Болезнь Кимура - клиника, диагностика, лечение
Добавил пользователь Евгений Кузнецов Обновлено: 21.01.2026
Болезнь Кимуры - редкое заболевание, представляющее собой псевдоопухолевый ангио- и лимфопролиферативный процесс, сопровождающийся эозинофилией и повышением уровня иммуноглобулинов Е в сыворотке крови. Преимущественно поражаются мягкие ткани лица, особенно веки и ткани орбиты. В процесс часто вовлекаются слезная и слюнные железы, региональные лимфатические узлы. Этиология заболевания не известна.
Впервые заболевание было описано китайскими врачами в 1937 г. как «гиперпластическая эозинофильная гранулема». Свое название болезнь получила по имени японского исследователя Кимура, который внес большой вклад в изучение «необычного гранулематоза, сочетающегося с гиперпластическими изменениями в лимфоидной ткани и эозинофилией» [1].
Представляем описание редкого клинического наблюдения болезни Кимуры с вовлечением тканей верхнего века правого глаза и правой орбиты у 7-летнего мальчика - ребенка мигрантов из стран Средней Азии.
Месяц назад родители мальчика заметили уплотнение в толще верхнего века правого глаза, которое постепенно увеличивалось. Лечили прогреванием, без эффекта. Ребенок был направлен в нашу клинику. При поступлении общее состояние мальчика удовлетворительное, температура тела 36,50. Обращал на себя внимание полный птоз верхнего века справа. Верхнее веко было резко утолщено, уплотнено. Кожа не изменена. Образование в толще верхнего века без четких границ уходило вглубь орбиты. Пальпация его была безболезненной. Правый глаз экзофтальмирован на 2 мм и смещен книзу на 50. Глаз спокоен (рис. 1).
При магнитно-резонансной томографии в толще верхнего века с переходом на верхне-медиальный отдел правой орбиты определялось образование негомогенной структуры с полостями, накапливающее контраст (рис. 2).
В клиническом анализе крови отмечено снижение сегментоядерных нейтрофилов, небольшой лимфоцитоз и умеренная эозинофилия. Было высказано предположение о глистной инвазии. Образование в псевдокапсуле было удалено из толщи верхнего века и переднего отдела правой орбиты. При гистологическом исследовании удаленного материала выявлена так называемая эозинофильная гранулема: вокруг некротизированных эозинофилов виден гистиоцитарный вал (рис. 3).
Типичный воспалительный инфильтрат, состоял из эозинофилов и плазматических клеток (рис. 4). Определялись очаги гиперплазии лимфоидной ткани и пролиферация сосудов с эпителиоидной трансформацией эндотелия. При иммуногистохимическом исследовании была отмечена мембранная экспрессия CD 31+ вэндотелии с эпителиоидной трансформацией (рис. 5).
На основании данных гистологического исследования биоптата был установлен диагноз болезни Кимуры с вовлечением мягких тканей верхнего века и переднего отдела правой орбиты. Учитывая возраст пациента, от дополнительной лучевой терапии и системного лечения глюкокортикоидами было решено воздержаться. Внешний вид ребенка через 2 мес. после операции представлен на рис. 6. Длительность ремиссии составляет 1,5 года без рецидива удаленного образования и других проявлений заболевания. Пациент находится под динамическим наблюдением.
Клинические проявления болезни Кимуры разнообразны, но преимущественно поражаются мягкие ткани лица и шейные лимфатические узлы [4]. Возможно двустороннее поражение век в виде множественных мелких подкожных узелков [5]. M. Mishima et al. описали выраженную двустороннюю инфильтрацию обоих век. Отек век был настолько значительным и плотным, что молодой мужчина не мог открывать глаза [6]. Часто поражаются ткани орбиты и параорбитальной области с вовлечением слюнных и слезных желез [7].
Существует точка зрения, что лечение болезни Кимуры должно быть комплексным и включать системную терапию глюкокортикоидами и дистанционную лучевую терапию [8]. По мнению других авторов, при лечении болезни Кимуры у детей можно ограничиться только локальным иссечением подкожного образования и оставить ребенка под наблюдением [9].
По данным ряда авторов, при отсутствии эффекта от лечения медленно прогрессирующий рост образований в мягких тканях лица приводит к изъязвлению кожи и обезображиванию [7].
Таким образом, болезнь Кимуры - редкое заболевание на территории европейской части России. Однако с учетом миграции и увеличением числа лиц азиатского происхождения, можно прогнозировать рост встречаемости этого заболевания в повседневной практике врача-офтальмолога. У детей болезнь Кимуры необходимо дифференцировать с воспалительными заболеваниями век, глистной инвазией, рабдомиосаркомой.
Гемолитико-уремический синдром
Гемолитико-уремический синдром - острое патологическое состояние, характеризующееся одновременным развитием микроангиопатической гемолитической анемии, тромбоцитопении и азотемии. Гемолитико-уремический синдром может проявляться кровавой диареей, абдоминальными болями, бледностью и иктеричностью кожи и склер, пастозностью лица, петехиями на коже, анурией, поражением ЦНС, печени, поджелудочной железы и сердца. Диагноз гемолитико-уремического синдрома основан на характерных клинических признаках, результатах общего и биохимического анализа крови и мочи, коагулограммы, бакпосева кала. Лечение гемолитико-уремического синдрома включает патогенетическую, симптоматическую и заместительную терапию.
