эндотелиальными клетками сосудов. Активаторы плазминогена и плазминоген (в плазме) связываются с фибрином, и активаторы плазминогена расщепляют плазминоген до плазмина (см. рисунок Фибринолитический путь [Fibrinolytic pathway]). Затем плазмин образует растворимые продукты распада фибрина, которые поступают в циркуляцию.
Фибринолитический путь
Отложение фибрина и фибринолиз должны быть сбалансированы во время восстановдения поврежденной стенки кровеносных сосудов. Поврежденные эндотелиальные клетки высвобождают активаторы плазминогена (тканевой активатор плазминогена, урокиназа), активирующие фибринолиз. Активаторы плазминогена расщепляют плазминоген в плазмин, который растворяет сгустки. Фибринолиз контролируется с помощью ингибиторов активатора плазминогена (PAI, например, PAI-1) и ингибиторов плазмина (например, α2-антиплазмина).
Существует несколько активаторов плазминогена:
- Тканевой активатор плазминогена(ТАП), производное эндотелиальных клеток, имеет низкую активность, когда находится в свободной форме в растворе, но его эффективность возрастает при взаимодействии с фибрином в непосредственной близости от плазминогена.
- Урокиназасуществует в одноцепочной и двухцепочной формах с различными функциональными возможностями. Одноцепочная урокиназа не способна активировать свободный плазминоген, но, как и ТАП, способна активировать плазминоген при взаимодействии с фибрином. Микроконцентрации плазмина расщепляют одноцепочечную урокиназу на двухцепочечную, которая активирует плазминоген в растворе, так же как и плазминоген, связанный с фибрином. Эпителиальные клетки в экскреторных протоках (например, почечные канальцы, протоки молочной железы) высвобождают урокиназу, которая в этих каналах является физиологическим активатором фибринолиза.
- Стрептокиназа, представляющая бактериальный продукт, не присутствующий в организме человека, является другим потенциальным активатором плазминогена.
Стрептокиназа, урокиназа и рекомбинантный ТАП (альтеплаза) используются в терапевтической практике с целью индукции фибринолиза у больных с острыми тромбозом.
Регулирование фибринолиза
Фибринолиз регулируется ингибиторами активатора плазминогена (ИАП) и ингибиторами плазмина, которые замедляют фибринолиз. ИАП-1, являясь наиболее важным ИАП, высвобождается из эндотелиальных клеток сосудов, инактивирует ТАП, урокиназу и активирует тромбоциты. Основным ингибитором плазмина является альфа-2-антиплазмин, который быстро инактивирует любой свободный плазмин, высвобождаемый из сгустка. Часть альфа2-антиплазмина также связывается с полимерами фибрина под действием фактора XIIIa во время образования сгустка. Это сшивание может предотвратить чрезмерную активность плазмина внутри сгустков.
Урокиназа и ТАП быстро выводятся печенью, что является другим механизмом предупреждения чрезмерного фибринолиза.
Тромбоцитарный сгусток. Ингибиторы факторов свертывания
Ткани и органы. Кровь
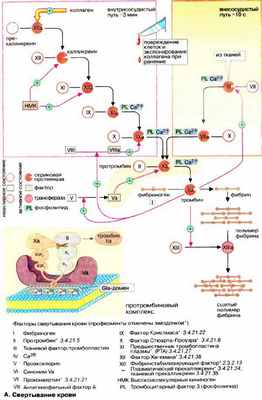
При нарушении целостности кровеносной системы уменьшение кровопотери обеспечивает система гемостаза. Гемостаз поддерживается двумя путями: остановкой кровотечения с помощью тромбоцитов и свертыванием крови . В данном разделе основное внимание уделено ферментативным реакциям свертывания крови. Повторное растворение сгустков крови, фибринолиз , рассмотрен на с. 284.
Номенклатура факторов свертывания крови несколько запутана. Факторы нумеруются римскими цифрами, при этом активированная форма фактора в наименовании содержит дополнительно букву «а» после римской цифры. Многие факторы являются протеиназами. На схеме неактивные предшественники протеиназ представлены в виде окружностей, а активные ферменты — окрашенными кружочками с вырезанным сектором. Вспомогательные факторы показаны в виде прямоугольников.
При свертывании крови происходит ферментативное превращение растворимого белка плазмы фибриногена (фактора I, см. рис. 71) в фибриновый полимер, сеть волокон нерастворимого белка. В этой реакции принимает участие фермент тромбин (фактор IIа), который протеолитически отщепляет от молекулы фибриногена небольшой пептидный фрагмент, в результате чего освобождаются участки связывания, что позволяет молекуле фибрина агрегировать в полимер . Затем с помощью глутамин-трансферазы (фактора XIII) образуются изопептидные связи боковых цепей аминокислот фибрина, что приводит к формированию нерастворимого фибринового сгустка (тромба).
Свертывание крови может запускаться двумя различными путями: вследствие нарушения целостности ткани ( внесосудистый путь, на схеме справа) или процессами, которые начинаются на внутренней поверхности сосуда ( внутрисосудистый путь , на схеме слева). В обоих случаях запускается каскад протеолитических реакций: из неактивных предшественников ферментов (зимогенов, условно обозначаемых на схеме окружностями) путем отщепления пептидов образуются активные сериновые протеиназы (обозначаемые на схеме окрашенными кружочками с вырезанным сектором), которые в свою очередь действуют на другие белки. Оба реакционных пути нуждаются в ионах Са 2+ и фосфолипидах [ФЛ (PL)] и оба завершаются активацией фактором Ха протромбина (фактора II) с образованием тромбина (IIа).
Внутрисосудистый путь инициируется коллагеном, который в норме не экспонирован на внутренней поверхности кровеносных сосудов; его контакт с кровью приводит к активации фактора XII. Внесосудистый путь активации начинается с освобождения фактора III (тканевого тромбопластина) из поврежденных клеток ткани. В течение нескольких секунд этот фактор приводит к свертыванию крови в области раны.
Факторы свертывания II, VII, IX и X содержат необычную аминокислоту, γ-карбоксиглутаминовую (Gla). Остатки Gla, которые образуются в результате посттрансляционного карбоксилирования остатков глутаминовой кислоты, группируются в особых белковых доменах. Они присоединяют ионы Са 2+ и вследствие этого связывают соответствующие регуляторные факторы с фосфолипидами на поверхности плазматической мембраны. На рисунке это схематически представлено на примере протромбинового комплекса (Va, Ха и II). Вещества, способные связывать свободные ионы Са 2+ в виде комплекса, например цитрат, предотвращают это взаимодействие с фосфолипидами и тормозят свертывание. Для синтеза остатков Gla необходим в качестве кофактора витамин К (см. с. 342). Антагонисты витамина К, такие, как дикумарин, подавляют синтез активных факторов коагуляции и действуют поэтому также как ингибиторы свертывания.
Генетически обусловленный дефицит отдельных факторов свертывания приводит к кровоточивости (гемофилия).
Контроль за свертыванием крови (не показан на схеме). Процесс свертывания крови находится в постоянном равновесии между активацией и торможением. Для торможения в плазме имеются очень эффективные ингибиторы протеиназ. Сериновые протеиназы системы свертывания инактивируются антитромбином. Его действие усиливается сульфатированным глюкозаминогликаном — гепарином (см. с. 336). Тромбомодулин, расположенный на внутренней стенке кровеносных сосудов, инактивирует тромбин, образуя с ним стехиометрический комплекс. За протеолитическое разрушение факторов V и VIII в плазме отвечает белок с. Этот белок в свою очередь активируется тромбином и, тем самым, реализуется самотормозящийся механизм свертывания крови.
Публикации в СМИ
Плазменные факторы свёртывания крови — различные компоненты плазмы, реализующие образование сгустка крови. Недостаточность плазменных факторов свёртывания может быть изолированной или комбинированной.
