Болезни Пелицеуса-Мерцбахера и Александера у детей
Добавил пользователь Владимир З. Обновлено: 22.01.2026
ЛЕЙКОДИСТРОФИИ (греч. leukos белый + дистрофия) — группа наследственных болезней, при которых нарушается процесс миелинизации. Термин был предложен в 1928 г. М. Бильшовским и Геннебергом (R. Henneberg) вместо термина «диффузные склерозы», применявшегося ранее.
Основной тип наследования Л.— аутосомно-рецессивный, возможно и сцепленное с полом рецессивное наследование. Наряду со спорадическими нередко описываются семейные случаи, в отдельных семьях болеют только мальчики. Встречаются случаи кровного родства родителей больных. Родители заболевших Л. фенотипически здоровы.
В основе патогенеза Л. лежит генетически детермированный энзиматический дефект, обусловливающий нарушение миелогенеза и обмена аномально построенного миелина; первоначально нарушается синтез миелина (дисмиелинизация).
Для этих заболеваний характерна атрофия мозга нередко с очагами размягчения различной давности. Общей патоморфол, особенностью Л. является обширное, относительно симметричное дистрофическое поражение белого вещества обоих полушарий головного мозга, мозжечка, спинного мозга. Первично страдают и наибольшим изменениям подвергаются миелиновые оболочки, часто, по в меньшей степени изменяются осевые цилиндры. При гистол. исследовании обнаруживаются множественные сливающиеся очаги демиелинизации пирамидных, экстрапирамидных, мозжечковых проводящих путей и ассоциативных волокон. Демиелинизация может сочетаться со спонгиозом. В белом веществе при биохим, исследовании обнаруживается уменьшение липидов. Продукты мелкозернистого распада миелина — липиды откладываются внутри и вне нервных клеток разных отделов ц. н. с., выявляется реактивная гиперплазия глиозной ткани с образованием глиозных рубцов. Липиды накапливаются во внутренних органах, гл. обр. в почках и печени.
В меньшей степени, чем белое вещество, страдает серое вещество, в к-ром обнаруживаются участки гибели и перерождения нервных клеток. В той или иной степени в патол, процесс вовлекаются периферические нервные волокна. В отличие от лейкоэнцефалитов (см.), ори лейкодистрофии отсутствуют воспалительные инфильтраты.
Чаще всего Л. начинаются в раннем детском возрасте, реже в юношеском, иногда наблюдаются случаи заболевания и у взрослых. До заболевания дети обычно развиваются нормально. Его первым проявлением может быть изменение поведения ребенка — он становится вялым, плаксивым, утрачивает интерес к окружающему, временами отмечается повышенная возбудимость. Вскоре появляются и быстро развиваются очаговые расстройства нервной системы в соответствии с ее множественным поражением. Клин, картина Л. характеризуется большим разнообразием симптоматики. У большинства больных наблюдаются разнообразные двигательные расстройства — спастические, вялые парезы, параличи (см. Параличи, парезы), атаксия (см.), координаторные расстройства, интенционное дрожание (см.), нистагм (см.), эпилептические припадки — общие и джексоновские (см. Эпилепсия, Джексоновская эпилепсия), Экстрапирамидные расстройства — синдром паркинсонизма (см.), гиперкинезы (см.) и часто миоклонические подергивания в разных мышечных группах, в т. ч. в языке. Одним из характерных симптомов является частичная или полная атрофия сосков зрительного нерва (см.), как правило, сопровождающаяся потерей зрения; нарушается слух, нарастает деменция. Наблюдаются разнообразные вегетативные расстройства, приступы гипертермии. В терминальной стадии заболевания возникает децеребрационная ригидность (см.), бульбарные расстройства. Больные погибают в разные сроки от начала заболевания (большей частью через несколько месяцев), находясь в состоянии кахексии, чаще всего от аспирационной пневмонии и других интеркуррентных заболеваний, иногда во время эпилептического статуса.
При Л. в крови, моче, цереброспинальной жидкости обнаруживаются выраженные биохим, изменения, из которых особенно важно изменение содержания липидов, а также аминокислот.
По данным некоторых авторов, в сыворотке крови могут быть обнаружены антитела к миелину, в цереброспинальной жидкости — нерезко выраженная белково-клеточная диссоциация. В материале биопсии кожных нервов возможен распад миелина.
При электроэнцефалографии (см.) выявляются выраженные неспецифические изменения — отсутствие или дезорганизация альфа-ритма, наличие тета и дельта волн. При электромиографии (см.) отмечается замедление скорости проведения импульсов по нервным стволам.
Л. иногда сочетаются с другими наследственными заболеваниями — фенилкетонурией, амавротической идиотией.
Диагноз представляет большие трудности, особенно на ранних стадиях заболевания, нередко ставится только на секции. Вопрос о возможности Л. возникает в случаях многоочагового прогрессирующего поражения ц. н. с. у ребенка при наличии в семье больного этим заболеванием.
В диагностике Л. необходимо комплексное обследование больного, включающее биохим, и морфол, исследование крови, мочи, цереброспинальной жидкости, исследование материала биопсии кожных нервов.
Лечение симптоматическое. Назначают переливание крови и плазмы, тканевые экстракты, противосудорожные, дегидратационные средства, витаминотерапию. При болезни Галлервордена—Шпатца заметное улучшение дает применение альфа-ДОФА.
Если в семье есть больные Л., рекомендуется воздерживаться от деторождения. В отдельных случаях применяется внутриутробная диагностика посредством амниоцентеза (см.).
В зависимости от возраста, в к-ром началось заболевание, неврол. симптоматики, данных биохимического и патологоанатомического исследования выделяют несколько нозол, форм Л., из которых наиболее изучены метахроматическая Л. Гринфилда—Шольца, глобоидно-клеточная Л. (болезнь Краббе), суданофильная Л. (болезнь Пелицеуса—Мерцбахера), болезнь Галлервордена—Шпатца.
Отдельные формы лейко дистрофий
Метахроматическая лейкодистрофии Гринфилда—Шольца (син. поздняя инфантильная лейкодистрофия). Эту болезнь описали Шольц (W. Scholz) в 1925 г. и Гринфилд (Y. G. Greenfield) в 1933 г. Она наследуется по аутосомно-рецессивному типу. В основе патогенеза лежит врожденный дефицит активности энзима цереброзидсульфатазы (арилсульфатазы А), что приводит к прогрессирующей обширной демиелинизации и спонгиозной дистрофии ткани мозга и к отложению в виде гранул продуктов обмена — сульфатидов — в нервных клетках, нервных волокнах, глие разных отделов ц. н. с., гл. обр. подкорковых узлов, а также и в периферических нервах, сетчатке глаз, канальцах почек, лейкоцитах крови.
В ткани мозга уменьшено содержание липидов и относительно повышен уровень сульфатидов. При лаб. исследованиях обнаруживается резкое уменьшение или отсутствие арилсульфатазы в лейкоцитах, увеличение экскреции сульфатидов с мочой.
Характерная морфол, особенность болезни Гринфилда—Шольца состоит в том, что при гистол, исследовании сульфатидные гранулы окрашиваются в коричневый цвет, хотя применяется краситель голубой толуидин (феномен метахромазии); при пробе Аустина, имеющей важное значение для диагностики метахроматической Л., осадок мочи больного при добавлении голубого толуидина окрашивается в коричневый цвет.
Болезнь чаще всего начинается в возрасте 1—3 лет, хотя бывают случаи более раннего и более позднего начала. Первыми симптомами болезни являются атактические расстройства, мышечная атония со снижением сухожильных рефлексов. В дальнейшем появляются спастические парезы, параличи, эпилептические припадки, нарастает деменция, развивается децеребрационная ригидность, кахексия.
Иногда болезнь проявляется в форме полиневропатии (см. Полиневрит) без симптомов поражения ц. н. с. В этих случаях с целью диагностики исследуется материал биопсии кожных нервов, в к-ром обнаруживаются распад миелина и метахроматические гранулы.
Продолжительность жизни большинства больных — от нескольких месяцев до 1 года, иногда до 10 и больше лет. В зависимости от возраста, в к-ром начинается заболевание, выделяют врожденную, детскую, юношескую и взрослую формы метахроматической Л.
Глобоидно-клеточная лейкодистрофия была описана Краббе (К. H. Krabbe) в 1916 г.; названа по имени автора (болезнь Краббе).
Болезнь передается по аутосомно-рецессивному типу и рецессивному, сцепленному с полом. Болеют исключительно мальчики.
Патогенез связывают с недостаточностью или отсутствием бета-галактозидазы, чем обусловливается нарушение метаболизма цереброзидов.
Мелоун (N. I. Malone) с соавторами (1975) исследовал содержание бета-галактозидазы у членов одной семьи. У детей с болезнью Краббе она отсутствовала, а у их родителей — фенотипически здоровых гетерозиготов по гену этой болезни — ее активность была снижена по сравнению с контрольной группой. Это наблюдение представляет интерес с точки зрения выявления гетерозиготности. На аутопсии обнаруживается значительная атрофия головного мозга с участками уплотнения в подкорковом белом веществе вследствие астроцитарного глиоза. При микроскопическом исследовании выявляются наряду с распространенной демиелинизацией очаги спонгиозной дистрофии, гл. обр. в верхних слоях мозговой коры и в U-образных ассоциативных волокнах. Гистопато л. особенностью болезни Краббе являются крупные одно- и много-ядерные глобоидные (шарообразные) клетки адвентициально гистиоцитарного происхождения.
Болезнь начинается в первые месяцы после рождения, в отдельных случаях отмечалось более позднее начало (в 3—6-летнем возрасте). Первые клин, проявления болезни выражаются частыми кратковременными приступами ригидности с опистотонусом (см.), быстро развивается атрофия дисков (сосков) зрительных нервов с утратой зрения, наблюдаются эпилептические припадки, миоклонии в разных мышечных группах, нарушается глотание, развивается деменция, к-рая может доходить до степени идиотии, в терминальной стадии возникает децеребрационная ригидность. Больные обычно погибают спустя несколько месяцев от начала болезни во время эпилептического статуса или от аспирационной пневмонии.
Суданофильная лейкодистрофия была описана Пелицеусом (F. Pelizaeus) в 1885 г., Мерцбахером (L. Merzbacher) в 1908 г.; названа по имени авторов (болезнь Пелицеуса — Мерцбахера). Тип наследования болезни — аутосомно-рецессивный. Оба пола заболевают одинаково часто. Имеются указания о возможности доминантного наследования. При гистол, исследовании мозга обнаруживается наряду с разреженными миелиновыми волокнами немало вполне сохранившихся. Характерным гистопатол. признаком болезни Пелицеуса—Мерцбахера является окрашивание Суданом III в красный цвет периваскулярных отложений распавшегося миелина в разных отделах мозга. При биохим, исследовании отмечается снижение в ткани мозга холестерина, сфингомиелина, цереброзидов.
Болезнь начинается в разных возрастах, чаще в грудном, иногда может быть врожденной. Ее первыми симптомами являются атаксия, нистагм, дрожание головы, интенционное дрожание, скандированная речь, косоглазие. В дальнейшем присоединяются центральные парезы, параличи конечностей, атрофия сосков зрительных нервов, часто только височных половин, в большинстве случаев интеллектуальные нарушения выражены нерезко.
Болезнь Пелицеуса—Мерцбахера имеет наиболее благоприятное течение по сравнению с другими Л. Больные могут жить десятки лет, процесс развивается медленно, возможны ремиссии.
Болезнь Галлервордена — Шпатца (син. прогрессирующая ригидность). В 1922 г. Галлерворден и Шпатц (J. Hallervorden, H. Spatz) описали семью, в к-рой было 5 больных этой болезнью. Тип наследования — аутосомно-рецессивный. Патологоанатомические изменения при болезни Галлервордена—Шпатца наиболее выражены в подкорковых узлах. Их особенностью является увеличенное количество железосодержащего пигмента в бледном шаре и черном веществе головного мозга, благодаря чему эти образования имеют коричневую окраску. В гиперплазированной глиозной ткани встречаются клетки с крупным ядром, напоминающие глию Альцгеймера. При биохим, исследовании ткани мозга обнаруживается нарушение обмена липидов, железа, катехоламинов.
Болезнь чаще начинается в 9 — 10 лет, однако заболевают и взрослые. В клин, картине болезни основное место занимают Экстрапирамидные двигательные расстройства. Первыми симптомами являются гиперкинезы атетозного, торсионнодистонического характера, акинетико-ригидный синдром (см. Дрожательный паралич), в части случаев присоединяются атактические расстройства. Могут наблюдаться изменения в эмоциональной и интеллектуальной сфере различной степени выраженности, большей частью умеренные. Течение болезни медленно прогрессирующее.
Спонгиозная дегенерация белого вещества (син.: болезнь Канавана, болезнь Канавана—Ван-Богарта — Бертрана).
Тип наследования — аутосомно-рецессивный и рецессивный, сцепленный с полом: заболевают почти исключительно мальчики. Болезнь возникает внутриутробно и клинически проявляется уже в первые дни после рождения. При патологоанатомическом исследовании обнаруживается отечность мозговой ткани, спонгиоз и демиелинизация белого вещества головного мозга со снижением цереброзидов и сфингомиелина.
Часто первыми симптомами болезни являются эпилептические припадки; ребенок вял, сонлив, быстро развивается деменция, парез ног, появляется нистагм, косоглазие,, утрачивается слух, а также зрение вследствие атрофии дисков зрительных нервов, расстраивается глотание.
Течение заболевания быстро прогрессирующее, в терминальной стадии наблюдается децеребрационная ригидность.
Продолжительность жизни не превышает двух лет. Больные погибают во время эпилептического статуса или от интеркуррентного заболевания.
Лейкодистрофии с диффузной волокнистой формацией Розенталя (болезнь Александера) встречается очень редко.
При патол, исследовании ц. п. с. обнаруживается увеличение мозга, распространенная демиелинизация с очаговыми размягчениями разной величины, особенно характерно описанное Розенталем (С. Rosenthal) скопление гиалина.
Болезнь начинается в раннем детском возрасте, основными клин, проявлениями ее являются гидроцефалия, быстро нарастающая деменция, эпилептические припадки, спастические параличи.
Продолжительность жизни больных не более двух лет.
Библиография: Бадалян Л. О., Таболин В. А. и Вельтищев Ю.Е. Наследственные болезни у детей, с. 90, М., 1971; Гусев Е. И. Клиническое и биохимическое изучение некоторых наследственных болезней обмена веществ с поражением нервной системы, Журн, невропат, и психиат., т. 71, № 10, с. 1475, 1971; Калмыкова Л. Г. Наследственная гетерогенность болезней нервной системы, с. 52, М., 1976; Маккьюсик В. А. Наследственные признаки человека, пер. с англ., с. 445, М., 1976; Handbook of clinical neurology, ed. by P. J. Vinken a. G. W. Bruyn, v. 10, Amsterdam a. o., 1975; P o s e r G. M. Leukodystrophy and the concept of dysmyelination, Arch. Neurol. (Chic.), v. 4, p. 323, 1961, bibliogr.
Лейкодистрофия
Лейкодистрофия — нейродегенеративное заболевание, обусловленное наследственным нарушением обмена веществ с накоплением в головном и спинном мозге метаболитов, провоцирующих разрушение миелина. Манифестирует в основном в детском возрасте задержкой психомоторного развития, двигательными расстройствами, поражением зрительных и слуховых нервов, гидроцефалией, эпилептическими приступами. Диагностируется лейкодистрофия по данным неврологического статуса, анамнеза, генетических исследований, МРТ или КТ картины головного мозга, биохимических анализов. Лечение симптоматическое. При раннем выявлении и медленном прогрессировании возможна трансплантация пуповинной крови или костного мозга.
МКБ-10
Общие сведения
Лейкодистрофия получила свое название в связи с поражением белого вещества мозга (с греческого leukos — белый). Различают около 60 разновидностей лейкодистрофии, определяющихся видом генной аномалии и возрастом манифестации клинических проявлений. Наряду с отдельными воспалительными поражениями ЦНС (например, лейкоэнцефалитом Шильдера) лейкодистрофия относится к синдрому диффузного склероза мозга. При этом доминирующее поражение миелина сближает ее с демиелинизирующими заболеваниями (рассеянным склерозом, РЭМ и пр.), а отдельные формы можно отнести к липидозам.
К основным формам лейкодистрофии относятся метахроматическая, суданофильная, глобоидно-клеточная, дегенерация Ван-Богарта-Бертрана, болезнь Александера, вариант Галлервордена-Шпатца. Наиболее распространены первые 3 вида лейкодистрофии. Их встречаемость колеблется от 0,4 до 1 случая на 100 тыс. новорожденных. Ряд форм лейкодистрофии являются настолько редкими, что в мировой литературе по неврологии описано всего несколько сотен их клинических наблюдений. В зависимости от возрастного периода, в котором дебютирует лейкодистрофия, каждая ее форма может подразделяться на инфантильный, поздний инфантильный, ювенильный и взрослый вариант.
Причины возникновения лейкодистрофии
В своей основе каждая лейкодистрофия имеет генетическую аномалию определенного фермента. Вид аномалии и локализация генной мутации пока установлены лишь для наиболее встречающихся форм патологии. В большинстве случаев лейкодистрофия имеет аутосомно-рецессивный путь наследственной передачи, однако отдельные ее формы могут наследоваться сцеплено с полом. Кроме того, не одиноки случаи спонтанных мутаций. Генетически детерминированный энзимный дефект ведет к обменным нарушениям (чаще в метаболизме липидов) с отложением определенного метаболита в нервных структурах и отдельных соматических органах, в первую очередь в печени и почках.
Следствием метаболической аномалии является разрушение миелина оболочек нервных стволов и проводящих путей, гибель нейронов с замещением их разрастающейся глиальной тканью. Морфологически лейкодистрофия характеризуется диффузными и симметрично расположенными в полушариях головного мозга зонами гибели миелина, скоплением продуктов миелинового распада, усиленной пролиферацией глии. В отдельных нозологических вариантах лейкодистрофия имеет специфическую морфологическую картину — метахроматическое или суданофильное окрашивание продуктов миелинового распада, скопление в зонах демиелинизации глобоидных клеток и т. п.
Симптомы лейкодистрофии
В большинстве случаев лейкодистрофия дебютирует в раннем детском возрасте. Новорожденные, как правило, выглядят здоровыми. Определенный период они нормально развиваются, а затем постепенно возникают различные неврологические симптомы, отличающиеся неуклонным прогрессированием. Скорость нарастания симптомов тем выше, чем раньше манифестировала лейкодистрофия. Ведущими проявлениями выступают прогрессирующая олигофрения, ухудшение зрения, тугоухость, эписиндром, спастические парезы. Первыми симптомами лейкодистрофии могут быть атаксия, мышечно-тонические расстройства (гипо- или гипертонус, мышечные подергивания), экстрапирамидные проявления, изменения поведения. Затем возникают эпиприступы, бульбарные проявления, снижается слух и зрение, отмечается интеллектуальное снижение с постепенной утратой ранее приобретенных навыков. Сенсорные расстройства не характерны. На поздних этапах развития болезни наблюдаются параличи, выраженная олигофрения, грубое расстройство глотания, амавроз, глухота. В терминальной фазе обычно отмечается децеребрационная ригидность.
Виды лейкодистрофии
Метахроматическая лейкодистрофия в зависимости от манифестации имеет 4 варианта. Врожденный вариант дебютирует в первые 1-3 мес. жизни задержкой развития и судорожным синдромом; дети не достигают возраста 1 года. Позднедетский вариант метахроматической лейкодистрофии начинается в период от 1 до 3 лет с мышечной гипотонии и слабости, атаксии, задержки психического развития (ЗПР). Затем формируется спастическая тетраплегия, афазия, псевдобульбарный синдром. В редких случаях пациенты доживают до 10-летнего возраста. Ювенильный вариант манифестирует в 4-6 лет и длится в среднем 7 лет. Взрослый вариант дебютирует в третьей декаде жизни, иногда позднее, продолжительность жизни пациентов от начала клиники варьирует в пределах 10-20 лет.
Суданофильная лейкодистрофия наследуется сцеплено с Х-хромосомой и имеет несколько разновидностей. Лейкодистрофия Пелицеуса-Мерцбахера может стартовать на 1-ом году жизни или в 3-4 года. Первым признаком является крупноразмашистый нистагм, позже возникает ЗПР, мозжечковая атаксия, гиперкинезы, парезы. Наибольшее прогрессирование происходит в возрасте до 10 лет, затем заболевание принимает замедленное течение с длительными ремиссиями. Пациенты могут жить до зрелого возраста. Адренолейкодистрофия — вариант, при котором лейкодистрофия сочетается с надпочечниковой недостаточностью. Характеризуется прогрессирующим течением с летальным исходом спустя 6-8 лет от начала клиники.
Глобоидно-клеточная лейкодистрофия (болезнь Краббе) — липоидоз с накоплением в очагах демиелинизации галактоцереброзида и образованием больших округлых глобоидных клеток. Раннедетский вариант развивается в первом полугодии жизни с гипервозбудимости и периодической гипертермии, задерживается психомоторное развитие, нарастает тонус мышц, затем развивается спастический тетрапарез, олигофрения, эписиндром, возможен опистотонус. В годовалом возрасте наступает летальный исход. Позднедетский вариант более редкий, манифестирует ухудшением зрения.
Спонгиозная дегенерация Ван-Богарта-Бертрана характеризуется эписиндромом, гиперсомнией, выраженной гидроцефалией с увеличением размеров головы, вызывающей амавроз атрофией зрительных нервов. Резкая внутричерепная гипертензия приводит к расхождению черепных швов, регистрируемому при рентгенографии черепа. Пациенты с этой формой лейкодистрофии погибают до 3-летнего возраста.
Болезнь Александера (лейкодистрофия с волокнистой формацией) обусловлена мутацией гена, ответственного за синтез GFAP белка. В результате происходит накопление в клетках глии аномального GFAP белка, содержащего волокна Розенталя. Неонатальный вариант имеет тяжелое течение с летальным исходом к концу 1-го года. Инфантильный вариант встречается примерно в половине случаев, проявляется в первые 1-2 года жизни ЗПР, затем присоединяются спастические парезы, атаксия, гидроцефалия. Дети погибают спустя несколько лет. Ювенильная лейкодистрофия Александера дебютирует в период от 4-х до 10-летнего возраста, протекает с преимущественно стволовой симптоматикой. Продолжительность жизни колеблется в пределах 10-30 лет. Взрослый вариант отличается поздней манифестацией и относительно медленным течением в пределах 10 и более лет.
Лейкодистрофия Галлервордена-Шпатца чаще всего стартует в 10-летнем возрасте. Проявляется дисфункцией стриопаллидарной системы, затем на фоне гиперкинезов прогрессирует тетрапарез, развивается гемералопия и пигментный ретинит, наблюдается снижение интеллекта, возникают эпиприступы.
Диагностика лейкодистрофии
Диагностический поиск требует привлечения ряда специалистов: невролога, педиатра, медицинского генетика, для диагностики расстройств зрения и слуха — отоларинголога и офтальмолога. Важное значение имеет изучение анамнеза болезни (возраст и симптомы дебюта, последовательность развития клиники) и семейного анамнеза (наличие лейкодистрофии у родственников). Нейросонография через родничок и эхо-энцефалография у пациентов более старшего возраста, как правило, выявляет повышение интракраниального давления. Лейкодистрофия сопровождается существенным увеличением концентрации белка, обусловленным разрушением церебральных клеток, что определяется при исследовании цереброспинальной жидкости.
С целью диагностики вида метаболической аномалии проводится целый ряд биохимических тестов с определением уровня ферментов и накапливающихся метаболитов. Очаги демиелинизации хорошо визуализируются при помощи МРТ, могут быть обнаружены и на КТ головного мозга. Обычно демиелинизация видна на МРТ головного мозга еще до клинической манифестации лейкодистрофии. Благодаря развитию генетики, лейкодистрофия имеет разработанную ДНК-диагностику, а отдельные ее формы (метахроматическая, адренолейкодистрофия, глобоидно-клеточная) — возможность пренатального диагностирования.
Лечение лейкодистрофии
На сегодняшний день лейкодистрофия не имеет эффективных способов терапии, позволяющих купировать прогрессирование симптомов. Проводится симптоматическое лечение — в основном дегидратационная и антиконвульсантная терапия. Единственным методом, способным увеличить продолжительность жизни пациентов с лейкодистрофией и улучшить качество их жизни, является трансплантация пуповинной крови или пересадка костного мозга. Трансплантация приводит к нормализации метаболизма. Однако этот процесс занимает длительное время (от 12 до 24 мес.), в течение которого продолжается прогрессирование лейкодистрофии. Поэтому зачастую тяжелая инвалидизация или гибель пациента наступает даже после успешной трансплантации.
Следует подчеркнуть, что трансплантация никак не влияет на уже развившийся неврологический дефицит, она лишь позволяет приостановить его дальнейшее прогрессирование. В связи с тем, что эффект такого лечения наступает спустя 1-2 года, оно целесообразно в случае ранней доклинической диагностики лейкодистрофии (при соответствующей настороженности родителей рожденного ребенка в связи с наличием подобной патологии в семье) или при медленно прогрессирующем варианте течения. Кроме того, необходимо учитывать, что трансплантация связана с риском ряда серьезных осложнений, таких как отторжение, реакция «трансплантат против хозяина», развитие инфекций.
Лейкодистрофия головного мозга — врожденное заболевание нервной системы. Википедия дает следующее определение данной патологии: «это группа тяжелых наследственных заболеваний, которые характеризуются поражением белого вещества головного мозга спинного». Это нейродегенеративные, достаточно редкие заболевания, для которых характерно нарушение обмена веществ, связанного с врожденным дефектом того или иного фермента, что сопровождается накоплением метаболитов, которые вызывают разрушение миелина белого вещества мозга и прогрессирующую его дегенерацию.
Группа лейкодистрофий включает более 60 форм, которые отличаются видом мутации и возрастом проявления болезни, что влияет на прогноз заболевания. В зависимости от возраста формы могут подразделяться на варианты: инфантильный, ювенильный и взрослый. Все эти заболевания сопровождаются нарушением передачи нервных импульсов в ЦНС, в связи с чем у больных возникают сначала двигательные, а затем и интеллектуальные расстройства, задержка психомоторного развития. По мере разрушения миелина расстройства прогрессируют и за несколько лет у больных развивается тяжелая физическая и психическая деградация. Некоторые формы имеют отличительные симптомы, связанными с поражением и других органов кроме ЦНС. Многие формы настолько редко встречаются, что даже в мировой медицинской литературе описано несколько сотен случаев и наблюдений за больными.
Патогенез
Генетически обусловленный дефект приводит к нарушению обмена (чаще нарушается метаболизм липидов). Это влечет отложение того или иного вещества (метаболита) в тканях. Прежде всего поражается головной мозг и спинной, почки и печень. Токсические метаболиты вызывают разрушение миелина, гибель или атрофию нейронов (клетки нервной ткани), при этом погибшие нейроны замещаются глиальной тканью (соединительная ткань), которая разрастается. Любая лейкодистрофия характеризуется основными патогенетическими процессами: диффузная гибель миелина в полушариях, скопление продуктов, которые образуются после его распада и усиленное разрастание глии. При отдельных формах отмечается специфическая картина — метахроматическое (суданофильное) окрашивание распавшихся продуктов миелина или скопление в зонах распада характерных глобоидных клеток. При болезни Александера в результате мутации в нервной ткани накапливается генетически измененный (мутантный) белок GFAP.
Классификация
Наследственные лейкодистрофии по характеру являются гипомиелинизирующими — миелин изначально не образуется или образуется в недостаточном количестве. Большинство из этих заболеваний наследуется по аутосомно-рецессивному признаку — это значит, что вероятность заболевания у ребенка составляет 25%, если оба родителя-носители гена. В таких случаях с одинаковой частотой в семьях заболевают мальчики и девочки, рожденные от близкородственных браков.
Есть форма, которая характеризуется наследованием, сцепленным с Х хромосомой (адренолейкодистрофия) — она передается Х-хромосомой матери-носительницы болезни. Чаще всего встречаются метахроматическая лейкодистрофия, Пелицеуса-Мерцбахера (или суданофильная), адренолейкодистрофия, болезнь Краббе, Александера и Канавана. Кратко остановимся на этих формах.
Метахроматическая лейкодистрофия — одна из частых и изученных форм. Девочки и мальчики поражаются одинаково часто. При данной патологии отмечается дефицит лизосомного фермента арилсульфатазы. Этот дефект приводит к тому, что в белом веществе головного мозга, периферических нервов и органах (печень, легкие, почки, сердце) накапливаются специфические метаболиты сульфатиды, которые при гистохимическом исследовании дают специфическое метахроматическое окрашивание. Функция внутренних органов не страдает, а в мозговом веществе прогрессируют дегенеративные изменения. Метахроматическая лейкодистрофия по клинике делится на типы: врожденный, поздний инфантильный, ювенильный (ранний и поздний) и взрослый. Все типы протекают с ухудшением двигательной и психической функции, но эти нарушения возникают в разный возрастной период и степень прогрессирования тоже разная.
Врожденная метахроматическая форма развивается до 3-х месяцев и проявляется эпилептическим синдромом и задержкой развития. К ним присоединяется парез и прогрессирующие расстройства глотания. Малыши умирают на первом году жизни.
Ведущий симптом при ювенильной форме (6-10 лет) — нарушение координации движений (мозжечковая атаксия). Больные неустойчивы, ходят неуверенно с широко расставленными ногами, отмечается шаткость при походке. С поздней ювенильной формой больные доживают до взрослого возраста. Взрослая форма проявляется нарушением поведения, из-за чего пациентов часто принимают за психиатрических больных, и снижением когнитивной функции. Постепенно развиваются нарушения движений. Дети редко доживают до десяти лет.
Взрослая форма начинается после пубертата в период до 50-60-лет. Начинается с психических расстройств (психопатия, шизофреноподобный синдром). Также развиваются полинейропатии. Если сравнивать с другими возрастными вариантами, то изменения прогрессируют медленно и заканчивается заболевание тетрапарезом и возрастной деменцией.
Лейкодистрофия Пелицеуса-Мерцбахера связана с генетическим дефектом синтеза апопротеина — белка, который важен для функции клеток олигодендроцитов, участвующих в миелинизации аксонов. Передается по аутосомно-рецессивному признаку: если оба родителя носители мутантного гена в 25% рождается больной ребенок. Второй тип наследования — сцепленная с полом передача (только мальчикам в семье или девочкам).
Проявляется болезнь рано — с 5 до 10 месяцев и имеет медленное развитие. В течение болезни появляется «светлый» промежуток, очень долго длящийся. В связи с этим больные доживают до зрелого возраста и летальный исход возможен только в возрасте 20-30 лет. Болезнь Пелицеуса-Мерцбахера в младенчестве проявляется кивательными движениями головы, блуждающими движениями глаз нистагмом, задержкой развития. Когда малыш начинает ходить, у него появляется атаксия и спастичность конечностей, хореоатетоз (сочетание быстрых и порывистых движений с медленными судорожными). Постепенно развивается нарушение речи и атрофия зрительных нервов. Умственное развитие детей не страдает.
Адренолейкодистрофия связана с мутациями гена АВСD1 и изменением белка ALDP. При генетической диагностике обнаруживают мутации гена ABCD1 (это могут точечные мутации, делеции нуклеотидов и крупные делеции). При этом нарушается транспорт нормальных жирных кислот в миелиновую оболочку и образуются аномальные сверхдлинноцепочечные жирные кислоты в большом количестве, оказывающие токсическое действие на миелин. В результате токсического действия концентрация миелина резко снижается. Помимо поражения ЦНС и периферической нервной системы отмечается поражение надпочечников в виде их недостаточности. В зависимости от преобладания симптомов в клинике выделяют церебральную форму, периферическую и форму только с надпочечниковой недостаточностью.
Церебральная Х-сцепленная адренолейкодистрофия может проявляться в детском возрасте (пик 7-8 лет, детская форма), юношеском (манифестирует в 10-21 лет, юношеская форма) и во взрослом возрасте (30-50 лет, встречается редко). Детская и юношеская формы характеризуются быстрым прогрессированием двигательных нарушений, интеллектуальных и поведения. В детском возрасте наиболее часто встречаются гиперактивное поведение или противоположность ему — аутистическое поведение. У детей возникает агрессивность, снижается память и внимание, возникают проблемы с обучением, нарушается походка и прогрессирует слабоумие.
Значительно реже бывают нарушения зрения и слуха, а также надпочечниковая недостаточность (низкое давление, гиперпигментация кожи, слабость, рвота и тошнота, возникающие периодически). В юношестве прогрессирует умственная отсталость, прогрессивно ухудшается память, развивается спастический тетрапарез (нарушается двигательная функция рук и ног), снижается зрение и слух, появляются судороги. У взрослых заболевание проявляется деменцией и шизофренией. Также у больных нарушается функция глотания и зрения (выпадают поля зрения). Чаще всего прогноз неблагоприятный: несколько лет от начала первых симптомов заболевание прогрессирует и приводит к смерти.
Лейкодистрофия Краббе связана с дефицитом определенного лизосомного фермента, что обусловлено мутациями в гене GALC. При недостатке этого фермента в нервной ткани накапливается галактозилсфингозин — высокотоксичное вещество, вызывающее демиелиенизацию и гибель клеток с образованием в очагах больших глобоидных образований. Болезнь Краббе бывает нескольких подтипов: инфантильный, поздний инфантильный, взрослый и ювенильный. Самая частая из всех форм — инфантильная, она считается классической и составляет до 90% всех случаев.
Первые проявления заболевания наблюдаются в 3-6 месяцев. Сначала у малышей появляется гипервозбудимость, повышенный мышечный тонус, рвота, гастроэзофагеальный рефлюкс, плохая прибавка веса и задержка развития (до этого периода ребенок развивался нормально). На втором этапе заболевания отмечается регресс психомоторного развития (теряются все навыки), появляются судороги и опистотонус, наступает атрофия зрительных нервов (слепота) и прогрессирует гипотрофия (нарушение питания). На третьей стадии заболевания ребенок полностью теряет произвольные движения, и у него возникает децеребрационная поза, которая свидетельствует о тяжелом поражении головного мозга.
У детей развивается слепота, глухота, он не реагирует на внешние раздражители. Поздняя инфантильная развивается не так быстро, но проявления сходны. Ювенильная формы проявляется в 3-8 лет и характеризуется быстрым психомоторным регрессом всех навыков. Взрослая форма проявляется после 8 лет периферической нейропатией (парестезии, снижение силы мышц, повышение их тонуса). Затем развивается нарушение походки и парезы, отмечается регресс психомоторного развития, потеря зрения. Смертельный исход наступает через 3 года после появления первых симптомов.
Лейкодистрофия Канавана обусловлена мутацией гена ASPA, ответственным за синтез фермента аспартоацилазы. Этот фермент расщепляет токсичный N-ацетиласпартат. Накопление токсичного вещества вызывает дегенерацию серого и белого вещества мозга. Тип наследования аутосомно-рецессивный с передачей гена от обоих родителей.
Болезнь Кэнэвэн-ван-Богарта-Бертрана развивается постепенно и сначала ребенок развивается нормально. Через несколько месяцев навыки начинают угнетаться, снижается мышечная активность, увеличивается спастичность мышц, взгляд становится нефиксированным, затрудняется вскармливание из-за нарушений глотания, а также постепенно увеличивается объем головы. Болезнь прогрессирует стремительно, но произвольные движения пока сохраняются. Потом ребенку становится трудно фиксировать голову, развиваются спастические парезы рук и ног. По мере прогрессирования глотательный рефлекс утрачивается и ребенка кормят через пищеводный зонд. Развиваются судорожные припадки, реакции на раздражители отсутствуют и утрачивается зрение — эти симптомы являются пиком заболевания. Развитие симптомов может быть быстрым (в течение нескольких месяцев) и длительным (до 10 лет). Бактериальные или вирусные инфекции — частые осложнения этого заболевания.
Причины
Как было указано выше, причиной этой группы заболеваний являются генные мутации и наследование патологии от родителей. Вид мутаций установлен для наиболее распространенных форм. Мутации выявляются в генах, кодирующих различные лизосомальные ферменты:
- при адренолейкодистрофии выявляются мутации гена АВСD1;
- болезнь Краббе — в гене GALC длинного плеча 14 хромосомы;
- метахроматическая лейкодистрофия — в гене 22 хромосомы, отвечающего за синтез фермента арилсульфатазы А;
- болезнь Канавана-ван-Богарта-Бертранда — аномальный ген ASPA;
- болезнь Пелициуса-Мерцбахера — мутации ген PLP1.
Симптомы
В зависимости от формы лейкодистрофия у ребенка проявляется или в грудном возрасте (первые месяцы) или раннем детстве. До этого времени ребенок развивается нормально и не отстает от сверстников. Постепенно развивающиеся изменения в ткани мозга (головного или спинного) проявляются неврологическими расстройствами.
Первыми появляются двигательные нарушения: ухудшается координация, ребенку трудно удерживать тело в равновесии, он перестает сидеть и ходить. Развивается мышечная слабость, изменяется тонус мышц (чаще всего он повышается, но может снижаться), появляются мышечные подергивания, а потом судорожные приступы. К двигательным расстройствам присоединяются психические нарушения (меняется поведение), ухудшается интеллект и память. Вышеперечисленные симптомы неуклонно прогрессируют. Постепенно ухудшаются слух и зрение. На поздних стадиях развиваются параличи, выраженная олигофрения, слепота, глухота, ребенок теряет возможность глотать. В терминальной стадии наблюдается децеребрационная ригидность. Больной лежит в позе разгибания. При этом шея и позвоночник напряжены, а голова запрокинута, ноги разогнуты и повернуты внутрь, могут быть скрещены. Стопы и руки разогнуты, а пальцы сжаты в кулак. Скорость прогрессирования симптомов заболевания больше, чем раньше проявились первые симптомы. Периферическая нейропатия у ребенка — важная черта метахроматической лейкодистрофии и болезни Краббе, при которых нарушается метаболизм липидов миелина.
Лейкодистрофия краббе и другие виды болезни: симптомы и лечение

Недуг относится к группе тяжелых генетических заболеваний, которое характеризуется прогрессирующим поражением белого вещества в головном мозге.
Вид наследования находится в прямой зависимости от определенной категории лейкодистрофии, которых может быть несколько.
Большая часть видов патологии (например, метахроматическая и глобоидно-клеточная лейкодистрофия) передаются по наследству по аутосомному и рецессивному типу.
Это говорит о том, что возможность появления недуга у человека будет равна 25% в том случае, когда каждый из родителей является носителем болезни.
Для всех типов болезни свойственно начало в детском или даже юношеском возрасте, при этом чаще всего с ним сталкиваются мальчики.
О сути заболевания
У человека, который столкнулся с лейкодистрофией, нарушен нормальный обмен миелина, что приводит к распаду оболочки мозга.
Миелин важен, потому что он формирует оболочку из нервных отростков и является гарантией эффективной передачи сигналов в ЦНС. К тому же, именно благодаря миелину у белого вещества мозга сохраняется его цвет.
Распад оболочки, которая покрывает не только головной мозг, но и нервные волокна, при болезни имеет прогрессирующий и необратимый характер.
При этом, как правило, происходит симметричное поражение полушарий, как головного мозга, так и мозжечка. Серое же вещество мозга оказывается пораженным в гораздо меньшей степени.
Причины и факторы риска
Подобные заболевания чаще проявляются у мальчиков, чем у девочек.
Специалисты отмечают, что в 85% случаев они проявляются в тех сообществах, где обычным явлением стали браки между близкими родственниками. Кроме того, лейкодистрофия могут встречаться с различной частотностью у каждой из народностей.
Например, такая форма как адренолейкодистрофия определяется Х-сцепленным наследованием и поэтому образуется у мальчиков.
Если же мать оказалась носительницей болезни, то вероятность появления недуга у ее сына равна 50%.
Именно поэтому тем семьям, где уже сталкивались с появлением на свет детей с любой формой болезни, перед их рождением в дальнейшем необходимо проконсультироваться с генетиком.
Виды и симптомы заболевания
В настоящее время различают следующие основные формы лейкодистрофии:
- метахроматическая Шольца;
- Краббе;
- Галлевордена - Шпатца;
- Пелицеуса - Мерцбахера;
- болезнь Канавана - ван Богарта - Бертранда;
- болезнь Александера.
Для первого типа характерно форсированное разложение миелина с излишне высоким накоплением активных в токсическом плане продуктов в ЦНС.
Так, недуг напрямую связан с нарушением обмена липидов, последующим их накоплением их в той же ЦНС, а также в периферических нервах и внутренних органах.
Определяют три формы лейкодистрофии в зависимости от времени возникновения симптомов.
Это поздняя инфантильная форма, при которой симптомы возникают в возрасте от одного до двух лет, ювенальная - в возрасте от трех до 10 лет и взрослая - после 16 лет.
Болезнь Краббе
Говоря о лейкодистрофии Краббе, следует отметить, что это острая детская форма недуга. При этом возникает повышенная степень возбудимости, плаксивости, а также могут наблюдаться приступы громкого крика.
В это время также могут появляться и судороги. Очень часто отмечается и повышение температурного режима тела (до 38 градусов и более).
Помимо инфантильной формы, в возрасте от трех месяцев, может образовываться и взрослая форма заболевания.
Галлевордена—Шпатца
Лейкодистрофия Галлевордена—Шпатца представляет собой одну из форм диффузного склероза мозга. На поздних стадиях диагностируется расстройство дыхательной функции и кровообращения.
- носит медленно прогрессирующий характер;
- может продолжаться на протяжении многих лет.
Болезнь Пелицеуса - Мерцбахера

лейкодистрофия пелицеуса мерцбахера
Четвертая из форм недуга может быть передана либо по аутосомно-рецессивному, либо по сцепленному с полом виду наследования.
Начинает проявляться болезнь Пелицеуса - Мерцбахера в раннем возрасте: от пяти до 10 месяцев.
Характеризуется она медленным развитием. В дальнейшем иногда отмечается «светлый» промежуток, который может длиться очень долго. Наиболее редко наблюдаются специфические не прогрессирующие типы заболевания.
Болезнь Канавана
Болезнь Канавана - ван Богарта - Бертранда также следует причислить именно к лейкодистрофиям.
Специалисты уверены, что все процессы, связанные с разложением миелиновой оболочки, начинаются еще во время существования ребенка в утробе. Первичные признаки болезни в 90% случаев проявляются уже на этапе рождения.
Болезнь Александера — сложно выявить и невозможно вылечить, но реально облегчить страдания умирающего человека.
Чем может быть вызвана головная боль напряжения и какие факторы ее провоцируют больше всего? Какой подход к купированию боли даст результат быстрей всего?
Болезнь Александера
Болезнь Александера представляет собой наиболее редкое проявление лейкодистрофии.
Характерными симптомами следует считать прогрессирующую гидроцефалию, то есть развивающееся увеличение размеров головы, которое происходит по причине накопления значительного количества жидкости.
Также следует отметить слабоумие, расстройство двигательных функций и судороги.
Общие проявления для всех видов
Несмотря на то, что у каждой из форм лейкодистрофии есть самостоятельные признаки, возможно отметить и некоторые общие симптомы:
- в первые дни или недели после рождения дети кажутся абсолютно здоровыми и развиваются в соответствии с возрастной группой, симптомы же проявляются постепенно;
- нарушение двигательных функций: усугубление координации движений, проблемы с поддержанием равновесия;
- возникновение мышечной слабости, излишне увеличенный или уменьшенный тонус мышц, подергивания мышц и судороги;
- изменение поведения, постепенное ухудшение памяти и интеллекта.
При этом, чем раньше возникают симптомы, тем быстрее болезнь развивается. Поэтому очень важно правильно поставить диагноз.
Диагноз
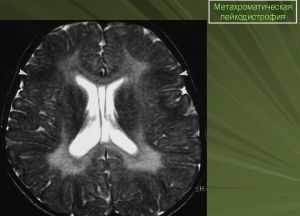
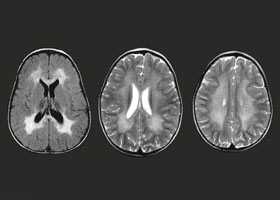
Поражение белого вещества в головном мозгу выявляется при помощи магнитно-резонансной томографии — МРТ.
С целью уточнения вида болезни могут быть использованы различные биохимические тесты. Например, определение соотношения ферментов, синтез или передача которых нарушены.
Может возникнуть необходимость и в других типах исследования, в том числе и молекулярном или генетическом.
Для метахроматической лейкодистрофии и некоторых других форм разработали способы пренатальной диагностики.
Виды лечения
Всего существует два типа лечения лейкодистрофии головного мозга:
- Первый из них - это аллогенная пересадка костного мозга, а также крови из пуповины от донора. В случае успешной пересадки это может привести к стабилизации соотношения дефицитного белка и, как следствие этого, к увеличению не просто продолжительности, но и качества жизни.
- Второй из способов лечения — симптоматический. Он подразумевает избавление или облегчение судорог. При этом используются препараты местного или общего воздействия, которые необходимо подбирать с особенной тщательностью, потому что неадекватное их применение может привести к серьезным осложнениям.
Осложнения
При лейкодистрофии могут возникать и осложнения, в частности ухудшение состояния миелиновых оболочек. Это приводит к:
- замедлению обрабатывания нервных сигналов;
- появлению не просто двигательных расстройств, но и проблем с интеллектом;
- усугублению восприятия сигналов от каждого из органов чувств.
Кроме того, осложнения могут возникать и в рамках трансплантации костного мозга. Речь идет о реакции отторжения трансплантата, что чревато гибелью больного.
Прогноз и выживаемость
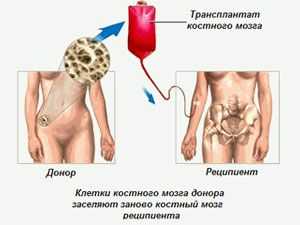
При лейкодистрофии прогноз неблагоприятный. Особенно это касается форм заболевания с ранним возникновением и быстрым развитием признаков.
Однако достаточно часто может оказаться спасительной пересадка костного мозга или, как отмечалось ранее, пуповинной крови.
В случае успешного проведения операции она дает возможность или делает более медленным развитие недуга. Также она дает возможность сохранить не только двигательные, но и интеллектуальные функции.
Как избежать болезни
Профилактические меры сводятся к медицинскому и генетическому консультированию на стадии планирования беременности.
Целью этого является определение риска появления на свет ребенка с подобной патологией. Кроме того, не следует забывать и о пренатальной профилактике, то есть проводимой во время беременности.
Это дает возможности выявить определенные формы патологии, например, метахроматическую.
Что следует понимать?
Лейкодистрофия - это очень сложное заболевание, которое, к тому же быстро прогрессирует. Именно поэтому необходимо уделять особенное внимание диагностике при планировании беременности и на всех ее этапах.
Не менее важно в случае сохранения проблемы осуществить максимально раннюю трансплантацию.
Таким образом, к лечению любой формы болезни следует обходить с особенным вниманием, чтобы сохранить качество жизни ребенка.
Нарушения миелинизации мозга и их отображения на МРТ
В целом, нарушения метаболизма составляют группу многочисленных редко встречающихся дефектов энзимов, часто с аутосомно - рецессивным типом наследствания. Некоторые нарушениям метаболизма приводят к нарушениям миелинизации, называемым дисмиелогенными заболеваниями. Дисмиелогенные заболевания классифицируются по дефекту энзимов следующим образом:
- Нарушения лизосомального накопления - метахроматическая лейкодистрофия, болезнь Краббе (глобоидно-клеточная лейкодистрофия), болезнь Сала, мукополисахаридоз
- Пероксисомальные нарушения - адренолейкодистрофия, связанная с Х-хромосомой; адреномиелонейропатия, синдром Зелвегера
- Нарушения метаболизма аминокислот и органических кислот - болезнь Канавана
- Демиелинизации с неясным метаболическим дефектом - болезнь Пелицеуса-Мерцбахера, болезнь Александера, синдром Съёгрена-Ларссона, лейкоэнцефалопатии (митохондриальная, мегалоцефальная и др.)
Нарушение миелинизации является одним из главных проявлений перечисленных заболеваний. При некоторых из них, например болезни Пелицеуса-Мирцбахера, в процесс вовлекается также серое вещество подкорковых ядер. Клинически дисмиелогенные заболевания проявляются в детском возрасте и начинаются с диффузных симптомов. Затем быстро прогрессируют от небольших парезов и легких нарушений интеллекта до спастичности и деменции. До появления МРТ, диагностика во многих случаях была очень затруднена, так как биопсия мозга выполняется в исключительных случаях. При МРТ в СПб нарушения метаболизма встречаются, в основном, в детской практике. В открытом МРТ дисмиелогенные процессы также могут быть видны.
МРТ головного мозга необычайно высоко чувствительна к дисмиелогенным нарушениям, но не очень специфична в плане дифференциальной диагностики внутри группы. При всех этих патологиях на Т2-взвешенных МРТ и МРТ, отражающих протонную плотность, видны гиперинтенсивные поля в белом веществе.
При адренолейкодистрофии на МРТ процесс быстро распространяется из центра затылочных долей вперед через внутреннюю и наружную капсулы, полуовальные центры и на подкорковое белое вещество. При МРТ головного мозга с контрастированием с контрастировании по краям поражения иногда бывает усиление, что, соответствует активному процессу разрушения миелина. Низкий сигнал на МРТ в связи с депонированием железа дают зрительные бугры, скорлупа и хвостатые ядра. При КТ могут наблюдаться участки кальцификации. Вариант заболевания, встречающийся у взрослых, адреномиелонейропатия, течет медленнее и сопровождается разрушением миелина (демиелинизацией) спинного мозга, что отображается на МРТ, периферических нервов, а также дисфункцией надпочечников.
Морфологические признаки болезни Краббе (глобоидный тип лейкодистрофии) отличается поражением зрительных бугров и лучистого венца. При МРТ описано также распространение на базальные ганглии.
Болезнь Александера (лейкодистрофия Розенталя) преимущественно поражает центральные участки лобных долей и сочетается с энцефаломегалией. В лобных долях нередко встречаются кисты. Часто поражается продолговатый мозг, где очаг на МРТ надо отличать от опухоли.
При болезни Канавана (спонгинозная дегенерация белого вещества, болезнь Канавана - ван Богарта - Бертрана) поражение начинается с затылочных долей и распространяется вплоть до кортико-медуллярного перехода . Последними вовлекаются височные доли. Наблюдаются также энцефаломегалия и атрофия мозжечка.
МРТ находки плохо коррелируют с клинической тяжестью течения заболевания. Роль МРТ заключается не в высокоспецифическом диагнозе (что требует биопсии) или оценке тяжести процесса, а в дифференциальной диагностике с заболеваниями, имеющими сходную клиническую симптоматику.
Читайте также:
- УЗИ при варфариновой (кумадиновой) эмбриопатии у плода
- Оси ЭКГ отведений от конечностей. Векторный анализ потенциалов ЭКГ
- Терапия низкого артериального давления (гипотензии) при отравлении
- Перитонеовенозный шунт LeVeen. Описание особенностей перитонеовенозного шунта.
- Общее описание идиопатических интерстициальных пневмоний