МКБ-10

Общие сведения
Гемолитико-уремический синдром (болезнь Гассера) - тяжелое полиэтиологическое расстройство, проявляющееся сочетанием неиммунной гемолитической анемии, тромбоцитопении и острой почечной недостаточности. Гемолитико-уремический синдром наблюдается преимущественно у детей грудного и младшего возраста (с 6 мес. до 4 лет), но также встречается у детей старшего возраста и редко у взрослых. Ежегодно в расчете на 100 тыс. детского населения регистрируются 2-3 случая гемолитико-уремического синдрома у детей до 5 лет и 1 случай у детей до 18 лет. Поскольку гемолитико-уремический синдром - одна из частых причин острой почечной недостаточности у детей, то от своевременности его диагностики и лечения зависит исход заболевания.

Причины
У детей частыми причинами гемолитико-уремического синдрома являются острая кишечная инфекция (90%) и инфекции верхних дыхательных путей (10 %).
Основное значение в развитии Д+ гемолитико-уремического синдрома имеет энтерогеморрагическая Е. coli, продуцирующая специфический шига-подобный веротоксин, способный избирательно повреждать эндотелиальные клетки сосудов почек и головного мозга. Наибольшее сродство веротоксина с эндотелием капилляров почек наблюдается у детей первых 3 лет жизни. Веротоксин вызывает эндотелиальный апоптоз и лейкоцитозависимое воспаление, а также запускает цепь патологических реакций, приводящих к гемолизу эритроцитов, агрегации и деструкции тромбоцитов, локальной активации процесса коагуляции и внутрисосудистого отложения фибрина, развитию ДВС-синдрома.
Такими же свойствами обладает шигатоксин S. dysenteriae I типа. Развивающиеся микроциркуляторные нарушения (микроангиопатическая гемолитическая анемия, тромбоцитопения и микротромбозы) приводят к ишемическим изменениям в органах мишенях. При гемолитико-уремическом синдроме на фоне ОКИ наиболее часто поражаются капилляры клубочков почек, что может приводить к снижению скорости гломерулярной фильтрации, ишемии или некрозу клубочков, вторичной дисфункции или некрозу почечных канальцев, при массивном поражении - к ОПН.
Заражение энтерогеморрагической Е. coli может произойти при контакте с животными (кошками, крупным рогатым скотом) или инфицированным человеком; употреблении недостаточно термически обработанных мясных изделий, непастеризованных молочных продуктов, фруктовых соков, загрязненной воды. Для гемолитико-уремического синдрома характерна сезонность: на фоне ОКИ - преимущественно теплое время года (июнь-сентябрь), на фоне вирусных инфекций - зимне-весенний период.
Д- гемолитико-уремический синдром может быть постинфекционным, лекарственным, поствакцинальным, наследственным, связанным с системными заболеваниями соединительной ткани, идиопатическим. В 40% случаев развитие Д- гемолитико-уремического синдрома обусловлено респираторной инфекцией, возбудителем которой является S. pneumoniae, разрушающий мембраны эритроцитов, тромбоцитов и эндотелиоцитов с помощью фермента нейраминидазы. Вирусы ветряной оспы, ВИЧ, гриппа, Эпштейна-Барра, Коксаки также могут быть причиной гемолитико-уремического синдрома.
Установлена связь между развитием гемолитико-уремического синдрома у взрослых и употреблением некоторых медикаментов (циклоспорина А, митомицина С, эстроген - содержащих контрацептивов, противоопухолевых препаратов), трансплантацией костного мозга, злокачественными новообразованиями, системной красной волчанкой и антифосфолипидным синдромом, беременностью. Выявлены семейные случаи гемолитико-уремического синдрома с аутосомным типом наследования обусловленные дефектом системы комплемента, нарушением обмена простациклина, недостаточностью антитромботических факторов и др.
В основе гемолитико-уремического синдрома может лежать активация тромбоцитов иммунными комплексами (например, комплексом антиген - антитело после прививок живыми вакцинами против полиомиелита, против ветряной оспы, против кори, АКДС).
Классификация
В зависимости от этиологии и клинических особенностей разделяют гемолитико-уремический синдром диареяассоциированный - Д+ (типичный) и не ассоциированный с диареей - Д- (спорадический или атипичный). Д+ гемолитико-уремический синдром чаще встречается у детей раннего и младшего возраста, является эндемическим (распространен в Поволжье, Московском регионе); недиарейный - более свойственен детям старшего возраста и взрослым.
По тяжести течения выделяют легкую и тяжелую формы гемолитико-уремического синдрома.
- Легкая форма гемолитико-уремического синдрома подразделяется на тип А (анемия, тромбоцитопения и азотемия) и тип Б (триада симптомов в сочетании с судорожным синдромом или артериальной гипертензией);
- Тяжелая форма - на тип А (триада симптомов в сочетании с анурией длительностью более суток) и тип Б (триада симптомов в сочетании с анурией, артериальной гипертензией и судорожным синдромом).
Симптомы гемолитико-уремического синдрома
В клинической картине гемолитико-уремического синдрома различают продромальный период, разгар заболевания и восстановительный период. Продолжительность продромального периода составляет от 2 до 7 суток. Для него характерно появление признаков поражения ЖКТ или дыхательных путей.
Гемолитико-уремический синдром на фоне ОКИ, вызванной энтеропатогенной Е. coli, имеет ярко выраженную симптоматику. Развиваются симптомы гастроэнтерита или колита (часто кровавая диарея), тошнота, рвота, абдоминальные боли, лихорадка. Постепенно общее состояния ребенка ухудшается, повышенная возбудимость сменяется вялостью.
В период разгара гемолитико-уремического синдрома превалируют проявления гемолитической анемии, тромбоцитопении и ОПН: бледность и иктеричность кожного покрова, склер и слизистых оболочек; пастозность век, голеней; кожный геморрагический синдром в виде петехий или экхимозов, иногда - носовые кровотечения, в тяжелых случаях - снижение диуреза (олигурия или анурия). Тяжесть и продолжительность дизурии зависит от степени и глубины повреждения почек.
Гемолитико-уремический синдром может проявляться полиорганной патологией: поражением ЦНС, печени, поджелудочной железы, сердца, артериальной гипертензией. В 50% случаев гемолитико-уремического синдрома наблюдаются неврологические нарушения: подергивания мышц, гиперрефлексия, децеребрационная ригидность, гемипарезы, судороги, ступор, кома (особенно выраженные у детей первых лет жизни). Выявляются гепатоспленомегалия, кардиомиопатия, тахикардия, аритмия.
Продолжительность гемолитико-уремического синдрома обычно составляет 1-2 недели, затем наступает стабилизация и в 70% случаев - постепенное восстановление нарушенных функций: улучшение выделения мочи, повышение уровня тромбоцитов, нормализация уровня гемоглобина. При тяжелом течении наступает либо летальный исход вследствие экстраренальных поражений, либо формирование ХПН.
Диагностика
Диагноз гемолитико-уремического синдрома основан на выявлении характерных клинических признаков, осложняющих течение ОКИ или ОРВИ: гемолитической анемии, тромбоцитопении, ДВС-синдрома, азотемии.
При гемолитико-уремическом синдроме в крови обнаруживаются анемия, анизоцитоз и полихроматофилия эритроцитов (наличие фрагментированных форм), присутствие свободного гемоглобина, снижение количества тромбоцитов, лейкоцитоз, умеренная непрямая гипербилирубинемия, возрастание уровня мочевины и креатинина, гипонатриемия, гиперкалиемия, ацидоз (в олигоанурической стадии ОПН), гипоальбуминемия.
Моча приобретает коричневато-ржавый цвет, в ней могут появиться фибриновые комки, отмечается гематурия, протеинурия, гемоглобинурия. У детей с ОКИ выполняют бактериологическое исследование кала на выявление штаммов энтеропатогенной Е. coli. При тяжелых неврологических нарушениях возможно проведение КТ головного мозга и люмбальной пункции для исключения кровотечения и менингита.
Дифференциальная диагностика гемолитико-уремического синдрома проводится с неотложными хирургическими состояниями (аппендицитом, кишечной непроходимостью, окклюзией мезентериальных сосудов, перфорацией кишечника, дивертикулом подвздошной кишки), ишемическим колитом, септицемией с ДВС-синдромом, вирусным или бактериальным гастроэнтеритом, тяжелой степенью дегидратации при кишечных токсикозах, тромботической тромбоцитопенией.
Лечение гемолитико-уремического синдрома
Лечение гемолитико-уремического синдрома определяется периодом развития заболевания и тяжестью поражения почечной ткани. Чем раньше ребенок с гемолитико-уремическим синдромом поступает в стационар, тем выше вероятность его успешного и полного излечения. Патогенетическая терапия включает нормализацию агрегатного состояния крови с использованием антиагрегантов, гепаринотерапии; улучшение микроциркуляции (трентал, эуфиллин); коррекцию антиоксидантного статуса (витамины А и Е).
При бактериальной этиологии гемолитико-уремического синдрома назначаются антибиотики широкого спектра действия; при инфекции, вызванной энтеропатогенной Е. coli, прием антибиотиков и препаратов, замедляющих моторику кишечника, не рекомендуется. При олигоанурии показана коррекция водно-электролитных расстройств, подавление реакций метаболического распада и инфекционного процесса. Для коррекции тяжелой анемии используется инфузия эритроцитарной массы.
В половине случаев типичного гемолитико-уремического синдрома необходимо раннее проведение заместительной терапии: обменного плазмафереза, перитонеального диализа или гемодиализа. Гемодиализ проводится ежедневно в течение всего олигоуремического периода. В случае развития терминальной стадии ХПН показана трансплантация почки.
Прогноз
Гемолитико-уремический синдром имеет серьезный прогноз, летальность у маленьких детей во время острой фазы заболевания составляет 3-5%, у 12% развивается терминальная ХПН, у 25% происходит снижение клубочковой фильтрации. Плохой прогноз имеют атипичные наследственные, аутоиммунные и связанные с беременностью формы гемолитико-уремического синдрома.
Классическая форма гемолитико-уремического синдрома у детей раннего возраста с преимущественным поражением почечных клубочков протекает более благоприятно. В случае Д+ гемолитико-уремического синдрома наблюдается лучший исход по сравнению с недиарейным синдромом, сопровождающимся частыми рецидивами и высокой летальностью.
Болезнь Кимура - клиника, диагностика, лечение
Поликлиника №1 ГУП "Медицинский центр управления делами мэра и правительства Москвы"
Болезнь Кимуры: типичный случай редкого заболевания
Журнал: Клиническая дерматология и венерология. 2011;9(1): 21‑25
Чистякова И.А., Соснина Е.А., Манукьян Т.Е. Болезнь Кимуры: типичный случай редкого заболевания. Клиническая дерматология и венерология. 2011;9(1):21‑25.
Chistiakova IA, Sosnina EA, Manuk'ian TE. Kimura's disease: a typical case of a rare disease. Klinicheskaya Dermatologiya i Venerologiya. 2011;9(1):21‑25. (In Russ.).
Авторы наблюдали мужчину 61 года с клинически и гистологически доказанным диагнозом: болезнь Кимуры (синонимы: эозинофильная гранулема мягких тканей, эозинофильная гиперпластическая лимфогранулема, эозинофильный лимфофолликулез, эозинофильная лимфофолликулярная гранулема, эозинофильная лимфоидная гранулема). Приводится дифференциальная диагностика со сходным, но самостоятельным заболеванием - ангиолимфоидной гиперплазией с эозинофилией (Angiolymphoid hyperplasia with eosiniphilia).
Болезнь Кимуры (БК) — хроническое воспалительное заболевание неизвестной этиологии, проявляющееся безболезненной унилатеральной шейной лимфаденопатией и (или) подкожными узлами в области головы и шеи. Первое описание заболевания принадлежит H. Kimm и C. Szeto (1937) под названием «эозинофильная гиперпластическая лимфогранулема». В 1948 г. Т. Кимура отметил выраженные изменения сосудов, и из множественных синонимов у заболевания осталось название — «болезнь Кимуры».

В 1969 г. G. Wells и соавт. [1—5] сообщили о заболевании, которое они назвали ангиолимфоидной гиперплазией с эозинофилией (Angiolymphoid hyperplasia with eosiniphilia - ALHE), отметив его сходство с БК. Многие авторы долгое время считали (а некоторые считают и до сих пор), что речь идет об одном и том же заболевании. Однако на основании клинических и гистопатологических данных ряд исследователей рассматривают их как различные нозологии [6]. Хотя для каждого из этих заболеваний характерно вовлечение сосудов, при ALHE артериовенозная патология имеет вторичный воспалительный характер, а при БК первичным является воспалительный процесс, а пролиферация сосудов вторична [7] (см. таблицу).
Этиология и патогенез БК остаются неизвестными, заболевание манифестирует нарушением пролиферации лимфоидных фолликулов и эндотелия сосудов. Эозинофилия в крови и наличие эозинофилов в воспалительном инфильтрате позволяют предположить, что БК может быть проявлением гиперчувствительности замедленного типа [3].
В результате многочисленных исследований японскими и китайскими учеными [6—9] выявлен ускоренный апоптоз эозинофилов и фагоцитоз их макрофагами в эпителиоидных гранулемах, наличие идентичных клонов Т-клеток в первичных и вторичных очагах поражения, активное участие цитокинов Th-2 и подавление их активности циклоспорином. Роль вирусов Эпштейна—Барра и HHV-8 в патогенезе БК большинством авторов отвергнута. В качестве возможной причины развития БК рассматривается нарушение иммунной регуляции. Предполагается наличие персистирующей антигенной стимуляции после укуса насекомых, существование паразитарной или грибковой инфекции. Однако ни одно из этих предположений не нашло своего подтверждения.
Обычно БК ограничивается поражением кожи, лимфатических узлов и слюнных желез. Сообщалось о корреляции с нефротическим синдромом, особенно у пациентов, находящихся на гемодиализе, и у больных ювенильным артритом. Болезнь Кимуры является доброкачественной, о чем свидетельствуют более чем 10-летние наблюдения А. Kawada [10]. Спонтанные инволюции в злокачественные формы крайне редки, они объясняются или ошибкой в диагнозе или сопутствующими неопластическими заболеваниями. Основной проблемой является медленный прогрессирующий рост очагов и возможный обезображивающий вид элементов, особенно локализующихся в области лица. Большинство описанных случаев заболевания касаются монголоидной расы. Наибольшее количество наблюдений БК имеется в Японии (более 250 случаев в год). Однако БК встречается у людей всех рас, у европейцев - редко. Чаще она появляется у мужчин (соотношение 6:1), причем превалируют молодые пациенты: средний возраст больных 28 лет. Описаны также случаи заболевания детей.
А. Kawada [10], проанализировав данные мировой литературы и собственные наблюдения, выделил несколько типов проявлений на коже и в подкожной жировой клетчатке. На коже встречаются туморозные и узелковоподобные элементы, а в подкожной клетчатке - узлы и узловатости. Узлы встречаются как единичные, так и множественные.
Дифференциальный диагноз БК в первую очередь необходимо проводить с ангиолимфоидной гиперплазией с эозинофилией. Он часто затруднителен из-за наличия сходных симптомов (см. таблицу). Следует исключить также цилиндрому, выбухающую дерматофибросаркому Дарье—Феррана, саркому Капоши, пиогенную гранулему, лимфогранулематоз, лимфому кожи. В 98% случаев при БК выявляется эозинофилия в периферической крови. Количество эозинофилов колеблется от 6 до 80,5%, в большинстве случаев составляет 30—40% и параллельно клиническим проявлениям болезни. Эозинофилия встречается также при исследовании костного мозга. Отмечается увеличение общего IgE в сыворотке крови.
Для подтверждения диагноза необходима биопсия кожи. Дополнительные исследования включают биопсию лимфатических узлов, компьютерную томографию увеличенных лимфоузлов, рентгенографию грудной клетки. Гистологическая картина изменений кожи характеризуется густым эозинофильным инфильтратом и эозинофильными абсцессами в дерме. Пролиферация капилляров не является характерной, но может присутствовать, манифестируя в виде канализированных сосудов с плоскими эндотелиоцитами. В очаге поражений отмечается фиброз разной степени выраженности. Количество коллагеновых волокон увеличено, они гиалинизированы, местами отечны. Эластические волокна не изменяются. Эпидермис, как правило, не поражен, возможно наличие незначительного гиперкератоза.
Изменения, характерные для дермы, могут наблюдаться в подкожной жировой клетчатке. При гистологическом исследовании пораженного лимфатического лимфоузла выявляются отдельные герминативные центры, обнаруживаются гиперпластические лимфоидные фолликулы и интерфолликулярная эозинофилия разной степени выраженности с пролиферацией тонкостенных сосудов. Для исключения сопутствующей почечной недостаточности и/или нефротического синдрома следует контролировать азот мочевины крови, креатинин и уровень белка в моче.
Прогноз заболевания quo ad vitаm благоприятный. Несмотря на проводимое лечение, очаги на коже и в лимфатических узлах часто рецидивируют и медленно увеличиваются в размерах. Не исключается возможность спонтанного регресса.
Нам удалось наблюдать пациента, страдающего БК, которому было проведено лечение с выраженным положительным эффектом.
Больной К., 61 года, поступил в отделение клинической дерматологии «ФГУ ГНЦД Росмедтехнологий» 07.07.08 с направительным диагнозом: «лимфома кожи?» с жалобами на высыпания на коже лица и туловища, незначительный зуд. Первый элемент появился после сильного переохлаждения в октябре 2007 г. на коже груди в виде пятна багрового цвета. В течение 2 мес заболевание приняло распространенный характер: появились новые элементы на коже лица, шеи, в заушной области, на груди, спине, верхних конечностях. Высыпания медленно увеличивались в диаметре, становились плотнее, эритематозные элементы трансформировались в узловатые. Параллельно было выявлено двустороннее увеличение околоушных и передних шейных лимфоузлов справа и слева. При появлении высыпаний в КВД по месту жительства был поставлен диагноз аллергический дерматит, наружное лечение не дало эффекта. В республиканском КВД была взята биопсия кожного узла с плеча, однако результаты гистологического и иммуногистохимического исследований не позволили установить диагноз.
В противотуберкулезном диспансере туберкулез и саркоидоз были исключены. Сопутствующая патология: ИБС. Стенокардия напряжения. ФК. Гипертоническая болезнь III стадии, риск 4.
Локальный статус при поступлении: процесс распространенный (рис. 1). Рисунок 1. (а, б). Множественные плотные узловатости неправильной формы на коже груди и боковых поверхностях туловища у пациента К. Элементы расположены как поверхностно, так и глубоко. На коже лица, спины, груди, верхних конечностей имеются плотные неправильной формы узловатости бурого цвета диаметром до 7 см с нечеткими границами, несколько напоминая саркому Капоши, некоторые элементы располагаются скученно (рис. 2). Рисунок 2. Сливающиеся узловатости на коже живота. Отмечено двустороннее увеличение передних шейных, подмышечных и затылочных лимфоузлов. Они безболезненны, размеры их достигали 2 см в диаметре. Лимфоузлы не спаяны с окружающими тканями и друг с другом.
Данные лабораторных исследований. При поступлении в общем анализе крови эозинофилы составляли 13%, после лечения их количество снизилось до 3%. Общий уровень IgE в сыворотке крови был повышен (более 2000 МЕ/мл при норме 0—87 МЕ/мл). Изменения в общем и биохимическом анализах крови, мочи не определялись. При УЗИ органов брюшной полости обнаружены признаки диффузных изменений паренхимы печени и деформация желчного пузыря.
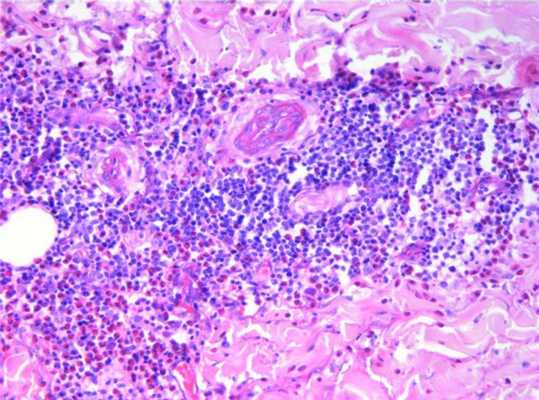
Гистологическое исследование кожи от 15.07.08 (рис. 3): Рисунок 3. Гистологическая картина в пораженной коже: пролиферация сосудов дермы, стенки их утолщены, густые сливающиеся инфильтраты из лимфоцитов и гистиоцитов со значительной примесью эозинофильных гранулоцитов. эпидермис обычной толщины, небольшой кератоз, в устье волосяного фолликула рыхлая роговая пробка. В дерме наблюдается очаговая пролиферация сосудов с утолщенными стенками, вокруг сосудов в разных ее отделах обнаруживаются густые, сливающиеся инфильтраты из лимфоцитов, гистиоцитов со значительной примесью эозинофильных гранулоцитов. Заключение: выявленные признаки носят характер ангиолимфоидной гиперплазии с эозинофилией.
Лечение. Больной получал дезинтоксикационные, антигистаминные препараты, трентал по 100 мг 2 раза в сутки, реаферон в суммарной дозе 18 млн МЕ, неотигазон по 35 мг в сутки, преднизолон 30 мг в сутки по 10 дней. Применялась мазь дермовейт на очаги поражения под окклюзионную повязку. Лечение вызвало значительное улучшение, новых высыпаний не появлялось, все элементы уплостились, плотность узлов уменьшилась, более мелкие регрессировали, оставив гиперпигментированные пятна. Пациент был выписан под наблюдение дерматолога по месту жительства с рекомендацией постепенного снижения дозы преднизолона до полной отмены.
Наблюдавшемуся больному был поставлен диагноз «болезнь Кимуры». В приведенной таблице указаны признаки, на основании которых можно было его подтвердить. В частности, в пользу болезни Кимуры свидетельствовали клиническая картина с узлами, расположенными глубоко в мягких тканях, лимфоаденопатия, эозинофилия в крови и в ткани, значительное повышение сывороточного IgE, гистологическая картина с пролиферацией сосудов в дерме и скоплением эозинофилов вокруг них. Хотя в биоптатах кожи мы не обнаружили структур по типу лимфоидных фолликулов, это не противоречило диагнозу, так как известно, что имеется несколько типов гистологической картины заболевания [10]. В нашей практике это уже пятое наблюдение данного заболевания. Это свидетельствует о наличии его в нашей стране и о необходимости проводить в случае сходных проявлений дифференциально-диагностический поиск.
Болезнь Кимуры
Болезнь Кимуры - заболевание неясной этиологии, сопровождающееся образованием эозинофильных инфильтратов мягких тканей. Они представляют собой плотные узлы различной величины и формы, локализующиеся на голове и шее. Могут иметь красно-бурый оттенок и достигать 5-7 см в диаметре. Заболевание протекает с увеличением лимфатических узлов, без нарушения общего состояния пациента. Болезнь Кимуры диагностируется на основании анализа крови на эозинофилы, определения уровня IgE, исследования биоптатов кожи и лимфоузлов (гистология, РИФ). Опухоль имеет тенденцию к саморазрешению, но по показаниям может быть удалена хирургически. Возможно применение радиотерапии, кортикостероидов, цитостатиков.
Болезнь Кимуры (эозинофильный лимфофолликулез кожи) - редкое дерматологическое заболевание, характеризующееся лимфоидной инфильтрацией дермы доброкачественного генеза с ангиопролиферативной составляющей. Относится к большой группе заболеваний, протекающих с иммунными нарушениями. Впервые с лимфоидно-подобной псевдоопухолью столкнулись китайские врачи в 1937 году. Тогда её расценили как своеобразную гранулёму - локальный островок продуктивно-экссудативного воспаления в соединительной ткани кожи. В 1948 году группа японских учёных впервые подробно описала клинические проявления болезни, не установив ее причину. По имени одного из авторов этого исследования заболевание известно сегодня во всём мире, как болезнь Кимуры.
Позднее на протяжении десятилетий, вплоть до настоящего времени, заболевание изучали английские, голландские, французские, швейцарские дерматологи, относя его то к разновидности ангиолимфоидной гиперплазии с эозинофилией (АЛГЭ), то считая самостоятельной патологией. Научные споры продолжаются по сей день. Доподлинно известно, что относительно высокая заболеваемость эозинофильным лимфофолликулёзом кожи наблюдается в странах Азиатско-Тихоокеанского региона. Среди азиатов болеют преимущественно молодые мужчины (средний возраст 30-32 года). Европейцы практически не страдают этой разновидностью лимфофолликулёза кожи (описаны единичные случаи).
Причины болезни Кимуры
Этиология и патогенез заболевания мало изучены. В настоящее время наиболее распространено мнение о том, что болезнь Кимуры развивается по сценарию реакции гиперчувствительности замедленного типа. Центральным звеном здесь является взаимодействие сенсибилизированных лимфоцитов и антигена, в качестве которого могут выступать как вирусы, так и ткани собственной дермы. Специальные структуры клеточной мембраны лимфоцитов выступают в качестве антител, связывающих эти чужеродные антигены. Чем больше антигенов, тем активнее процесс их связывания.
В ходе реакции антиген-антитело сенсибилизированные лимфоциты меняются морфологически и биохимически. Они приобретают способность инфильтрировать область, где идёт иммунокомплексная реакция через клетки-мишени, которыми в данном случае оказываются клетки кожи. Сопровождается это продуктивно-пролиферативным воспалением. Воспаление прогрессирует, нарушая функции органов, тканей, проницаемость сосудов, способствуя возникновению и развитию болезни Кимуры - доброкачественного лимфопролиферативного процесса с иммунными нарушениями. На иммунозависимый характер патологии указывает нередкое сочетание болезни Кимуры с атопическим дерматитом, поллинозом, бронхиальной астмой, лекарственной аллергией.
Симптомы болезни Кимуры
При болезни Кимуры чаще всего страдают мягкие ткани шеи, головы (околоушной области, глазной орбиты, века), паховой области, подмышечных впадин. Опухолевидные инфильтраты в виде плотных папул шарообразной формы с чёткими контурами, величиной от 1 до 20 мм, или в виде узлов размерами до 7 см располагаются в поверхностных и глубоких слоях кожи, подкожно-жировой клетчатки. Папулы имеют розовато-фиолетовый цвет, гладкую поверхность, иногда покрытую небольшими корочками, склонны к кровотечению. Узлы имеют буроватый оттенок, не кровоточат. После разрешения возможна небольшая остаточная пигментация.
С течением времени в процесс вовлекаются регионарные лимфоузлы, слюнные железы. Кожа над ними не изменена. Лимфатические узлы плотноэластической консистенции, подвижны при пальпации, безболезненны. Отмечены случаи геморрагического поражения слизистых. Общее самочувствие не нарушено. Внутренние органы не страдают. Эстетическая проблема состоит в том, что солитарные или множественные узлы медленно растут, естественным образом обезображивая внешний вид пациента.
Диагностика и лечение болезни Кимуры
Диагностируется патология на основании консультаций врача-дерматолога и аллерголога-иммунолога с привлечением патоморфолога. Исследуют общий анализ крови, в котором выявляется эозинофилия и возможный лейкоцитоз. Характерным иммунологическим маркером болезни Кимуры служит повышение уровня сывороточного IgE. Методом РИФ определяется отложение иммунных комплексов, IgA, IgM, фракции комплемента С3 в стенках сосудов. Гистологически исследуют биоптат кожи и пунктат лимфатических узлов, в которых диагностируют наличие эозинофильных инфильтратов, гранулём с микроабсцессами, пролиферацию сосудов. Дифференциальный диагноз проводят с саркоидозом, лимфомой, саркомой Капоши, сосудистыми опухолями, метастазами в лимфатические узлы, болезнью Микулича.
Тактику терапии диктует распространённость и локализация опухоли. С учётом доброкачественного течения процесса и возможности самостоятельного регресса опухоли, единичные небольшие узлы (до 1 см) подлежат наблюдению у дерматолога. Солитарные и множественные инфильтраты больших размеров, нарушающие качество жизни пациента, удаляются хирургическим путём или воздействуют на них с помощью радиотерапии. При необходимости используют реконструктивно-пластические операции. В лечении узлов до 5 см в диаметре применяют лазер.
При отсутствии показаний к хирургическому вмешательству используют консервативное лечение: кортикостероиды по индивидуальной схеме, включая инъекционную блокаду очагов (бетаметазон), цитостатики (циклоспорин), наружно - кортикостероидные мази. Прогноз благоприятный для жизни, возможны рецидивы заболевания.
Аномалия Кимерли
Аномалия Кимерли — наличие в структуре первого шейного позвонка дополнительной костной дужки, ограничивающей движения позвоночной артерии и вызывающей синдром ее сдавления. Аномалия Кимерли характеризуется головокружением, шумом в ушах, шаткостью походки и расстройством координации, «мушками» и потемнением в глазах, приступами потери сознания и внезапной мышечной слабости. Возможны двигательные и чувствительные расстройства, возникновение ТИА и ишемического инсульта. Диагностируется аномалия Кимерли при рентгенографическом исследовании краниовертебрального перехода, проведении магнитно-резонансной ангиографии, дуплексного сканирования и УЗДГ сосудов головы и шеи. Сосудистые нарушения, которыми сопровождается аномалия Кимерли, подлежат комплексному консервативному лечению. Операция по резекции аномальной дуги производится лишь в тяжелых случаях.


Наряду с аномалией Киари, платибазией и ассимиляцией атланта аномалия Кимерли относится к так называемым краниовертебральным мальформациям — врожденным нарушениям строения области сочленения черепа с первыми шейными позвонками. По некоторым данным аномалия Кимерли встречается у 12-30% людей. Вызывая сдавление позвоночной артерии, аномалия Кимерли сопровождается хронической ишемией в задних отделах мозга. Однако такая ситуация возникает далеко не всегда. Сама по себе аномалия Кимерли не является заболеванием и ее наличие не говорит о том, что именно она вызывает сосудистые нарушения в бассейне позвоночной артерии. При обследовании пациентов, у которых имеется синдром позвоночной артерии и аномалия Кимерли, лишь у 25% обнаруживается причинно-следственная связь между наличием аномалии и развитием синдрома.
Патогенез
Правая и левая позвоночные артерии отходят от соответствующих подключичных артерий. Каждая позвоночная артерия проходит вдоль шейного отдела позвоночника, находясь в канале, образованном отверстиями поперечных отростков его позвонков. Затем она входит в большое затылочное отверстие, попадая таким образом в полость черепа. Позвоночные артерии и их ветви образуют так называемый вертебро-базилярный бассейн, кровоснабжающий часть спинного мозга в шейном отделе позвоночника, мозжечок и ствол мозга. Выходя из шейного канала позвоночная артерия огибает шейный позвонок и горизонтально проходит в широкой костной борозде, где она может свободно перемещаться при движениях головы. Костная дужка, наличием которой характеризуется аномалия Кимерли, расположена над костной бороздой и ограничивает движения позвоночной артерии в этом месте.
Аномалия Кимерли может приводить к развитию синдрома позвоночной артерии двумя путями: за счет срабатывания периваскулярных вегетативно-ирритативных механизмов симпатической иннервации и за счет уменьшенного поступления крови в вертебро-базилярный бассейн из-за механического сдавления позвоночной артерии. Факторами, приводящими к тому, что аномалия Кимерли становиться клинически значимой, являются атеросклероз, поражение сосудистой стенки при васкулитах, шейный спондилоартроз, остеохондроз шейного отдела позвоночника, артериальная гипертензия, наличие других краниовертебральных мальформаций, рубцовый процесс, черепно-мозговая травма или травма позвоночника с повреждениями в области краниовертебрального перехода. К возникновению клинической картины синдрома позвоночной артерии у пациентов с аномалией Кимерли могут приводить травмы плеча, вызывающие повреждение ограниченной костной дужкой позвоночной артерии по хлыстовому механизму.
В неврологии выделяют 2 вида аномалии Кимерли. Первая характеризуется наличием костной дужки, соединяющей суставной отросток атланта с его задней дугой. Во втором варианте аномалия Кимерли представлена костной дужкой между суставным отростком атланта и его поперечным отростком.
Аномалия Кимерли может иметь односторонний характер и ли наблюдаться с обоих сторон первого шейного позвонка. Кроме того, аномалия Кимерли может быть полной и неполной. Полная аномальная костная дужка имеет вид полукольца, неполная костная дужка представляет собой дугообразный вырост.
Симптомы аномалии Кимерли
Клинические проявления, которыми сопровождается аномалия Кимерли, обусловлены уменьшенным притоком крови к задним отделам головного мозга. В результате пациенты испытывают шум в ухе или обоих ушах (свист, звон, гул, шипение), мелькание «мушек» или мерцание «звездочек» перед глазами, внезапное преходящее потемнение в глазах. Указанные симптомы усиливаются при поворотах головы. Поскольку аномалия Кимерли сопровождается нарушением кровоснабжения мозжечка, то возникают головокружение и шаткость походки, которые также могут усугубляться при поворотах головой. На фоне некомфортного положения головы или перенапряжения мышц шеи при аномалии Кимерли у пациентов могут наблюдаться приступы потери сознания. Возможна внезапно возникающая мышечная слабость, приводящая к падению больного без потери сознания.
В случаях более тяжелого течения аномалия Кимерли может сопровождаться головной болью, тремором рук и ног, нистагмом, нарушениями координации, гипестезией и/или мышечной слабостью части лица или туловища, чувствительными и двигательными расстройствами одной или нескольких конечностей. Могут наблюдаться транзиторные ишемические атаки в вертебро-базилярном бассейне. Особо тяжелым осложнением наличия аномалии Кимерли является ишемический инсульт.
При обращении пациента с симптомами недостаточности кровообращения в вертебро-базилярном бассейне головного мозга в первую очередь производят рентгенографию черепа и рентгенографию позвоночника в шейном отделе. Аномалия Кимерли, как правило, достаточно четко визуализируется на боковых рентгенограммах области краниовертебрального перехода. При наличие ушного шума для исключения лор-патологии (кохлеарный неврит, хронический средний отит, лабиринтит) может потребоваться консультация отоларинголога, проведение аудиометрии и других исследований слуха. Производится также исследование вестибулярного анализатора (вестибулометрии, электронистагмографии, стабилографии).
Рентгенография ШОП (боковая проекция). Дополнительное костное кольцо в области задней дужки С1 (аномалия Кимерли)
Поскольку выявленная аномалия Кимерли может не являться причиной синдрома позвоночной артерии, неврологу необходимо исключить другие возможные причины вертебро-базилярной недостаточности. Выявить тромбоз, артерио-венозную мальформацию или аневризму сосудов головного мозга, сдавление сосуда объемным образованием (опухоль, киста или абсцесс головного мозга) способна контрастная ангиография. Определить насколько клинически значима аномалия Кимерли, т. е. степень ее влияния на кровообращение в вертебро-базилярном бассейне, позволяет применение целого ряда гемодинамических исследований: УЗДГ экстракраниальных сосудов, транскраниальной допплерографии, дуплексного сканирования и магнитно-резонансной ангиографии сосудов головного мозга. С их помощью при аномалии Кимерли возможно выявить локализацию сдавления позвоночной артерии и ее зависимость от положения головы и шеи.
Лечение аномалии Кимерли
Аномалия Кимерли требует лечения в случае наличия клинических и гемодинамических признаков нарушения кровообращения в вертебро-базилярном бассейне, связанного именно с данной патологией. Пациенты, у которых имеется аномалия Кимерли, должны соблюдать некоторые меры предосторожности в рамках охранительного режима. При аномалии Кимерли следует избегать форсированных физических нагрузок, резких околозапредельных поворотов головой, стоек на голове, кувырков, спортивных занятий и игр, связанных с ударами головой (борьба, футбол, спортивная гимнастика и пр.). При прохождении массажа или мануальной терапии шейного отдела позвоночника пациенту необходимо предупреждать массажиста и мануального терапевта о том, что у него аномалия Кимерли. Ухудшение состояния пациента является поводом к незамедлительному обращению к врачу.
В большинстве случаев аномалия Кимерли, приводящая к клиническим проявлениям сосудистой недостаточности, подлежит консервативному лечению. Проводится сосудистая терапия направленная на улучшение мозгового кровотока (ницерголин, винпоцетин, винкамин, циннаризин). По показаниям под контролем коагулограммы при аномалии Кимерли применяются препараты, улучшающие реалогические свойства крови (пентоксифиллин). В комплексную терапию включают также антиоксиданты, ноотропы, нейропротекторы и метаболические препараты (пирацетам, препараты гинкго билоба, никотиноил-гамма-аминобутировая кислота, мельдоний).
Аномалия Кимерли на сегодняшний день не является показанием для проведения хирургического лечения. Необходимость в оперативном лечении может возникнуть при декомпенсированном течении синдрома позвоночной артерии, приводящем к выраженной недостаточности кровообращения в вертебро-базилярном бассейне при отсутствии достаточного коллатерального кровоснабжения. Операция при аномалии Кимерли заключается в резекции аномальной дуги и мобилизации позвоночной артерии. В послеоперационном периоде пациентам необходимо ношение воротника Шанца сроком от 2 до 4 недель.
Читайте также:
- Воспалительные гинекологические заболевания. Боли при воспалительных заболеваниях у женщин.
- Гингивит беременных. Диабетический гингивит. Отек десен при гипотиреозе.
- Синдром Охары (Ohara)
- Лучевые признаки дисплазии трикуспидального клапана сердца плода
- Трансплантационный веноокклюзионный синдром (ВОС)