Болезни экспансии нестабильных нуклеотидных повторов. Примеры
Добавил пользователь Валентин П. Обновлено: 28.01.2026
Увеличение числа триплетных повторов (экспансия) в нестабильных участках генома является причиной развития гентингтоноподобных заболеваний: гентингтоноподобное заболевание (ГПЗ) 2 типа (выявление экспансии в гене JPH3), ГПЗ 4 типа (выявление экспансии в гене TBP), дентаторубро-паллидолюисова атрофия (выявление экспансии в гене АТN1).
Синонимы русские
Гентингтоноподобное заболевание 2 типа (ГПЗ 2), ген JPH3, гентингтоноподобное заболевание 4 типа (ГПЗ 4), ген TBP, дентаторубро-паллидолюисова атрофия (ДРПЛА), ген ATN1, экспансия триплетных повторов, генетическое обследование.
Синонимы английские
Huntington disease-like 2 (HDL2), junctophilin 3 (JPH3) gene; huntington disease-like 4, TATA box-binding protein (TBP) gene; dentatorubral-pallidoluysian atrophy (DRPLA), gene atrophin 1 (ATN1), expansion of triplet repeats.
Название гена
Гены JPH3, TBP, ATN1.
Локализация гена на хромосоме
Ген JPH3 расположен в локусе 16q24.2 (ГПЗ2), ген TBP- в локусе 6q27 (ГПЗ4), ген ATN1-в локусе 12p13.31 (ДРПЛА).
Метод исследования
Фрагментный анализ генов JPH3, TBP, ATN1.
Какой биоматериал можно использовать для исследования?
Как правильно подготовиться к исследованию?
- Не курить в течение 30 минут до исследования.
Общая информация об исследовании
Экспансия нуклеотидных повторов в нестабильных участках генома может приводить к развитию болезней экспансии нуклеотидных повторов (БЭНП). Примером БЭНП являются гентингтоноподобные заболевания. Экспансией называется патологическое увеличение количества нуклеотидных последовательностей в нестабильных участках генома, приводящих к нарушению функции кодируемого белка и других протеинов.
Для БЭНП характерен ряд специфических клинических признаков. Для многих из них существует прямая корреляция между количеством повторов в участке экспансии и тяжестью течения болезни, а также обратная корреляция со временем начала первых симптомов. Это связывают с более глубоким нарушением функции белка и с его повышенной нейротоксичностью при увеличении количества нуклеотидных повторов. В связи с этим для каждой формы БЭНП существуют референтные значения количества повторов. При нормальном количестве повторов диагноз исключен; риск развития болезни у пациента отсутствует, а риск развития болезни у последующего поколения крайне низок. При умеренном увеличении количества повторов диагноз исключен; риск развития болезни у пациента отсутствует, но имеется риск развития болезни у последующего поколения. При умеренной экспансии диагноз болезни подтвержден, но наблюдается легкая форма болезни с поздней манифестацией или классическая форма; имеется высокий риск развития заболевания в будущем у бессимптомных носителей, а также высокий риск развития болезни у последующего поколения. При выраженной экспансии диагноз болезни подтвержден; болезнь обязательно разовьется у бессимптомных носителей, а также имеется высокий риск развития болезни у последующего поколения.
Кроме этого для БЭНП характерен феномен антиципации, что связано с динамическим увеличением количества повторов в последующих поколениях при наличии у предка экспансии или умеренного увеличения количества повторов. Антиципацией называют утяжеление клинических проявлений заболевания в последующих поколениях (более ранняя манифестация, быстрое прогрессирование, появление более тяжелых симптомов).
Гентингтоноподобные заболевания наследуются по аутосомно-доминантному типу, то есть имеется 50% риска их развития у потомков.
Гентингтоноподобное заболевание 2 типа (ГПЗ 2) - наследственное прогрессирующее нейродегенеративное заболевание, вызванное экспансией CТG- повторов, во 2А-экзоне гена JPH3, располагающегося на длинном плече 16 хромосомы и кодирующего белок юнктофилин-3 (ЮФ-3).
Клинические проявления и признаки:
ГПЗ 2 является фенокопией болезни Гентингтона.
- Моторные нарушения - гиперкинезы: хорея, дистония, атетоз, тремор, миоклония; окуломоторные нарушения; брадикинезия и ригидность; дисфагия; нарушение равновесия, координации и походки.
- Когнитивные нарушения - снижение исполнительных повседневных функций; деменция.
- Нейропсихические нарушения - депрессия; раздражительность, агрессивность; психозы (бред, галлюцинации, паранойя, сверхценные идеи); апатия; персеверация и обсессивно-компульсивное расстройство.
- У некоторых пациентов с ГПЗ 2 наблюдается повышенное количество акантоцитов в общем анализе крови.
- Инструментальное обследование - МРТ головного мозга: атрофия головки хвостатого ядра, проявляющаяся увеличением фронтальных рогов латеральных желудочков головного мозга. Отношение ширины передних рогов к расстоянию между хвостатыми ядрами (по их краям), измеряемое в поперечной плоскости, уменьшается с 2,2-2,6 до значений, близких к 1,0.
Гентингтоноподобное заболевание 4 типа (ГПЗ 4) - прогрессирующее нейродегенеративное заболевание, вызванное экспансией CAG/CAA-повторов в 3 экзоне гена TBP, располагающегося на коротком плече 6 хромосомы и кодирующего ТАТА-бокс-связывающий белок (ТБС).
- Моторные нарушения - хорея, дистония (блефароспазм, тортиколлис, писчая судорога, дистония стопы).
- Мозжечковая атаксия.
- Паркинсонизм.
- Пирамидальные знаки, ригидность.
- Когнитивные нарушения - снижение исполнительных повседневных функций; деменция.
- Нейропсихические нарушения - депрессия; раздражительность, агрессивность; психозы (бред, галлюцинации, паранойя, сверхценные идеи); апатия; персеверация и обсессивно-компульсивное расстройство.
- Инструментальное обследование - МРТ головного мозга: атрофия коры головного мозга, ствола головного мозга и мозжечка. Отсутствует атрофия хвостатых ядер.
Дентаторубро-паллидолюисова атрофия (ДРПЛА) - наследственное нейродегенеративное прогрессирующее заболевание, вызванное экспансией CAG-повторов в гене ATN1, располагающегося на длинном плече 12 хромосомы и кодирующего белок атрофин-1 (АТФ1).
В детском возрасте:
- миоклонус и эпилептические припадки (тонические, атонические, клонические или тонико-клонические приступы);
- когнитивные нарушения - снижение исполнительных повседневных функций; деменция;
- Инструментальное обследование - МРТ головного мозга: атрофические изменения в мозжечке и стволе головного мозга (особенно в покрышке моста). У пациентов с длительным течением ДРПЛА с началом во взрослом возрасте на МРТ на Т2-взвешенных изображениях выявляются очаги диффузного повышения МР-сигнала в глубоком белом веществе.
Для чего используется исследование?
В соответствии с международными клиническими рекомендациями, генетическое обследование на гентингтоноподобные заболевания проводится при наличии у пациента клинической симптоматики, характерной для данного заболевания, а также родственникам и детям больного.
Когда назначается исследование?
- При подозрении на гентингтоноподобные заболевания;
- при дифференциальной диагностике гиперкинезов;
- при дифференциальной диагностике хореи и дистонии;
- при когнитивных и нейропсихических нарушениях;
- при выявлении характерных изменений при проведении МРТ-исследования головного мозга;
- при раннем выявлении заболевания у родственников;
- при планировании семьи.
Что означают результаты?
Генетическое обследование является основным методом подтверждения одного из перечисленных диагнозов и основано на подсчете числа тройных повторов с помощью метода фрагментного анализа в генах JPH3, TBP, ATN1. Диагностическая значимость обнаруженного числа триплетных повторов представлена в таблице:
Критерии оценки CTG-повторов при ГПЗ 2 типа
Количество CTG-повторов
Диагностическая значимость
6-28 CTG-повторов - норма
29-39 CTG-повторов - клиническая значимость неизвестна
В ряде исследований данное количество CТG-повторов расценивается как норма, но склонное к увеличению в последующих поколениях. Были описаны случаи с атипичной симптоматикой и очень поздним началом ГПЗ 2
≥ 40 CTG-повторов - выраженная тринуклеотидная экспансия
Диагноз "ГПЗ 2" подтвержден
Критерии оценки CAG/CAA-повторов при ГПЗ 4 типа
Количество CAG/CAA-повторов
25-40 CAG/CAA-повторов -норма
41-48 CAG/CAA-повторов - умеренная экспансия
Диагноз "ГПЗ 4" подтвержден. Заболевание развивается позднее и проявляется более легкой симптоматикой
≥ 49 CAG/CAA-повторов - выраженная экспансия
Диагноз "ГПЗ 4" подтвержден
Критерии оценки CAG-повторов при ДРПЛА
Количество CAG-повторов
6-35 CAG-повторов - норма
36-47 CAG-повторов - норма, риск развития у потомков
Норма. Существует риск появления заболевания у последующих поколений
≥ 48 CAG-повторов - выраженная экспансия
Диагноз "ДРПЛА" подтвержден
Что может влиять на результат?
Хотя генетический тест является точным методом лабораторной диагностики, время клинических проявлений заболевания (пенетрантность болезни) зависит от внешней среды, индивидуальных генетических факторов. Для оценки характера наследования у детей и родственников, характера развития заболевания в последующем, назначения лечения рекомендуется получить консультацию специалиста.
Важные замечания
- Для получения заключения по результату обследования необходимо проконсультироваться у клинического генетика.
Кто назначает исследование?
Невролог, психиатр, врач-генетик.
Также рекомендуется
58 Медь в крови
55 Медь в моче
43 Обследования на частые генетические причины мозжечковой атаксии (СЦА 1,2,3,6,7, АФ)
43 Определение экспансии триплетов при спиноцеребеллярной атаксии 1 типа (в гене ATXN1)
43 Определение экспансии триплетов при спиноцеребеллярной атаксии 2 типа (в гене ATXN2)
45 Определение экспансии триплетов при спиноцеребеллярной атаксии 3 типа (в гене ATXN3)
46 Определение экспансии триплетов при спиноцеребеллярной атаксии 6 типа (в гене CACNA1A)
47 Определение экспансии триплетов при спиноцеребеллярной атаксии 7 типа (в гене ATXN7)
Много букв про много букв: Болезни экспансии тринуклеотидных повторов
В основном, наследственные генетические болезни характеризуются однократной мутацией в гене или генах. К таким заболеваниям относят, например, митохондриальные болезни, болезни соматических клеток и другие. Болезни, о которых мы сейчас будем говорить, характеризуются несколько иными мутациями - динамическими. Под словом «динамические» подразумевается то, что с каждым поколением возрастает число копий этих самых тринуклеотидных повторов. Число этих повторов прямо пропорционально тяжести заболевания и обратно пропорционально возрасту начала заболевания. Здесь же мы сталкиваемся с таким термином, как антиципация (нарастание тяжести течения заболевания в ряду поколений, усугубление течения наследственных заболеваний от одного поколения к другому). Прежде чем говорить о самих болезнях, стоило бы поговорить о молекулярных механизмах, лежащих в основе этих заболеваний.
Механизмы
- ДНК-Полимераза сталкивается с прямым повтором и реплицирует его.
- ДНК-полимераза приостанавливает свою работу по какой-либо причине (отсутствие нужного нуклеотида, например).
- Происходит отделение вновь синтезированной цепочки, и на ней образуется «шпилька» из повторов.
- ДНК-Полимераза возобновляет свою работу, но из-за образовавшейся шпильки повторяет введение дезоксирибонуклеотидов в позиции, к которым присоединила их до этого. Именно этот механизм лежит в основе экспансии тринуклеотидных повторов , . Смотри Рис. 1.
Болезни, вызываемые экспансией тринуклеотидных повторов.
В первую очередь следовало бы поговорить о самой известной и уже упомянутой выше болезни - Хорея Гентингтона.
Хорея Гентингтона - одно из самых тяжелых прогрессирующих нейродегенеративных наследственных заболеваний головного мозга. Хорея (chorea; от греческого слова "choreia" - пляска) - форма гиперкинеза, характеризуется непроизвольными, быстрыми, нерегулируемыми движениями, возникающими в различных мышечных группах. Для этого заболевания выяснено соотношение числа повторов CAG в гене IT-15 . Этот ген кодирует белок гентингтин, наибольшая концентрация которого обнаружена в ЦНС. Функция его неизвестна, но многие исследования подтверждают его участие в таких процессах, как взаимодействие с транскрипционными факторами, везикулярный транспорт, заякоривание цитоскелета, а также предотвращение апоптоза в нейронах . Увеличение числа повторов триплетов CAG (Рис. 3), кодирующих аминокислоту глутамин, приводит к образованию полиглутаминовой последовательности - из-за этого происходит неправильное сворачивание белка гентингтина, тем самым, приводит к нарушению всех функций, где участвует этот белок . Рис. 3. Соотношение количества повторов CAG с тяжестью заболевания. По типу наследования Хорея Гентингтона является аутосомно-доминантным заболеванием. Болезнь Гентингтона поражает специфические области мозга. Наиболее заметные ранние изменения затрагивают область базальных ганглиев, называемую полосатым телом, которое состоит из хвостатого ядра и скорлупы . Другие повреждаемые области включают: чёрную субстанцию, 3, 5 и 6 слои коры головного мозга, гиппокамп, клетки Пуркинье в мозжечке, боковые туберальные ядра гипоталамуса и часть таламуса (Рис. 4). Рис. 4. Слева - пораженный Хореей Гентингтона мозг Симптомы. Первичная манифестация заболевания может произойти в любом возрасте, но чаще всего первые симптомы появляются в возрастном промежутке от 35 до 40 лет. Основным клиническим признаком на ранних этапах является хорея, которая проявляется неконтролируемыми и беспорядочными движениями. Изначально развивается по типу незначительных нарушений координации с внезапными, не поддающимися контролю движениями. Эти движения могут быть как резкими, так и замедленными. В последующем к данным признакам присоединяются симптомы нарушения речи, проблемы с процессом глотания и жевания. Из-за постоянного подергивания и нарушения тонуса мышц человек гримасничает, больной испытывает проблемы со сном. Также хотелось бы добавить, что с экспансией повторов CAG связана не только болезнь Гентингтона, но и другие нейродегенеративные заболевания: болезнь Кеннеди (спинобульбарная мышечная атрофия), болезнь Мачадо-Джозефа (спиноцеребеллярная атрофия типа 3).
Синдром Мартина-Белла (синдром ломкой X-хромосомы)
Развитие данного заболевания связано с увеличением числа повторов CGG в гене FMR1 (fragile X mental retardation 1). Белок, кодируемый этим геном, отвечает за нормальное когнитивное развитие . Также этот белок связывается со многими мРНК, отвечающими за синаптическую передачу. При увеличении числа повторов CGG происходит сначала уменьшение экспрессии белка FMR1 (состояние премутации), а затем транскрипционный сайленсинг гена: происходит метилирование участков CGG, а также промотора гена FMR1, гипоацетилирование ассоциированных гистонов и конденсация хроматина (Рис. 5). Рис. 5. Как предполагают, аномальное метилирование промотора гена FMR1 является причиной формирования сайта ломкости Х хромосомы (сайта, где с большей вероятностью произойдет разрыв). Обычно на таких сайтах образуются уже знакомые нам шпильки, которые либо препятствуют, либо замедляют репликацию. Синдром является X-сцепленным доминантным заболеванием с неполной пенетрантностью. Симптомы. Проявления могут быть от легких - отставания в обучении, задержки психомоторного развития до более серьезных когнитивных (или умственных) расстройств, приводящих к умственной отсталости. Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько. Возможно даже развитие аутизма. Наблюдается также гипотония (Рис. 6). Рис. 6. Ребенок с Синдромом Мартина-Белла.
Заключение
Эффективное лечение болезней экспансии тринуклеотидных повторов пока еще не разработано, однако уже есть потенциальные идеи, как эти болезни победить. Создание лекарств оказалось достаточно трудоемким, поэтому ученые обратили свое внимание на генную терапию - сайленсинг мутантных генов путем использования системы CRISPR/Cas9 , антисмысловых олигонуклеотидов и РНК-интерференции . Каждый из этих методов имеет свои достоинства и недостатки в лечении различных болезней экспансии тринуклеотидных повторов. Главным испытанием для них пока еще остается доставка в клетки in vivo. Но нужно сказать, что и в этой области наблюдается прогресс . Автор: Станислав Груздев
X Международная студенческая научная конференция Студенческий научный форум - 2018

Болезни экспансии тринуклеотидных повторов -это патологическое увеличение числа копий внутригенных тандемных последовательностей, состоящих из 3-х нуклеотидов; этот тип мутаций называется также динамическими мутациями.
Основной механизм возникновения мутаций — пробуксовывание (проскальзывание) при репликации. Случаются такие пробуксовки тогда, когда в зоне репликации оказываются тандемные повторы. В общем и целом этот процесс проходит в четыре стадии:
ДНК-Полимераза сталкивается с прямым повтором и реплицирует его.
ДНК-полимераза приостанавливает свою работу по какой-либо причине (отсутствие нужного нуклеотида, например).
Происходит отделение вновь синтезированной цепочки, и на ней образуется «шпилька» из повторов.
ДНК-Полимераза возобновляет свою работу, но из-за образовавшейся шпильки повторяет введение дезоксирибонуклеотидов в позиции, к которым присоединила их до этого. Именно этот механизм лежит в основе экспансии тринуклеотидных повторов [1]
Болезнь Гентингтона (синдром Гентингтона, хорея Гентингтона или Хантингтона) — генетическое заболевание нервной системы, характеризующееся постепенным началом обычно в возрасте 30—50 лет и сочетанием прогрессирующего хореического гиперкинеза и психических расстройств. Заболевание вызывается умножением кодона CAG в гене HTT. Ген HTT, присутствующий у всех людей, кодирует белок хантингтин (Htt). Ген HTT расположен на коротком плече 4-й хромосомы (4p16.3). [] Этот ген содержит в себе участок с повторяющейся последовательностью трёх азотистых оснований — цитозин-аденин-гуанин. Этот ген кодирует 350-kDa белок хантингтин с неизвестной функцией. В гене дикого типа (не мутантного) у разных людей присутствует разное количество CAG-повторов, однако, когда число повторов превышает 36, развивается болезнь. Нейроморфологическая картина характеризуется атрофией стриатумa, а на поздней стадии также атрофией коры головного мозга.
Частота встречаемости заболевания среди населения с европейскими корнями составляет примерно 3-7:100000, и 1:1000000 среди остальных рас
С момента появления первых симптомов продолжительность жизни составляет около 15—20 лет.Смерть обычно происходит не из-за болезни Хантингтона, а из-за сопутствующих ей осложнений, включая пневмонию, заболевания сердца и травмы. Частой причиной смерти является суицид.
Для проведения генетической диагностики болезни Хантингтона необходим забор крови с последующим определением количества повторов ЦАГ в каждом НТТ-аллеле . Положительный результат не подтверждает диагноз, поскольку может быть получен за несколько лет до появления первых симптомов. Однако отрицательный результат однозначно свидетельствует об отсутствии вероятности развития болезни Хантингтона
Болезнь Хантингтона неизлечима, но существует лечение, способное облегчить некоторые симптомы.[4]
Синдром Синдром Мартина-Белл (ломкой Х-хромосомы)
Развитие синдрома связано с экспансией единичных тринуклеотидов (ЦГГ) в Х-хромосоме и приводит к недостаточной экспрессии белка FMR1, который необходим для нормального развития нервной системы. Мутация гена FMR1 приводит к подавлению транскрипции белка FMR1. У здоровых индивидов FMR1, как считают, регулирует значительную популяцию мРНК: FMR1 играет важную роль в обучении и запоминании, а также принимает участие в развитии аксонов, формировании синапсов, появлении и развитии нервных связей.
Первым признаком, который заставляет заподозрить заболевание, является макроорхизм (увеличение размеров яичек) при отсутствии эндокринной патологии. Также есть определённые фенотипические признаки: большая голова с высоким и широким лбом, длинное лицо с увеличенным подбородком, несколько уплощённая средняя часть лица, тупой, слегка клювовидно загнутый кончик носа. Уши большие, иногда оттопыренные, низко расположенные. Кисти и стопы широкие, дистальные фаланги пальцев также широкие, суставы имеют повышенную подвижность. Кожа нередко гиперэластична. Часто встречаются светлоокрашенные радужные оболочки, светлые волосы. Не обязательно встречаются все признаки — могут быть один или несколько.
Неврологическая симптоматика неспецифична, определяется как и у всех детей с умственной отсталостью. Наблюдается некоторая мышечная гипотония, дискоординация движений. Также могут быть глазодвигательные, пирамидные и экстрапирамидные нарушения.
Распространённость непосредственно заболевания — приблизительно 1 из 4000 мужчин и 6000 женщин
Синдром ундины (врожденный центральный гиповентиляционный синдром) (ССHS)— редкое аутосомно-доминатное заболевание, характеризующееся отсутствием автономного контроля над процессом дыхания при отсутствии нервно-мышечных, лёгочных, кардиологических заболеваний или поражения ствола мозга. Впервые данное заболевание описано в 1970-х годах. Заболевание обусловлено мутацией в гене РНОХ2В в локусе 4р12. Описано два вида мутаций: экспансия триплетных повторов GCN (более 25 повторов) и экспансия триплетов, несвязанных с полиаланином. Число повторов триплетов и степень их экспансии коррелирует со степенью тяжести заболевания. Характер наследования аутосомно-доминантный с неполной пенетрантностью. Чаще заболевание обусловлено мутацией de novo, однако описаны семейные случаи. Первые симптомы заболевания как правило появляются сразу после рождения. В случае более мягких форм заболевание может дебютировать в более позднем детском или зрелом возрасте. Кроме того, существует форма заболевания (синдром ROHHAD), при которой помимо гиповентиляции отмечаются гормональные нарушения (ожирение, обусловленное дисфункцией гипоталамуса, гиповентиляцией и вегетативной дисфункцией). При данном заболевании нарушение автономного контроля дыхания приводит к неадекватному ответу на гиперкапнию и гипоксию. Во время бодрствования дыхание сохранно, однако во время сна наблюдается гиповентиляция с нормальной частотой дыхания и поверхностным дыханием во сне. В тяжёлых случаях гиповентиляция присутствует и во время бодрствования, и во время сна. Часто имеются физиологические и анатомические проявления генерализованной дисфункции автономной нервной системы: нарушение регуляции автономной нервной системы, нарушение развития структур, исходящих из «нейрального гребня», нейрокристопатии. Нейрокристопатии включают болезнь Гиршпрунга (врожденный аганглиоз кишечника), ассоциированную с синдромом ундины в 16% случаев.
Синдром ундины неизлечим и не отвечает на лекарственную терапию. Единственный доступный способ борьбы с ним — искусственная вентиляция лёгких. В первые месяцы после рождения ребёнку необходима постоянная вспомогательная вентиляция. Выполняется трахеостомия и СРАР-терапия. На первом году жизни возможно использование вспомогательной вентиляции только во время сна. В возрасте старше 18 месяцев — имплантация стимулятора диафрагмального нерва. [2]
Список литературы:
Острейков И.Ф. Врождённый центральный гиповентиляционный синдром (клинический случай синдрома Ундины) / И.Ф. Острейков, Ю.Ю. Соколов, Ю.Л. Мизерницкий, С.И. Козлова, В.Н. Шеин, А.Л. Заплатников, В.В. / /«Земский врач» №2, 2012.
Gellibolian, R. СTG Triplet Repeats Associated with Neurodegenerative Disease / R. Gellibolian, A. Bacolla, R.G. Wells // Biol. Chem. 1998, 273:5204-5210.
Болезни экспансии тринуклеотидных повторов как актуальная проблема генетики и медицины 21-го века
Болезни экспансии тринуклеотидных повторов (БЭТП) были открыты в конце 20-го века, и до настоящего времени мало изучены и трудно диагностируемы. БЭТП - особая группа наследственных заболеваний, объединенных общим молекулярным механизмом — наличием «динамических мутаций», характеризующихся увеличением определенного порогового уровня числа копий тринуклеотидных повторов в регуляторной или транслируемой части генов. Такой тип мутаций обнаружен пока только в генах человека и является результатом нарушения функции ДНК-полимеразы во время репликации ДНК в митозе или мейозе. Как правило, при возникновении этих мутаций сначала возникает состояние премутации - увеличенное по сравнению с нормой количество тринуклеотидных повторов, но не достаточное для развития заболевания. А затем аллель, содержащий такую «премутацию», становится нестабильным, что в ряде случаев приводит к возникновению полной мутации.
Болезни экспансии классифицируют на две группы в зависимости от локализации тринуклеотидных повторов: в кодирующей части гена (болезнь Кеннеди, хорея Гентингтона) или некодирующей (атаксия Фридрейха, синдром Мартина-Белл). Особенности наследования этих заболеваний, такие как доминантный либо полудоминантный характер, проявления геномного импритинга и процесса антиципации, обуславливают сложности в их анализе и прогнозировании. В настоящее время применяют метод молекулярной диагностики, а также полимеразной цепной реакции в режиме реального времени для диагностики данных заболеваний.
Большинство БЭТП развиваются быстро, характеризуются тяжёлыми гетерогенными проявлениями, что определяет необходимость их дальнейшего и более глубокого изучения.
R-петли: новый штрих нестабильности генома

R-петли представляют собой особые структурные конформации нуклеиновых кислот, которые образуются при гибридизации РНК с комплементарной цепью двухцепочечной ДНК. В результате вытесняется одна из исходных цепей ДНК в области гибридизации по типу петли.
Впервые этот феномен был описан у прокариот более 20 лет назад [1]. Долгое время R-петли рассматривались как побочный продукт транскрипции, не имеющий значительных эффектов и, соответственно, не представляющий особого интереса. Однако развитие геномики и транскриптомики с совершенствованием технических возможностей позволили по-новому взглянуть на забытые области науки. В течение последнего десятилетия все большее число исследований выявили новые особенности строения и функций R-петель в ходе транскрипции (рис. 1) [2]. Будучи ранее «теневыми» структурами со смутным и неясным значением, R-петли все больше обнаруживают свою биологическую и клиническую ценность.
.
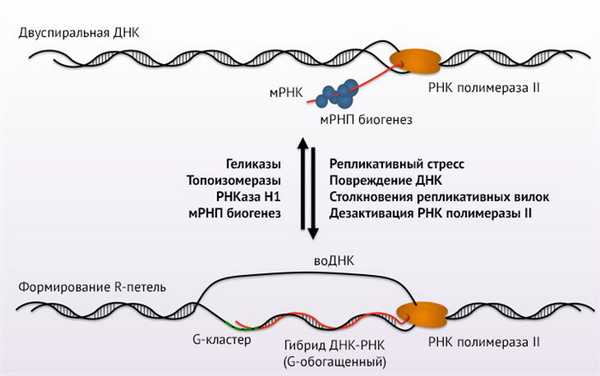
Рисунок 1 | Модель R-петли
Геномное картирование R-петель в клеточных линиях человека, мыши и дрожжей с помощью иммунопреципитации ДНК/РНК с последующим высокопроизводительным секвенированием (DRIP-seq) помогло понять, как и где они образуются. Обнаружено, что R-петли — весьма крупные комплексы, которые занимают до 5 % генома млекопитающих и 8 % генома почкующихся дрожжей [3]. «Территориально» петли предпочитают GC-богатые регионы, такие как типичные промоторы и 3'-концевые области, чтобы защитить эти области от метилирования ДНК. В участках терминации генов они способствуют более эффективной терминации транскрипции.
Многочисленные связи между формированием R-петель и состоянием геномной стабильности стали фундаментом для их роли в развитии патологических состояний. Когда при гибридизация РНК с ДНК-матрицей образуется петля, одинокая вытесненная одноцепочечная ДНК становится крайне уязвимой и нестабильной, повышается ее чувствительность к различным нуклеазам (например, индуцированная активацией цитидин-деаминаза). Это служит фундаментом для формирования двухцепочечных разрывов молекулы ДНК и гипермутаций таких регионов.
Клетки используют разнообразные механизмы для регулирования образования R-петель. К «противопетлевой» системе относится РНКаза Н, которая специфически расщепляет РНК-фрагмент у гибридов РНК-ДНК. Также геликазы, такие как сенатаксин, «разматывают» гибриды РНК-ДНК. Топоизомеры регулируют суперспирализацию (суперскручивание) ДНК: суперспиральная структура способствует компактной упаковке огромной молекулы. Потеря активности топоизомеразы I и ее релаксирующего действия на молекулу приводит к усилению отрицательной (против часовой стрелки) суперспирализации, что способствует раскручиванию двойной спирали. Это может привести к остановке как РНК-полимеразы, так и репликационных вилок, что приведет к разрывам цепей. Заблокированные репликационные вилки являются нестабильными структурами, склонными к рекомбинации. Все эти факторы усиливают нестабильность генома.
Увеличение числа нуклеотидных повторов (экспансия) в нестабильных участках генома способна приводить к различным нарушениям, таким как ингибирование транскрипции и экспрессия токсичных РНК, токсичных полиглутаминовых белков. Значительное количество геноспецифических нуклеотидных экспансий было ассоциировано с возникновением R-петель. Также репликационный стресс и нестабильность генома — фундамент для канцерогенеза. Наиболее вероятно, что этот механизм реализуется при аберрациях в генах репарации ДНК, например, генах-супрессорах BRCA 1 и 2, ответственных за гомологичную репарацию двунитевых разрывов ДНК. Наличие стабильной структуры R-петли позволяет поддерживать открытую локальную конформацию хроматина и усиливает связывание транскрипционного фактора со смещенной одноцепочечной цепью ДНК, что повышает экспрессию виментина (табл.1) 6.
.

Таблица 1 | Заболевания, ассоциированные с нарушением баланса R-петель 7
*Импринтированный ген — уровень экспрессии генетического материала зависит от материнского или отцовского происхождения аллели (например, материнская аллель экспрессируется, отцовская — нет, так как импринтирована) [7].
Молекулярная биология превращается в мощный инструмент медицинской науки. Помимо генома, все более доступны исследования транскриптома и их взаимосвязи. R-петли стали одним из пересмотренных факторов в геномной нестабильности. Множество тайн еще неразгаданы, а каждая новая степень в понимании порождает более глубокие вопросы. На сегодняшний момент все описанные R-петли являются котранскрипционными и, следовательно, образуются в цис-положении. R-петли, образующиеся в транс-положении, вдали от места их транскрипции, были описаны у дрожжей [8]. Возможность транс-положения R-петель и в геноме человека позволит расширить представление об их регуляторных функциях. Также увеличится спектр заболеваний, в патогенез которых вовлечены эти структуры. Кроме того, идентификация молекул-модуляторов R-петель может найти применение в диагностике и терапии таких нозологий.
Читайте также:
- Варианты нормы строения краниовертебрального сочленения
- Лучевые признаки поражения легких при эозинофильном гранулематозе с полиангиитом
- УЗИ при тромбозе воротной вены
- Векторы. Векторы на основе РНК-содержащих вирусов. Векторы на основе ДНК-геномных вирусов. Невирусные векторы.
- Трудоспособность после перенесенного отогенного менингита