• Изолированная недостаточность •• Фактор I (фибриноген, превращающийся в фибрин под влиянием тромбина). Недостаточность фактора I — см. Дисфибриногенемии •• Фактор II (протромбин; активная форма — фактор IIa, или тромбин, образуется под влиянием фактора Xa, мембран тромбоцитов и других клеток, Са2+ и фактора Va). Недостаточность фактора II (*176930, 11p11–q12, дефект гена F2, Â ) приводит к развитию диспротромбинемии и гипопротромбинемии. Клинически: геморрагический синдром •• Фактор III (тканевой фактор, или тромбопластин, запускает внешний механизм свёртывания крови путём активации фактора VII с образованием фактора VIIa). Единственный фактор свёртывания, изолированная врождённая недостаточность которого в литературе не описана (см. ниже
Недостаточность фактора CAR) •• Фактор IV (ионы кальция). Недостаточность фактора IV — см. Гипокальциемия •• Фактор V (проакцелерин; активированная тромбином форма — акцелерин, участвующий в активации протромбина путём образования рецептора к сериновой протеазе фактора Xa на поверхности тромбоцитов). Недостаточность фактора приводит к развитию редко встречаемой парагемофилии (болезнь Оврена, *227400, 1q23, дефект гена F5, r ), по клиническим проявлениям аналогичной гемофилии (см. Гемофилия); гетерозиготы имеют уменьшенное содержание фактора V, но у них отсутствует склонность к кровотечениям; точечная мутация гена фактора V (фактор VЛейден) — фактор риска венозных тромбозов •• Фактор VII (проконвертин).
Недостаточность может быть врождённой или приобретённой ••• Врождённая недостаточность фактора VII (*227500, 13q34, дефект гена F7, r ) протекает с пурпурой и кровоточивостью слизистых оболочек ••• Приобретённая недостаточность фактора VII (возникает при гиповитаминозе К, в период новорождённости, при введении протромбинопенических препаратов). Примечание. Гипопроконвертинемия также известна как болезнь Александера (В. Alexander). Другая болезнь Александера (Alexander WS) — одна из лейкодистрофий (203450) •• Фактор VIII (антигемофильный фактор; участвует в образовании комплекса с фактором IXa, тромбоцитами, Са2+, приводящего к активации фактора X). Недостаточность фактора VIII приводит к развитию классической гемофилии А у мужчин (см. Гемофилия). Недостаточность мультимерных форм фактора VIII-фон Виллебранда (VIII:R, ффВ), необходимых для агрегации тромбоцитов, приводит к развитию болезни фон Виллебранда (см. Болезнь фон Виллебранда) •• Фактор IX (Кристмаса фактор; активная форма — фактор IXa). Недостаточность приводит к развитию гемофилии В (см. Гемофилия) •• Фактор X (Стюарта фактор, Стюарта–Прауэр фактор, активная форма — фактор Xa). Недостаточность фактора X (*227600, 13q34, r ) характеризуется снижением активности внутреннего и внешнего путей активации свёртывания крови, повышением ПВ и ЧТВ. Клинически проявляется геморрагическим синдромом и спонтанными абортами. Синдром Скотта (262890, r ) клинически проявляется также, но развивается вследствие дефекта рецепторов тромбоцитов к фактору X •• Фактор XI (плазменный предшественник тромбопластина; активная форма — фактор XIa, сериновая протеаза, превращающая фактор IX в фактор IХa).
Недостаточность фактора XI (*264900, 4q35, дефект гена F11, r ) приводит к геморрагическому синдрому •• Фактор XII (фактор Хагемана; активная форма — фактор XIIa, активирует факторы VII и XI). Недостаточность фактора Хагемана (*234000, 5q33–qter, дефект гена HAF, r ) проявляется увеличением времени свёртывания, ЧТВ, но кровотечения наблюдают редко •• Фактор XIII (Лаки–Лорана фактор; фибринстабилизирующий фактор; активная форма [активированная тромбином] — фактор XIIIа) стабилизирует фибриновый сгусток, образуя нерастворимый фибрин. Состоит из 2 субъединиц — А и В. Недостаточность фактора XIII может проявляться недостаточностью субъединицы A (*134570, 6p25–p24, Â ) — геморрагический синдром, олигоспермия у гомозиготных мужчин, гемартрозы; недостаточностью субъединицы B (*134580, 1q31.2–q32.3, r ) — геморрагический синдром •• Изолированная недостаточность других факторов ••• Недостаточность кофактора 2 (*142360, лейсерпин 2, 22q11, дефект гена HCF2, ). Клинические проявления: ДВС, тромбофилия ••• Недостаточность тромбоцитарного фактора 3 (173450, Â ). Клинические проявления: геморрагический синдром. Лабораторные исследования: врождённая недостаточность фосфолипидов тромбоцитарной мембраны •••
Геморрагический синдром отсутствует, показатели свёртывания нормальные, повышено ЧТВ. Примечание: Флетчеры — семейство, в котором впервые обнаружен данный фактор ••• Недостаточность a 2 2-ингибитора плазмина (*262850, антиплазмин, 17pter–p12, дефект гена PLI, r ). Клинические проявления: геморрагический синдром ••• Недостаточность плазминогена (*173350, 6q26, дефект гена PLG, Â ) проявляется тромбофилией.
• Комбинированная недостаточность •• Недостаточность плазменных факторов свёртывания множественная (*277450, витамин К зависимый коагуляционный дефект, r ) — недостаточность затрагивает факторы II, VII, IX и X, а также белки S и С. Клинически: геморрагический синдром, гипоплазия носа и дистальных фаланг пальцев •• Тип I (227310, r ) проявляется геморрагическим синдромом вследствие недостаточности факторов V и VIII в сочетании с нормальным содержанием белков S и С •• Тип II (134510, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов VIII и IX •• Тип IV (134430, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов VII и VIII •• Тип V (134520, Â ) проявляется геморрагическим синдромом (повышение ЧТВ) вследствие недостаточности факторов VIII, IX и XI •• Тип VI (134540, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов IX и XI.
МКБ-10. D68.2 Наследственный дефицит других факторов свёртывания
Код вставки на сайт
Недостаточность плазменных факторов свёртывания
Плазменные факторы свёртывания крови — различные компоненты плазмы, реализующие образование сгустка крови. Недостаточность плазменных факторов свёртывания может быть изолированной или комбинированной.
• Изолированная недостаточность •• Фактор I (фибриноген, превращающийся в фибрин под влиянием тромбина). Недостаточность фактора I — см. Дисфибриногенемии •• Фактор II (протромбин; активная форма — фактор IIa, или тромбин, образуется под влиянием фактора Xa, мембран тромбоцитов и других клеток, Са2+ и фактора Va). Недостаточность фактора II (*176930, 11p11–q12, дефект гена F2, Â ) приводит к развитию диспротромбинемии и гипопротромбинемии. Клинически: геморрагический синдром •• Фактор III (тканевой фактор, или тромбопластин, запускает внешний механизм свёртывания крови путём активации фактора VII с образованием фактора VIIa). Единственный фактор свёртывания, изолированная врождённая недостаточность которого в литературе не описана (см. ниже
Недостаточность фактора CAR) •• Фактор IV (ионы кальция). Недостаточность фактора IV — см. Гипокальциемия •• Фактор V (проакцелерин; активированная тромбином форма — акцелерин, участвующий в активации протромбина путём образования рецептора к сериновой протеазе фактора Xa на поверхности тромбоцитов). Недостаточность фактора приводит к развитию редко встречаемой парагемофилии (болезнь Оврена, *227400, 1q23, дефект гена F5, r ), по клиническим проявлениям аналогичной гемофилии (см. Гемофилия); гетерозиготы имеют уменьшенное содержание фактора V, но у них отсутствует склонность к кровотечениям; точечная мутация гена фактора V (фактор VЛейден) — фактор риска венозных тромбозов •• Фактор VII (проконвертин).
Недостаточность может быть врождённой или приобретённой ••• Врождённая недостаточность фактора VII (*227500, 13q34, дефект гена F7, r ) протекает с пурпурой и кровоточивостью слизистых оболочек ••• Приобретённая недостаточность фактора VII (возникает при гиповитаминозе К, в период новорождённости, при введении протромбинопенических препаратов). Примечание. Гипопроконвертинемия также известна как болезнь Александера (В. Alexander). Другая болезнь Александера (Alexander WS) — одна из лейкодистрофий (203450) •• Фактор VIII (антигемофильный фактор; участвует в образовании комплекса с фактором IXa, тромбоцитами, Са2+, приводящего к активации фактора X). Недостаточность фактора VIII приводит к развитию классической гемофилии А у мужчин (см. Гемофилия). Недостаточность мультимерных форм фактора VIII-фон Виллебранда (VIII:R, ффВ), необходимых для агрегации тромбоцитов, приводит к развитию болезни фон Виллебранда (см. Болезнь фон Виллебранда) •• Фактор IX (Кристмаса фактор; активная форма — фактор IXa). Недостаточность приводит к развитию гемофилии В (см. Гемофилия) •• Фактор X (Стюарта фактор, Стюарта–Прауэр фактор, активная форма — фактор Xa). Недостаточность фактора X (*227600, 13q34, r ) характеризуется снижением активности внутреннего и внешнего путей активации свёртывания крови, повышением ПВ и ЧТВ. Клинически проявляется геморрагическим синдромом и спонтанными абортами. Синдром Скотта (262890, r ) клинически проявляется также, но развивается вследствие дефекта рецепторов тромбоцитов к фактору X •• Фактор XI (плазменный предшественник тромбопластина; активная форма — фактор XIa, сериновая протеаза, превращающая фактор IX в фактор IХa).
Недостаточность фактора XI (*264900, 4q35, дефект гена F11, r ) приводит к геморрагическому синдрому •• Фактор XII (фактор Хагемана; активная форма — фактор XIIa, активирует факторы VII и XI). Недостаточность фактора Хагемана (*234000, 5q33–qter, дефект гена HAF, r ) проявляется увеличением времени свёртывания, ЧТВ, но кровотечения наблюдают редко •• Фактор XIII (Лаки–Лорана фактор; фибринстабилизирующий фактор; активная форма [активированная тромбином] — фактор XIIIа) стабилизирует фибриновый сгусток, образуя нерастворимый фибрин. Состоит из 2 субъединиц — А и В. Недостаточность фактора XIII может проявляться недостаточностью субъединицы A (*134570, 6p25–p24, Â ) — геморрагический синдром, олигоспермия у гомозиготных мужчин, гемартрозы; недостаточностью субъединицы B (*134580, 1q31.2–q32.3, r ) — геморрагический синдром •• Изолированная недостаточность других факторов ••• Недостаточность кофактора 2 (*142360, лейсерпин 2, 22q11, дефект гена HCF2, ). Клинические проявления: ДВС, тромбофилия ••• Недостаточность тромбоцитарного фактора 3 (173450, Â ). Клинические проявления: геморрагический синдром. Лабораторные исследования: врождённая недостаточность фосфолипидов тромбоцитарной мембраны •••
Геморрагический синдром отсутствует, показатели свёртывания нормальные, повышено ЧТВ. Примечание: Флетчеры — семейство, в котором впервые обнаружен данный фактор ••• Недостаточность a 2 2-ингибитора плазмина (*262850, антиплазмин, 17pter–p12, дефект гена PLI, r ). Клинические проявления: геморрагический синдром ••• Недостаточность плазминогена (*173350, 6q26, дефект гена PLG, Â ) проявляется тромбофилией.
• Комбинированная недостаточность •• Недостаточность плазменных факторов свёртывания множественная (*277450, витамин К зависимый коагуляционный дефект, r ) — недостаточность затрагивает факторы II, VII, IX и X, а также белки S и С. Клинически: геморрагический синдром, гипоплазия носа и дистальных фаланг пальцев •• Тип I (227310, r ) проявляется геморрагическим синдромом вследствие недостаточности факторов V и VIII в сочетании с нормальным содержанием белков S и С •• Тип II (134510, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов VIII и IX •• Тип IV (134430, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов VII и VIII •• Тип V (134520, Â ) проявляется геморрагическим синдромом (повышение ЧТВ) вследствие недостаточности факторов VIII, IX и XI •• Тип VI (134540, Â ) проявляется геморрагическим синдромом вследствие недостаточности факторов IX и XI.
МКБ-10. D68.2 Наследственный дефицит других факторов свёртывания
Читайте также: