Болезнь Фабри на МРТ, КТ головного мозга
Добавил пользователь Евгений Кузнецов Обновлено: 28.01.2026
Сфинголипидозы - это группа липидозов, при которой имеет место нарушение обмена сфингомиелина с накоплением его в мозге и внутренних органов, прежде всего - в системе мононуклеарных фагоцитов. В стернальном пунктате выявляются специфические клетки Нимана-Пика.
Болезнь Фабри - форма липидоза, характеризующаяся накоплением гликолипида церамидтригексозида в стенках кровеносных сосудов, в периферических нервах, в нейронах мозга, в роговице, в почках. Развивается вследствие недостаточности фермента α-галактозидазы (А). Заболевание полисистемное. Проявляется наличием ангиом, кератом, поражением роговиц, сенсорной и вегетативной ганглиополиневропатией с пароксизмальными парестезиями и болями в конечностях. Отложение гликолипидов в сосудах может вызвать нарушение кровообращения и, в частности, развитие мозговых инсультов, почечную недостаточность, поражение других органов.
Крабе лейкодистрофия - проявляется в возрасте 2-6 месяцев. Сначала наблюдается остановка, а затем редукция физического и психического развития. При этом характерны приступы крика, центральный парез мышц конечностей, гиперестезия, позже - приступы мышечных спазмов, опистотонус с вытянутыми и перекрещенными ногами, клонические судороги; атрофия зрительных нервов, сопровождающаяся прогрессирующим снижением зрения; гипертермия. Прогноз плохой. В типичных случаях болезнь длится 5-8 месяцев. В терминальной стадии отмечается слепота, кахексия, судороги, децеребрационная ригидность, слабоумие.
Болезнь Ниманна-Пика - группа липидозов, при которых имеет место нарушение обмена сфингомиелина с накоплением его в мозге и внутренних органах. Классическая форма болезни проявляется в возрасте 5-6 месяцев расстройствами зрения и слуха, умеренных повышений мышечного тонуса, вскоре сменяющихся мышечной гипотонией, а затем атонией. Олигофрения, чаще в форме идиотии. Характерны желто-коричневый цвет кожи, гепатоспленомегалия, увеличение лимфатических узлов.
Лейкодистрофии (дегенеративное заболевание мозга с преимущественным поражением белого вещества) - это наследственные диффузные демиелинизирующие процессы в белом веществе головного мозга, спинного мозга и периферических нервов, имеющие прогрессирующее течение. В основе их - генетически детерминированное нарушение обменных процессов в миелиновых структурах и распад неправильно сформированного миелина (дисмиелинизация).
Метахроматическая лейкодистрофия (сульфатиоз, Шольца синдром) - чаще других встречающаяся форма лейкодистрофии, обусловленная редукцией активности лизосомного фермента арилсульфатазы (А) (цереброздсульфатаза) в связи с дефектом фермента на 22-й хромосоме. При этом происходят метахроматически окрашивающиеся вещества - в мозге, печени, почках, уменьшение в мозге количества цереброзидов. В результате происходит диффузная демиелинизация нервной ткани, накопление гранул сульфатидов в клетках глии, макрофагах, в нейронах, поражение ганглиозных клеток сетчатки, избыток липидов в печени, в почечных канальцах.
Заболевание проявляется в возрасте от 1 года до 30 лет (1-4 года - инфантильная форма, 5-20 лет - ювенильная форма, позже 20 лет - взрослая форма). Нарастает мышечный тонус, нарушается координация движений, изменяется походка. Появляются глазодвигательные расстройства, дизартрия, снижаются сухожильные рефлексы. Грубо нарушается психическое развитие, могут быть судорожные припадки. В далеко зашедшей стадии (через несколько лет) развиваются тетраплегия, децеребрационная ригидность, декортикация, грубые псевдобульбарные и бульбарные нарушения. На КТ головного мозга обнаруживают симметричные очаги пониженной плотности в белом веществе лобных и теменных долей мозга. В мозге выявляется почти полная демиелинизация с дегенерацией демиелинизированных волокон, особенно выраженная в каудальной части мозгового ствола. Смерть наступает через 2-10 лет после начала заболевания.
Лейкодистрофия Ван-Богарта - Шерара-Эпстайна - характеризуется нарастающим слабоумием, эпилептиформными приступами, экстрапирамидными симптомами, псевдобульбарными расстройствами. Церебральный холестериноз, сопровождающийся отложением холестерина в двигательной зоне коры большого мозга, ножках мозга и мозжечка, а также отеком клеток передних рогов спинного мозга. Проявляется задержкой физического и психического развития, сухостью кожи, слабостью лицевой мускулатуры, диазатрией, мозжечковой атаксией, бульбарными и пирамидными расстройствами, катарактой, остеопорозом, ксантомой.
Болезнь Вольмана - ранняя инфантильная органомегалия, рвота, диарея, различные повреждения нервной системы, желтуха. Дефектный фермент - кислотная липаза, накапливаемое вещество - эфир, холестерола, триглицириды.
Случай диагностики болезни Фабри у пациента, доставленного скорой помощью с предварительным диагнозом инсульт
В неврологическое отделение машиной скорой помощи доставлен мужчина 32 лет с жалобами на резкую боль в голове, спутанность сознания и нарушения походки. По клиническим проявлениям заподозрен инсульт.
Жалобы
На момент госпитализации больной предъявлял жалобы на интенсивную головную боль, ощущение тяжести в правой половине тела, нарушение координации при движении. Речь пациента спутанная, ориентация в пространстве и времени затруднена. При детальном расспросе пациента после стабилизации его состояния на фоне проводимого лечения удалось установить, что пациента с подросткового возраста беспокоит периодическое чувство жжения, "ползания мурашек" в области стоп и кистей. Больной плохо переносит высокую физическую нагрузку, так как быстро устаёт, появляется одышка. Дважды переносил тепловой удар в летнее время. С перечисленными жалобами многократно обращался к неврологу по месту жительства, где был поставлен диагноз "Болезнь Рейно".
Чувство жжения в области стоп и кистей рук провоцировалось длительными физическими нагрузками, стрессом, высокими температурами окружающей среды. Продолжительность болевых ощущений составляла 10-15 минут, после чего боль постепенно уменьшалась. Ухудшение общего самочувствия часто наблюдалось в жаркое время года. В покое описанные жалобы не возникали. Летом частота болезненных ощущений была выше.
Анамнез
Первые проявления заболевания — парестезии — появились в возрасте 13-14 лет, с возрастом постепенно симптомы усиливались, увеличивалась их частота до двух-трёх раз в месяц в летнее время года. За сутки до госпитализации появилась головная боль, слабость в правой половине тела, нарушение сознания. С 20 лет страдает артериальной гипертензией, постоянно принимает гипотензивные препараты.
Двоюродный брат матери пациента умер от ишемического инсульта в 36 лет, анамнез заболевания неизвестен.
Обследование
Больной повышенного питания, гиперстенического типа телосложения. При проведении осмотра обращают на себя внимание выступающие лобные бугры, пухлые губы. На животе, вокруг пупка единичные красные пятна-ангиокератомы. Со стороны органов дыхания и пищеварения патологии не выявлено. При аускультации сердца: сердечные тоны приглушены. При перкуссии сердца: смещение границ сердца влево. Наблюдается слабость скелетной мускулатуры, особенно выражена справа.
ЭКГ: гипертрофия левого желудочка, диффузные ишемические изменения миокарда. Общий анализ крови: гипохромная микроцитарная анемия лёгкой степени. Общий анализ мочи: умеренная протеинурия. МРТ головного мозга: множественные ишемические изменения. Эхо-КГ: гипертрофия левого желудочка. Биохимический анализ крови: без особенностей. Проведён поиск мутаций в гене GLA, отвечающем за развитие болезни Фабри. Выявлена мутация c.680G>A в гене GLA, что подтвердило диагноз "Болезнь Фабри".
Диагноз
Лечение
В стационаре была проведена тромболитическая, противоишемическая терапия. Применяемые препараты: ацетилсалициловая кислота, "Варфарин", бета-адреноблокаторы, "Актовегин", ингибиторы АПФ, антагонисты кальция. Проведён курс лечебной физкультуры, физиотерапевтическое лечение.
Пациент смог вернуться к обычной жизни, направлен в медико-генетический центр в Москве для решения вопроса о возможности применения заместительной ферментотерапии как единственного патогенетического метода лечения.
Заключение
Данный клинический случай показывает необходимость обследования на болезнь Фабри при жалобах на чувство жжения, онемения, "ползания мурашек" в дистальных отделах конечностей, непереносимости жары, холода и физических нагрузок. Иногда диагноз ставится слишком поздно, когда заместительная терапия не даст должного эффекта, а у пациента уже произошли необратимые изменения.
Диагностические маркеры поражения сердца при болезни Фабри
Болезнь Фабри - сцепленное с X-хромосомой прогрессирующее наследственное заболевание, вызванное недостаточностью или полным отсутствием активности фермента альфа-галактозидазы А, характеризующееся накоплением гликосфинголипидов в клетках различных органов и тканей.
Болезнь Фабри характеризуется мультисистемными проявлениями, затрагивающими в основном нервную систему (нейропатическая боль), почки (развитие в конечном итоге почечной недостаточности), кожу (ангиокератомы, гипогидроз) и сердце. Причем прогноз заболевания определяется степенью выраженности поражения сердца и почек.
Основными кардиальными проявлениями болезни Фабри являются прогрессивное развитие гипертрофии (болезнь Фабри должна обсуждаться у всех пациентов с необъяснимой гипертрофией левого желудочка), фиброз миокарда, нарушения ритма, сердечная недостаточность, а также внезапная сердечная смерть. В данной заметке приведены основные выдержки по диагностике поражения сердца при болезни Фабри из Экспертного консенсусного документа, посвященного ведению пациентов с поражением сердца в рамках болезни Фабри.
Электрокардиография. У взрослых с болезнью Фабри наиболее часто выявляемыми отклонениями при выполнении электрокардиографии являются: укорочение интервала PR, вольтажные критерии гипертрофии левого желудочка и соответствующие ей нарушения реполяризации. На более продвинутых стадиях может быть диагностирована брадикардия и задержка атриовентрикулярного проведения. Электрокардиография должна выполняться всем пациентам с болезнью Фабри на этапе установления диагноза, прогрессирования симптомов, а также каждые 6-12 месяцев взрослым пациентам.
Эхокардиография. Типичными находками для болезни Фабри являются: концентрическая гипертрофия левого желудочка (чаще без обструкции выносящего тракта левого желудочка в покое), гипертрофия папиллярных мышц, утолщение стенки правого желудочка. Фракция выброса чаще бывает нормальной и снижается по мере прогрессирования фиброза, зоны которого наиболее часто локализуются в области задней и нижней стенок левого желудочка. Также могут выявляться признаки диастолической дисфункции и снижение глобальной деформации левого желудочка. Еще одним признаком болезнь Фабри при эхокардиографии является утолщение створок митрального и аортального клапанов с развитием их недостаточности, тогда как стенотическое поражение клапанного аппарата не является характерным для болезни Фабри. Периодичность выполнения эхокардиографии аналогична таковой для электрокардиографии.
Магнитно-резонансная томография сердца (МРТ). МРТ позволяет более точно охарактеризовать размеры полостей сердца, его массу и геометрию, а также оценить выраженность фибротических изменений, которые нередко бывают и в отсутствии гипертрофии. МРТ сердца рекомендована всем пациентам с болезнью Фабри, однако периодичность ее выполнения в документе не обозначена.
Эндомиоркадиальная биопсия. Эндомиокардиальную биопсию следует рассмотреть как метод диагностики у пациентов с неопределенной активностью болезни Фабри.
Электрофизиологическое исследование. Учитывая относительно высокую частоту нарушений ритма сердца при болезни Фабри, электрофизиологическое исследование рекомендовано всем пациентам с персистирующей или рецидивирующей суправентрикулярной тахикардией, атриовентрикулярной узловой реципрокной тахикардией, атриовентрикулярной тахикардией, связанной с наличием дополнительного проводящего пути, а также пациентам с подозрением на поражение синоатриального узла или атриовентрикулярную блокаду.
Лабораторные тесты. Всем пациентам с болезнью Фабри рекомендован скрининг на другие патологии, ассоциированные с поражением сердца (болезни щитовидной железы, сахарный диабет). Другими обязательными лабораторными тестами являются: определение микроальбуминурии и скорости клубочковой фильтрации, определение уровня С-реактивного белка и интерлейкина-6 (повышены при болезни Фабри и ассоциированы с гипертрофией левого желудочка и наличием фиброза. Также рекомендуется определение уровня NTproBNP, повышение которого даже при отсутствии эхокардиографических признаков, может свидетельствовать о начальном этапе поражения сердца.
Кратко обсуждая терапию различных вариантов поражения сердца при болезни Фабри, следует отметить, что специальных исследований на этой категории пациентов не проводилось, однако в целом терапия схожа с таковой, применяемой в общей популяции. Также ввиду увеличения риска нарушения функции синоатриального узла и развития блокад, эксперты рекомендуют несколько осторожнее относиться к назначению пульсурежающих препаратов, а при развитии фибрилляции предсердий, учитывая высокий риск тромбоэмболических осложнений, вне зависимости от количества баллов по шкале CHA2DS2VASc рекомендовано назначение антикоагулянтной терапии.
Факоматозы и их диагностика
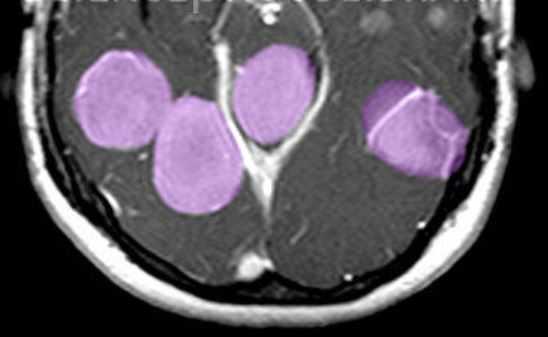
МРТ головного мозга. Т1-взвешенная МРТ с контрастированием. Множественные менингиомы при нейрофиброматозе 2 типа. Цветовая обработка изображения.
Факоматозы (нейрокожные нарушения) - это группа врожденных дисплазий эмбриональной эктодермы (кожи, центральной и переферической нервной систем, глаза) и эндодермы (эпителиальной выстилки желудочно-кишечного тракта).
К факоматозам относят:
- Нейрофиброматоз тип I (болезнь Реклингхаузена)
- Нейрофиброматоз тип II
- Туберозный склероз (синдром Бурневиля)
- Болезнь Гиппель-Линдау
- Синдром Стурге-Вебера
- Редкие заболевания - синдром Клиппеля-Треноне-Вебера, синдром Протея, синдром Ослера-Рандю-Вебера, синдром Вайбурна-Масона, болезнь Фабри, синдром кожной гемангиомы, атаксия-телеангиэктазия (синдром Луи-Бара), менингоангиоматоз (синдром Ульмана), нейрокожный меланоз, гипомеланоз Ито, синдром эпидермального невуса, базальноклеточный невус.
Все перечисленные заболевания выявляются при МРТ головного мозга.
Нейрофиброматоз I типа (болезнь Реклингхаузена) имеет аутосомно - доминантный тип наследования (50%), сцепленный с 17 парой хромосом, или с её спонтанными мутациями. Частота заболевания составляет один на 4000 населения. Диагностическими критериями служат (National Institutes of Health, 1988) не менее 6 кожных пятен (макулы цвета “кофе с молоком” размером не менее 5 мм в пре- и 15 мм в послепубертатном периоде), и не менее 2 любых нейрофибром, либо одна плексиформная (подкожная) нейрофиброма, множественные веснушки в подмышечных и паховых областях, костные дисплазии (истончение кортикального вещества длинных трубчатых костей с коксартрозом или без него, дисплазия клиновидной кости ), двухсторонние глиомы зрительных нервов, 2 или более пигментированные гамартомы радужной оболочки глаза (узлы Лиша) и наличие ближайшего родственника с этим заболеванием. Для постановки диагноза достаточно наличия 2 из перечисленных критериев. Кроме того, характерны следующие сопутствующие патологии: невриномы, кожные нейрофибромы, макроцефалия, астроцитомы, множественные менингиомы, кифосколиоз, саркома Юинга, сирингомиелия. Из опухолей головного мозга наиболее часто (в 5-15% случаев НФ I) встречается пилоцитарная астроцитома зрительного нерва и тракта . На МРТ астроцитома видна в виде утолщения зрительного нерва. Образование изоинтенсивно или немного гипоинтенсивно на Т1-взвешенных МРТ и гиперинтенсивно на Т2-взвешенных МРТ. Среди других локализаций астроцитома может быть в стволе, гипоталамусе, III желудочке. Редко обнаруживаются анапластические астроцитомы полушарий и мозжечка. У детей при НФ I встречаются очаги , напоминающие гамартомы. Они выявляются в ножках мозга, мосте, бледном шаре, среднем мозге, зрительном бугре, продолговатом мозге, реже белом веществе полушарий и мозжечка. На Т2-взвешенных МРТ очаги слегка гиперинтенсивны, на Т1-взвешенных МРТ изоинтенсивны белому веществу, за исключением бледного шара, где они тоже чуть гиперинтенсивны. Они не превышают 15 мм в размерах, не вызывают масс-эффекта и не контрастируются. Предположительно, очаги представляют собой участки изменённого миелина.
Третьим образованием диагностируемым при нейрофиброматозе I типа является нейрофиброма, обычно расподоженная в орбите и распространяющаяся в полость черепа и на кавернозный синус. Нейрофиброма слегка гиперинтенсивнее мышцы на Т1-взвешенных МРТ и выраженно гиперинтенсивнее на Т2-взвешенных МРТ. Кистозный компонент опухоли может давать сниженный сигнал в ее центре на Т1-взвешенных томограммах. При нейрофиброматозе I типа встречается также эктазия твердой мозговой оболочки в расширенном слуховом канале и стеноз водопровода. Эктазию важно не путать с невриномой. Стеноз водопровода не связан с опухолевой компрессией, но приводит к гидроцефалии.
Нейрофиброматоз II типа также имеет аутосомно-доминантный тип наследования, но сцепленный с 22 парой хромосом. Его частота составляет один на 100 тыс. населения. Диагностическими критериями служат (National Institutes of Health, 1988) двухсторонние невриномы слуховых нервов, выявляемые на КТ или МРТ, либо сочетание наследственной предрасположенности (наличие двухсторонних неврином у ближайшего родственника) с односторонней невриномой или двумя другими типичными опухолями (плексиформная нейрофиброма, менингиома, глиома, невринома любой локализации) плюс кожные пятна. В отличие от НФ I кожные пятна единичные и не служат главным критерием, а опухолевое поражение ассоциируется не с астроцитомами, а с невриномами и менингиомама. Сопутствующими патологиями являются менингоангиоматоз, глиальные узлы, эпендимальные эктопии, гипертрофический глиоз зрительного нерва, сирингомиелия, комплекс Арнольда-Киари. Типичная невринома развивается из шванновской оболочки слуховых нервов (VIII пара), обычно с обеих сторон , реже тройничного нерва или других. При МРТ невриномы гипо- или изоинтенсивны белому веществу на Т1-взвешенных МРТ и изо- или гиперинтенсивны на Т2-взвешенных МРТ. Хорошо усиливаются гадолинием. Менингиомы, как правило, сопутствуют невриномам. Локализация не отличается от случаев не связанных с нейрофиброматозом, но встречается также нетипичное поражение сосудистого сплетения. Картина менингиом при нейрофиброматозе II типа имеет все типичные признаки.
Туберозный склероз (синдром Бурневиля) встречается реже нейрофиброматоза. Его частота по данным литературы составляет около одного на 180 тыс. населения. От 20 до 40% случаев туберозного склероза унаследованы по аутосомно- доминантному типу, остальные возникли вследствии мутаций предположительно 9 и 11 пар хромосом (тип 1), либо 19 пары (тип 2). Поражение может затрагивать практически любые органы. Патогномоничными поражениями ЦНС являются корковые узлы в головном мозге и множественные субэпендимальные глиальные узлы , а также внутрижелудочковая гигантоклеточная астроцитома, встречаются сопутствующие аномалии - агенезия мозолистого тела, пахигирия, аневризмы. Характерны дерматологические проявления в виде множественных ангиофибром лица в форме «бабочки», бледные пятна на лице и груди, фибромы кожи, под ногтями и сетчатке глаза. Из других проявлений встречаются множественные ангиолипомы почек и печени, рабдомиомы сердца, лимфангиоматоз лёгких, костные склеротические и кистозные изменения. Диагноз туберозного склероза ставится при наличии у пациента 2 из перечисленных характерных признаков.
Корковые узлы - самое частое проявление туберозного склероза. Они расположены в коре головного мозга, деформируют её, захватывают прилегающее белое вещество и подвергаются кальцификации . При МРТ узлы изоинтенсивны серому веществу на Т1-взвешенных МРТ и чуть гиперинтенсивнее его на Т2-взвешенных. Контрастирование наблюдается в 5% случаев. В белом веществе обнаруживаются тяжи , отходящие радиально от желудочков. Корковые узлы и тяжи нередко называют «гамартомами», хотя они представляют собой скорее демиелинизацию и кальцификацию, чем истинную гетеротопию.
Туберозный склероз. Гамартомы. КТ, Т2-зависимая МРТ и FLAIR
Субэпендимальные, то есть проецирующиеся в желудочек, но растущие со стороны паренхимы мозга, узлы чаще расположены рядом с хвостатым ядром или гипоталамической бороздой сразу за отверстием Монро, реже в области III, IV желудочков и Сильвиева водопровода. На Т2-взвешенных томограммах субэпендимальные узлы умеренно гиперинтенсивны и часто содержат кальцинаты . От астроцитом их отличает не столь яркий сигнал и меньшие размеры. Контрастирование при введении препаратов гадолиния иногда наблюдается и в субэпендимальных узлах , и всегда в астроцитомах.
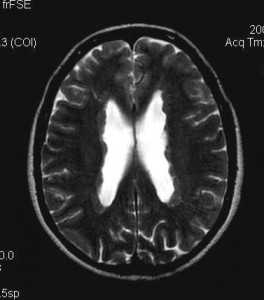
Туберозный склероз. Субэпендимальные узлы. Аксиальная Т-зависимая МРТ.
Болезнь Гиппеля - Линдау представляет собой симптомокомплекс, состоящий из гемангиобластом ЦНС (40% случаев) и сетчатки глаза (45%), и висцеральных проявлений в виде кист почек (75%), печени и поджелудочной железы. Диагноз ставится при наличии двух и более гемангиобластом или одной гемангиобластомы и висцеральных изменений, либо только висцеральных проявлениий при наличии семейной наследственности. Болезнь Гиппеля-Линдау в 20% случаев имеет врожденное семейное происхождение с аутосомно-доминантным типом наследования, в остальных связана с мутацией 3 пары хромосом. Частота примерно 1 случай на 36 тыс. населения. Кроме гемангиобластом и характерных висцеральных изменений при болезни Гиппель - Линдау встречается сопутствующие патологии: карцинома почки (более четверти наблюдений) и поджелудочной железы, феохромоцитома (около 10%, часто двухсторонняя), рабдомиома сердца, кисты лёгких, эпидидимит.
Гемангиобластомы при болезни Гиппеля - Линдау всегда множественные и примерно в половине случаев локализуются в мозжечке, реже стволе, спинном мозге и полушариях. При мозжечковой локализации опухоль чаще расположена поверхностно. При МРТ определяется неоднородный узел, гиперинтенсивный на Т2-взвешенных и изо- или гипоинтенсивный на Т1-взвешенных МРТ. В ряде случаев визуализируются патологически расширенные сосуды, имеющие характерное отсутствие сигнала. Кальцификации узлов не наблюдается. Гемангиобластомы хорошо контрастируются гадолинием. Чисто солидные гемангиобластомы наблюдаются только в 10% случаев. Окружающая узел киста гиперинтенсивна на томограммах обоих типов взвешенности, так как содержит примесь белка.
Синдром Стурге - Вебера ненаследуемое заболевание, ее морфологическим субстратом является ангиоматоз, связанный с тем, что сохраняются синусоидальные эмбриональные сосуды. Таким образом, синдром Стурге - Вебера представляет собой аномалию развития в “чистом” виде. Артериальный и венозный ангиоматоз приводят к избыточной васкуляризации оболочек мозга, кальцификации оболочечных артерий. Поражаются мягкие мозговые оболочки, обычно затылочной доли, причем с одной стороны (75% случаев). Кора мозга над ангиомой атрофируется и кальцифицируется . Нередко выявляется патологически расширенная кортикальная вена. Описано также увеличение сосудистого сплетения, гемиатрофия мозга на стороне поражения, ускорение и нарушение миелинизации, мегалэнцефалия и гидроцефалия. В постановке диагноза помогает наличие невуса кожи лба, который расположен по ходу первой ветви тройничного нерва, с той же стороны, что и очаг в мозге. Из других проявлений заболевания встречаются костные - ипсилатеральная гипертрофия черепа и синусов, глазные - ипсилатеральный экзофтальм, глаукома (30%), колобома радужной оболочки, гемангиома сосудистого сплетения глаза, висцеральные - ангиоматоз щитовидной железы, лёгких, поджелудочной железы, печени, почек, кишечника. Клинически синдром проявляется контрлатеральным гемипарезом, гомонимной гемианопией, судорогами (80% случаев) и умственной отсталостью (60%). При МРТ выявляется хорошо контрастирующийся ангиоматозный клубок сосудов, утолщенная оболочка, расширенная кортикальная вена и, иногда, расширенное сосудистое сплетение.
МРТ СПб дает место выбора выполнения МРТ головного мозга. При МРТ в СПб мы выступаем за комплексный подход к диагностике факоматозов с исследованием всех их проявлений. Обычно факоматозы лучше видны в высоких полях, но многие, особенно, опухоли видны и в низкопольных открытых МРТ.
Болезнь Фабри на МРТ, КТ головного мозга
Звоните нам по телефону 8 (812) 241-10-46 с 7:00 до 00:00 или оставьте заявку на сайте в любое удобное время
Ваша заявка принята!
Благодарим за обращение.
В ближайшее время с вами свяжется наш специалист.
Виды лейкодистрофии, классификация по МКБ, диагностика
Лейкодистрофия - группа заболеваний с поражением мозжечка, белого вещества, полушарий головного мозга с сохранностью корковых структур.
Нейродегенерация мозговой ткани сопровождается накоплением внутри спинного и головного мозга метаболические соединений, разрушающих миелин. Повреждение оболочки нейронов приводит к необратимым заболеваниям, сопровождающимся двигательными расстройствами, нарушением психомоторной функции, поражение слуха и зрении, эпилепсией, судорогами, неврологическими расстройствами, эпилептическими приступами.
Лейкострофии МРТ головного мозга
Классификация по МКБ 10
Международная классификация болезней 10 пересмотра относит лейкодистрофии к сфинголипидозам - заболеваниям, сопровождающимся избыточным отложением патологических жиров (липидов). Код нозологии - «E 75».
Нарушения обмена ганглиозидов кодируются «GM 2»:
- Ювенильная форма;
- Лейкодистрофия взрослых;
- Болезнь Сендхоффа;
- Синдром Тея-Сакса.
Другие ганглиозидозы («E 75.1»):
- Муколипидоз IV;
- Ганлиозидозы GM3, GM1.
Другие сфинголипидозы («E 75.2»):
- Недостаточность сульфатазы;
- Метахроматическая лейкодистрофия;
- Болезнь Нимана-Пика;
- Синдром Краббе;
- Синдром Фабера;
- Болезни Фабри-Андерсона.
Неуточненный сфинголипидоз - «E 75.3». К категории относятся все формы этиологические факторы, которых установить не удалось. Липофусциноз нейронов - «E 75.4». Избыточное образование атипичных жировых части приводит к нарушению передачи нервных сигналов. Неклассифицированные состояния («E 75.5»):
- Болезнь Волмана;
- Холестероз Ван-Богарта-Шерера.
Дисбаланс метаболических соединений внутри головного мозга обеспечивает атипичную клинику.
Неуточненная болезнь накопления липидов - «E 75.6».
Международная классификация МКБ 10 принята во всем мире для унификации перечная нозологических форм. Стандартизации тактики лечения.
Виды лейкодистрофии
Перечень биохимических изменений, приводящих к лейкодистрофии мозжечка, стволовых структур головного и спинного мозга, не выявлен. Ученые считают патологию вариантом повреждения лизосом. Научные исследования не выявили ферменты, отвечающие за клинические проявления нозологии.
Лизосомальные виды лейкодистрофий:
- Галлерводена-Шпатца;
- Краббе;
- Пелициуса-Мерцбахера.
Большинство форм лейкодистрофий возникает в раннем возрасте, но обнаруживается патология и у взрослых. При всех разновидностях возникают неврологические и пирамидальные расстройства, экстрапирамидальная ригидность, демиелинизация нервных волокон. Перечень лабораторных изменений при лейкодистрофиях - увеличение белка, усиленный плеоцитоз.
Метахроматическая лейкодистрофия
Проявляется у взрослых после 21 года. Преимущественно встречается нозология у мужчин. Наследуется по аутосомно-рецессивному механизму. Метахроматическая лейкодистрофия головного мозга развивается постепенно. До выраженных клинических симптомов может пройти более двадцати лет. Особенности проявлений психоза:
- Забывчивость;
- Снижение академических возможностей;
- Неразумные действия;
- Странности поведения;
- Излишняя подозрительность.
Аналогичные клинические симптомы возникают при шизофрении. Присоединение неврологических симптомов мозжечковой атаксии, пирамидальных расстройств, неловкость движений пациента провоцирует психическую деградацию личности. Беспомощность, отсутствие контакта с окружающими людьми, прикованность к постели обеспечивает быстрое прогрессирование клиники из-за ряда метаболических изменений:
- Падение активности лейкоцитарных ферментов (арилсульфатазы A);
- Усиленное выделение сульфатидов с уриной;
- Дисбаланс проведения нервного импульса по поврежденным волокнам;
- Перераспределение пигментного вещества.
Метахроматическая лейкодистрофия у детей (Гринфилда) сопровождается судорогами, атаксией, нистагмом. Признаки терминальной стадии лейкодистрофии у детей:
- Децеребрационная ригидность;
- Бульбарные расстройства;
- Тетраплегия.
Причиной метахромного вида является избыточное скопление липидов. Патогенетическим механизмом формирования патологии является недостаточность фермента цереброзидсульфатазы. Развивается нозология позже форм Краббе или Тея-Сакса. Примерно в 5 лет у ребенка нарушается походка из-за повышенного тонуса мускулатуры. Постепенно утрачивается рефлекторная активность, иннервация сухожилий.
Клинические симптомы лейкодистрофии
Большинство видов возникает в детском возрасте. Сразу после рождения патологических изменений у ребенка не прослеживается. Через несколько месяцев или лет прослеживается неврологическая или психическая симптоматика, которая постепенно усугубляется.
Признаки ранних стадий лейкодистрофии:
- Патология зрения;
- Олигофрения;
- Мышечный спазм;
- Подергивания конечностей;
- Гипертонус;
- Тонические судороги;
- Признаки экстрапирамидальной патологии (шаткая походка);
- Падение интеллекта.
Множественные чувствительные расстройства, патология глотания, глухота диагностируются у дошкольников.
Симптомы лейкодистрофии мозга у грудничков второго года жизни:
- Замедленное психомоторное развитие (олигофрения);
- Патология походки.
Клинические проявления, начинающиеся с третьего года жизни:
- Потеря слуха и зрения;
- Гипертермический синдром;
- Тетраплегия;
- Гипертермия (повышение температуры).
Тяжелая симптоматика появляется через 10 лет после начала первичных изменений головного мозга.
Первичные изменения мозга сопровождаются спастичностью, миоклонией, задержкой развития, мышечным тремором. У взрослых прогрессирующая форма сопровождается быстрой потерей свойств личности, расстройствами речи, патологическим мышлением. Постепенное прогрессирование сопровождается разнообразными изменениями слизистой оболочки с развитием спастичности, мышечными судорогами, гипертонусами.
Вариант метахроматической лейкодистрофии сопровождается психозом, деменцией, эмоциональной неустойчивостью, расстройством речи, мышлением.
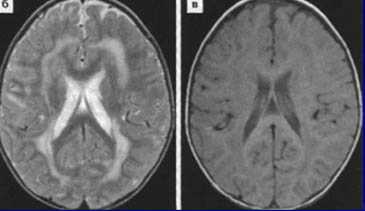
Томограммы метахроматической лейкодистрофии
Первые признаки лейкодистрофии у ребенка
При большинстве лейкодистрофий первые симптомы появляются на четвертом году жизни. Диагностировать нозологию удается по следующим признакам:
- Повышенный мышечный тонус;
- Сильная нервная возбудимость;
- Психомоторное развитие не соответствует возрасту;
- Кулаки ребенка сжаты.
- Атрофия зрительных нервов вплоть до слепоты;
- Усиление сухожильных рефлексов;
- Мышечный спастический тетрапарез;
- Миоклонические судороги;
- Общая двигательная реакция.
Периферическая нейропатия встречается только у отдельных детей. Летальный исход у детей прослеживается в возрасте от семи месяцев до трех лет.
Волокнистая лейкодистрофия Александера
Патогенетический механизм развития болезни Александера - дефект гена, отвечающего за выработку протеина GFAP. Дефект провоцирует избыточное скопление белка внутри глиальной ткани головного мозга. Уникальная структура протеина позволяет диагностировать нозологию посредством обнаружения специальных волокон Розенталя.
Неонатальная форма приводит к летальному исходу через 1 год после начала.
Менее опасен инфантильный вид, при котором возникают пороки развития, гидроцефалия, атаксия, парезы, спастическое сокращение мускулатуры. В большинстве случаев смерть возникает через пару лет.
Ювенильная дистрофия Александера появляется у школьников в возрасте 4-10 лет. Стволовая симптоматика длится долго. Симптоматика прогрессирует на протяжении 10-20 лет. Манифестация во взрослом периоде имеет медленное течение. Общая продолжительность заболевания свыше 10 лет.
Лейкодистрофия Галлервордена-Шпатца
Начинается заболевания у детей в возрасте 10 лет.
Клинические симптомы патологии:
- Эпилептические приступы;
- Тетрапарез;
- Дисфункция стриопаллидарной сферы;
- Ретинит пигментный;
- Гимералопия.
Поздняя форма, возникающая у детей в школьные годы. Длительность нозологии до полного появления клинических проявлений - около десяти лет.
- Эпилептические припадки;
- Судорожные подергивания;
- Ригидность мускулатуры;
- Гиперкинетические состояния.
Передается патология по аутосомно-рецессивному типу. Возникает у лиц женского и мужского пола. Сопровождается выраженным слабоумием, полной обездвиженностью пациентов. Патоморфологические изменения:
- Избыточное накопление железа внутри тканей;
- Инфильтративные скопления в глиальном слое;
- Дегенеративные поражения аксонов;
- Повышенная пигментация таламуса, мозжечка, коры большого мозга, субталамических структур;
- Расстройство пигментно-липидного обмена;
- Дисбаланс катехоламинов.
Паталогоанатомическое обследование выявляет морфологические признаки.
Наследуется по аутосомно-рецессивному механизму.
Болезнь Нимана-Пика
Сфингомиелиновые расстройства типов A и B возникают по причине недостаточности фермента - сфингомиелиназы. Соединение необходимо для разрушения сфингомиелина.
Симптомы болезни Нимана-Пика:
- Расширение селезенки, поджелудочной железы, печени;
- Покраснение внутриглазной сетчатки;
- Неврологические расстройства;
- Ожирение внутренних органов.
Сфингомиелиновый жировой липидоз приводит к постепенному поражению паренхиматозных структур (почки, печень, селезенка).
Болезнь Гоше
Нозология характеризуется липидозом, сопровождающимся недостаточностью фермента глюкозилцерамидазы. Ранние стадии сопровождаются гепатоспленомегалией. Болевых ощущений, другой симптоматики не возрастает до тех пор, пока размеры органов не станут огромными.
Прогрессирующие неврологические расстройства обуславливают ранний летальный исход.
Разновидность патологии у взрослых людей обусловлена аутосомно-рецессивным механизмом передачи. Передача из поколения в поколение не доказана, но практика показывает вероятность информации.
Болезнь Гоше относится к категории взрослых заболеваний, но первые изменения появляются у детей в возрасте 10 лет. В более раннем или позднем возрасте симптоматика возникает значительно реже. Гиперспления, патологические переломы, асептические некрозы головки бедренной кости, псевдоостеомиелит - распространенные вторичные состояния на фоне первичной лейкодистрофии Гоше.
При всех разновидностях нозологии в костномозговом пунктате выявляются специальные «нагруженные клетки».
Болезнь Фабри
Патология встречается из-за дефекта фермента альфа-галактозидазы. В тканях избыточно скапливается вещество - тригексозид. Наследуется нозология по Х-хромосоме, поэтому часто встречается у мужчин.
Обычно формируется патология в пожилом возрасте. Клиническое проявление нозологии - болевая нейропатия. Магнитно-резонансная томография головного мозга не выявляет патологических изменений до возникновения прогрессирующего поражения почек. Средний возраст пациентов - 20-40 лет.
Артериальные тромбозы при болезни возникают в детском возрасте. Летальный исход формируется из-за выраженной недостаточности почек.
Болезнь Вольмана
Развивается у детей раннего возраста. Вначале прослеживается гепатоспленомегалия, затем присоединяются вторичные проявления:
- Рвотный рефлекс;
- Анемический синдром;
- Кальцинация надпочечников;
- Повышение концентрации холестерина;
- Фиброз печени.
Болезнь Вольмана передается по аутосомно-рецессивному типу.
Болезнь Краббе-Бенеке
Наследственная болезнь - лейкодистрофия Краббе передается аутосомно-рецессивным путем. Формируется нозология в детском возрасте, характеризуется рядом клинических признаков:
- Снижение слуха, зрения вплоть до полной слепоты;
- Деменция;
- Спастический паралич;
- Судороги мускулатуры;
- Децеребрационная ригидность.
Морфологические проявления нозологии сопровождаются демиелинизацией нервных оболочек, нарушением выработки церебролизидов. Лейкодистрофия Краббе генетически детерминирована. Клинические симптомы:
- Слепота;
- Снижение слуха;
- Мышечные спазмы;
- Судорожные припадки.
Носительство аномального гена обнаружить не удается. Отсутствует эффективное лечение.
Синонимы: диффузный инфантильный склероз, болезнь Краббе-Бенеке, глобоидно-клеточная лейкодистрофия.
Суданофильная лейкодистрофия Пелицеуса-Мерцбахера
Возникает нозология преимущественно у мальчиков, так как локализуется патологический ген в Х-хромосоме. Ученые не изучили патогенетические механизмы патологии. Диффузная демиелинизация обуславливает клинические проявления на первом году жизни. Возникает поражение стволовых структур головного и спинного мозга, мозжечка. Повреждение миелиновой оболочки приводит к разрушению центральных и периферических нервных волокон. На первом году жизни у человека возникают специфические признаки:
- Внутриглазной нистагм;
- Кивательное подергивание головы;
- Мышечные гипо- и гиперклонии;
- Паркинсонический синдром;
- Дегенерация волокон зрительного нерва;
- Снижение интеллектуальной функции.
Диффузная демиелинизация Пелицеуса-Мерцбахера наследуется по аутосомно-рецессивному механизму. Изменения серого вещества сопровождается повреждением осевых цилиндров.
Диагностика патологии на ранней стадии основана на первичных признаках:
- Нистагм;
- Нарушение координации;
- Дрожание головы.
Позднее присоединяется атрофия зрительного нерва, снижение интеллекта, мышечный гипертонус, нарушение речи. Тяжелая стадия патологии сопровождается нарастающей деменцией, паркинсоническим синдромом, гиперкинезами.
Перивентрикулярная лейкомаляция
Заболевание сопровождается повреждением белого вещества головного мозга. Характеризуется появлением некротических очагов с локализацией в перивентрикулярных сегментах. Сопровождается возникновением очагов некроза в полушариях, перивентрикулярных областях. Причина морфологических нарушений - гипоксически-ишемическая энцефалопатия. Клинические проявления нозологии:
- Задержка дыхания сразу после рождения;
- Снижение артериального давления;
- Повреждение белого вещества.
Возникновению нозологии у детей способствуют ишемические изменения. Возникает гипоксия, гипокапния, ацидоз у новорожденных детей из-за внутриутробной инфекции, длительных родов. Недостаток кислорода приводит к формированию очагов некроза с локализацией между вентрикулопетальными и вентриклофагальными артериальными ветвями.
Болезнь Канавана-ван-Богарта-Бертрана
Прогрессирующее повреждение нервных клеток головного мозга приводит к нейродегенеративным заболеваниям. Заболевание относится к ряду генетических изменений, приводящих к разрушению оболочки нейронов. Демиелинизация запускается геном, расположенным в семнадцатой хромосоме.
Комплекс морфологических изменений болезни Канавана провоцируется накоплением дефектного белка ASPA из-за недостатка фермента аспартоацилазы.
- Умственная отсталость;
- Потеря моторной активности;
- Дефекты мышечного тонуса;
- Зрительная слепота;
- Трудности удержания головы в физиологической позиции.
Диагностика лейкодистрофии
Первоначальные признаки болезни выявляют клинические специалисты - педиатры, терапевты, неврологи, офтальмологи, отоларингологи.
Генетическое консультирование выявляет аномальные гены, провоцирующие сфинголипидозы головного мозга.
Клинические методы эхо-энцефалографии, нейросонографии выявляют увеличение внутричерепного давления. Исследование цереброспинальной жидкости проводится с целью обнаружения повышенной концентрации протеина.
Нарушение метаболизма выявляется биохимическими анализами крови.
МРТ головного мозга ребенку делают для определения очагов демиелинизации головного мозга. Исследование позволяет верифицировать патологические изменения ранней стадии.
Самый точный способ диагностики - инновационная ДНК-диагностика глобоидно-клеточной, метахроматической лейкодистрофии.
Читайте также: