Децитабин в лечении миелодиспластического синдрома (МДС) - эффективность
Добавил пользователь Владимир З. Обновлено: 02.02.2026
Вам поставили диагноз: Миелодиспластические синдромы
Наверняка Вы задаётесь вопросом: что же теперь делать?
Подобный диагноз всегда делит жизнь на «до» и «после». Все эмоциональные ресурсы пациента и его родных брошены на переживания и страх. Но именно в этот момент необходимо изменить вектор «за что» на вектор «что можно сделать». Очень часто пациенты чувствуют себя безгранично одинокими вначале пути. Но вы должны понимать - вы не одни.
Предлагаем вашему вниманию краткий, но очень подробный обзор данного заболевания.
Его подготовили высоко квалифицированные специалисты Отдела лекарственного лечения злокачественных новообразований МРНЦ имени А.Ф. Цыба и Отдела лекарственного лечения опухолей МНИОИ имени П.А. Герцена - филиалов ФГБУ «НМИЦ радиологии» Минздрава России под редакцией заведующих отделами, д.м.н. ФАЛАЛЕЕВОЙ Н.А. и д.м.н. ФЕДЕНКО А.А.
Филиалы и отделения, где лечат миелодиспластические синдромы
МНИОИ им. П.А. Герцена - филиал ФГБУ «НМИЦ радиологии» Минздрава России.
Отдел лекарственного лечения опухолей
Заведующий отделом, д.м.н. ФЕДЕНКО Александр Александрович
тел: 8 (494) 150 11 22
МРНЦ им. А.Ф. Цыба - филиал ФГБУ «НМИЦ радиологии» Минздрава России.
Отдел лекарственного лечения злокачественных новообразований
Заведующая отделом, д.м.н. ФАЛАЛЕЕВА Наталья Александровна
тел: 8 (484) 399 - 31-30
Миелодиспластические синдромы
(МДС) - разнородная группа заболеваний системы кроветворения, сопровождающаяся нарушением созревания кроветворных клеток с нарушениями их строения (дисплазией) и функции, а также повышенным риском развития острого лейкоза. МДС чаще всего сопровождаются снижением показателей общего анализа крови - цитопенией. Снижение уровня гемоглобина и числа эритроцитов обозначают термином анемия; числа лейкоцитов/нейтрофилов - лейкопенией/нейтропенией; уровня тромбоцитов - тробоцитопенией. Изредка может быть и увеличение числа лейкоцитов и/или тромбоцитов.
Ежегодная заболеваемость МДС в среднем составляет 3-4 случая на 100000 населения и увеличивается с возрастом. Основной контингент больных МДС представлен пожилыми людьми (средний возраст - 70 лет). В отдельную группу относят МДС, развившиеся после химиотерапии и/или лучевой терапии предшествующих заболеваний (преимущественно онкологических). Они составляют 10-15% от ежегодно выявляемых случаев МДС и называются вторичными МДС.
В связи с гетерогенностью заболевания возникает вопрос о выборе оптимальной терапии. Одним из основных факторов для решения этой задачи является определение прогностической группы (группы риска), к которой относится конкретный больной.
Каждый прогностический признак оценивается в баллах. В соответствии с суммарным числом баллов больных объединяют в 4 группы: низкого (0 баллов), промежуточного-1 (0,5-1,0 балл), промежуточного-2 (1,5-2,0 балла) и высокого (2,5 балла и выше) риска. Первые 2 группы (низкого и промежуточного-1 риска) характеризуются принципиально благоприятным прогнозом, а 2 остальные (промежуточного-2 и высокого риска) — неблагоприятным прогнозом. Определение прогноза необходимо для выбора лечения.
Существует несколько основных направлений в лечении МДС:
Химиотерапия: на протяжении последних 20 лет для лечения больных МДС с увеличенным числом бластных клеток, то есть преимущественно в группе принципиально неблагоприятного прогноза, используются схемы терапии острого миелоидного лейкоза. Применение стандартной химиотерапии позволяет получить высокую частоту полных ремиссий - 50-70%. Однако продолжительность ремиссий относительно короткая (как правило, менее 1,5 лет), а лечение сопровождается высокой токсичностью.
Трансплантация гемопоэтических стволовых клеток (трансплантация костного мозга): единственным методом лечения, позволяющим существенно увеличить продолжительность жизни больных МДС, является аллогенная трансплантация гемопоэтических стволовых клеток (аллоТГСК). Аллогенная ТГСК также позволяет получить наилучшие результаты по сравнению с другими методами лечения при вторичных МДС. Однако выполнение аллоТГСК не всегда возможно в связи с пожилым возрастом большинства больных и отсутствием идентичного родственного донора.
Иммуносупрессивная терапия: использование иммуносупрессивных препаратов, таких как циклоспорин А, демонстрирует наибольшую активность при гипопластическом варианте МДС, при отсутствии увеличения числа бластных клеток в костном мозге, нормальном кариотипе (без отклонений в наборе хромосом), наличии лимфоидных узелков (очаговых скоплений лимфоидных клеток) в трепанобиоптате, наличии клона клеток, составляющих субстрат пароксизмальной ночной гемоглобинурии (ПНГ-клон), и у HLA-DR-15 - позитивных молодых больных.
Терапия колониестимулирующими препаратами: эритропоэз-стимулирующие препараты - эритропоэтины (препараты, направленные на лечение анемии) фигурируют практически во всех рекомендациях по лечению МДС. Практически все специалисты сходятся во мнении о необходимости определения уровня эндогенного (собственного, вырабатываемого организмом) эритропоэтина до начала лечения. При уровне эндогенного эритропоэтина свыше 500 ед/л лечение эритропоэтином не показано, большая эффективность наблюдается при уровне эндогенного эритропоэтина в пределах 200 ед/л.
Сопроводительная (симптоматическая) терапия : к сопроводительной терапии при МДС относят гемотрансфузии (переливания) эритроцитов и тромбоцитов, антимикробную терапию, в том числе в комбинации с Г-КСФ или ГМ-КСФ, использование комплексонов (хелаторов) железа.
Программа лечения МДС основана на риск-адаптированном подходе в зависимости от групп прогноза, возраста, общего состояния больного. Иными словами, назначается индивидуальная терапия в зависимости от результатов полностью проведенного обследования. В некоторых случаях, когда проявления заболевания незначительны, может быть предложено только наблюдение у врача. Лечение некоторыми препаратами может проводиться как в условиях стационара, так и дома.
Филиалы и отделения Центра, в которых лечат миелодиспластические синдромы
ФГБУ «НМИЦ радиологии» Минздрава России обладает всеми необходимыми технологиями лучевого, химиотерапевтического и хирургического лечения, включая расширенные и комбинированные операции. Все это позволяет выполнить необходимые этапы лечения в рамках одного Центра, что исключительно удобно для пациентов.
Отдел лекарственного лечения злокачественных новообразований МРНЦ имени А.Ф. Цыба - филиал ФГБУ «НМИЦ радиологии» Минздрава России
Заведующая отделом, д.м.н. ФАЛАЛЕЕВА Наталья Александровна
8 (484) 399 - 31-30, г. Обнинск, Калужской области
Отдел лекарственного лечения опухолей МНИОИ имени П.А. Герцена -филиал ФГБУ «НМИЦ радиологии» Минздрава России
Заведующий отделом, д.м.н. ФЕДЕНКО Александр Александрович
Децитабин в лечении миелодиспластического синдрома (МДС) - эффективность
1 ГОУ ВПО «Красноярский государственный медицинский университет им проф. В.Ф. Войно-Ясенецкого», Красноярск, Россия
Приведенный опыт лечения больной с миелодиспластическим синдромом высокотехнологичным препаратом, ингибирующим метилирование ДНК, - децитабином (Дакоген) представляет собой интерес в связи с тем, что ведение больных с миелодиспластическим синдромом все еще является нерешенной задачей. Подход к лечению МДС должен быть индивидуальным и основываться на группе риска больного, возрасте, соматическом статусе. При выявлении цитогенетических поломок (в частности, делеции 5 q хромосомы) преимущество в отношении повышения качества жизни даёт эпигенетическая терапия децитабином. Лечение дакогеном действительно обладает клинической эффективностью, вызывая высокий уровень общего ответа на терапию у больной миелодиспластическим синдромом, так как позволяет исключить потребность в гемотрансфузиях, снизить количество осложнений, связанных с переливанием донорской крови, сократить потребность в химиотерапии и поддерживающей терапии, повысить выживаемость больной, в данном случае на 14 месяцев, и тем самым увеличить время до прогрессирования заболевания.
2. Грицаев С.В., Абдулкадыров К.М. Некоторые аспекты диагностики, прогнозирования и лечения миелодиспластического синдрома // Вестник гематологии. - 2008. - №3. - С. 45-53.
3. Грицаев С.В., Бессмельцев С.С. и др. Эффективность рекомбинантного эритропоэтина (Эпокрин) у больных миелодиспластическим синдромом // Вестник гематологии. - 2005. - № 9. - С. 27-32.
4. Грицаев С.В., Мартынкевич И.С., Абдулкадыров К.М. Прогностический потенциал морфологических и цитогенетических показателей у больных миелодиспластическим синдромом // Тер. Архив. - 2005. - № 7. - С. 22-27.
5. Грицаев С.В., Тиранова С.А., Мартынкевич И.С. и др. Особенности иммуноупрессивной терапии больных миелодиспластическим синдромом // Вестник гематологии. - 2005. - № 1. - С. 43-47.
6. Garcia-Manero G. Modifying the epigenome as a therapeutic strategy in myelodysplasia // Hematology (Am. Soc. Hematol. Educ. Program.). - 2007. - P. 405-411.
7. Germing U. et al. No increase in age-specific incidence of methylation // Hematologica. - 2004. - Vol. 89. - № 8. - P. 905-910.
9. Greenberg P., Cox C., LeBeau MM et al. International scoring system for evaluating prognosis in myeloblastic syndromes // Blood. - 1997. - Vol. 89. - P. 2088-2089.
10. Cazzola M., Malcovati I. Myelodysplastic syndromes - coping with ineffective hematopoiesis // Engl. J Med. - 2005. - Vol. 52. - № 3. - P. 536-538.
11. Kantarjian H., Issa J.-P., Rosenfeld C. et al. Decitabine improves patient outcomes in myelodysplastic syndromes. Results of a phase III randomized study // Cancer. - 2006. - Vol. 106. - P. 1794-1803.
13. Leone G., Teofili L., Voso M.T., Lubbert M. DNA methylation and demethylation drugs in myelodysplastic syndrome and secondary leukemias // Hematologica. - 2006. - Vol. 87. - P. 1324-1341.
Миелодиспластический синдром (МДС) - группа гетерогенных, клонально обусловленных, приобретенных заболеваний крови опухолевой природы, характеризующаяся нарушением функции костного мозга, сопровождающимся последствиями угнетения эритроидного, миелоидного и тромбоцитарного ростков, что выражается соответствующей цитопенией (анемия, нейтропения, тромбоцитопения или их комбинации). Диагностика и лечение МДС является до сих пор нерешённой проблемой в гематологии. Отличительной особенностью МДС является необратимый, опухолевый характер, выявленных при МДС изменений крови и отчётливая тенденция к трансформации в острый лейкоз. Риск перехода в острый лейкоз по данным разных авторов составляет до 30% [2; 4; 9].
В основе МДС лежат различные генетические аномалии, а также аномальное метилирование ДНК, приводящее к торможению экспрессии генов-онкосупрессоров, что в свою очередь приводит к множественным нарушениям клеточного цикла и дифференцировки клеток [1; 2].
Заболевание характеризуется низкой продолжительностью жизни пациентов и быстрой трансформацией в острый миелобластный лейкоз, что определяет его высокую социальную значимость. При отсутствии лечения общий срок выживаемости составляет в среднем 0,4 года для больных с высоким риском прогрессирования и 5,7 года при низком риске прогрессирования. Срок до перехода в ОМЛ у 25% больных этих групп составляет соответственно 0,2 и 9,4 года [9]. Причинами смерти больных с МДС являются последствия цитопенических нарушений - в частности инфекции, тяжелые кровотечения, а также трансформация в ОМЛ [4; 12].
С совершенствованием методов диагностики МДС становится все более актуальным заболеванием в онко-гематологической практике. По данным западных авторов, заболеваемость МДС составляет 5 случаев на 100 000 населения в год [7]. По оценкам ведущих специалистов в России, в настоящее время в РФ насчитывается около 2,5 тысяч пациентов с МДС, средний возраст которых приближается к 40 годам. Однако централизованного учета больных МДС не ведется, а выявляемость заболевания остается на низком уровне. Трудности выявления МДС связаны с отсутствием типичной клинической картины и со сложной диагностикой заболевания, которая, помимо обычного клинического обследования, проводящегося при подозрении на любое онкогематологическое заболевание, включает обязательный морфологический анализ и цитогенетическое исследование костного мозга [2; 4; 8].
По данным североамериканского исследования III Фазы медиана выживаемости и отсутствия прогрессии в ОМЛ среди ответивших на терапию дакогеном составила 17,5 месяцев против 9,8 месяцев в контрольной группе, получавшей поддерживающую терапию (трансфузии эритроцитарной массы, тромбоконцентрата, рекомбинантный эритропоэтин) [11].
Приводим собственный опыт лечения больной с миелодиспластическим синдромом препаратом, ингибирующим метилирование ДНК, - децитабином( Дакоген).
Пациентка Л., 58 лет, наблюдалась в поликлинике у гематолога в течение 6 лет (2002-2008 гг.). В 2002 году во время медосмотра выявлены низкие цифры гемоглобина (в пределах 100-90г/л. В течение 5 лет периодически назначались препараты железа, витамин В12. С 2005 года гемоглобин снизился до 70-80 г/л, на лечении цифры не корригировались. В стернальном пунктате выявлены умеренные изменения - пунктат гиперклеточный, за счет равномерного увеличения всех ростков миелопоеза с нарушением вызревания. Красный росток с выраженным мегалобластоидным компонентом, бластов 1,6%. Проводился поиск причины анемического синдрома (онкопоиск, аутоиммунный процесс и т.д.), но патологии, которая могла бы привести к упорной, рефрактерной анемии, не выявлено.
В апреле 2008 года для уточнения диагноза и лечения больная была госпитализирована в гематологическое отделение ГКБ № 7, где при повторном исследовании костного мозга на фоне сужения красного ростка найдено 30% сидеробластов, кольцевидные формы 18%. Проведена трепанобиопсия - жировая ткань и костномозговые элементы в соотношении 2:1, 3:1, т.е. отмечается уменьшение кроветворных элементов. Данных за миело- и лимфопролиферацию нет. У больной диагностирован миелодиспластический синдром по типу рефрактерной анемии с кольцевидными сидеробластами, промежуточная группа риска 1. Проведено лечение: переливание эритроцитарной взвеси № 2. Выписана с клинико-гематологическим улучшеним - анемический синдром купирован, гемоглобин повысился до 120г/л. Повторные госпитализации в июне, сентябре 2008 г., апреле, июле 2009 года - поступала с выраженным анемическим синдромом (гемоглобин снижался до 60г/л, эритроциты 1,81*109). В стационаре проводились переливания эритроцитарной взвеси, получала эпоэтин альфа по 10 000 млн. МЕ 3 раза в неделю. Амбулаторно периодически получала лечение рекомбинантными эритропоэтинами.
В сентябре 2009 г. впервые проведено цитогенетическое исследование костного мозга, выявлено: кариотип женщины, мозаичный вариант со структурными перестройками по типу делеции 5 q -del(5)(q31;q35). В миелограмме препарат умеренноклеточный, тип кроветворения преимущественно нормобластный с небольшим процентом мегалобластов. Белый росток несколько угнетен. Созревание гранулоцитов без особенностей, бластов 4,1%. Красный росток умеренно раздражен - представлен нормобластами 30,2%; мегалобластами 1%; соотношение белого и красного ростков 2,2:1; индекс созревания эритробластов 0,93; полихромазия. Мегакариоцитов 8/на100полей зрения.
Был диагностирован миелодиспластический синдром по типу рефрактерной анемии с изолированной делецией 5q, промежуточная группа риска - 1 (по IPSS), тяжёлое течение.
Учитывая отсутствие эффекта от паллиативной терапии, потребность в частых гемотрансфузиях, наличие делеции q 5 хромосомы, больной было решено провести лечение новым высокотехнологичным препаратом для эпигенетической терапии МДС - децитабином.
С 6.10. 2009 г. в гематологическом отделении ГКБ № 7 проведен первый курс лечения децитабином 50 мг № 5 в/в. После курса развилась глубокая панцитопения с присоединением фебрильной нейтропении, агранулоцитоз продолжался 12 дней. Проводилась терапия выхаживания в полном объеме: Г-КСФ Грасальва 30 млн. МЕ п/к № 18, переливание эритровзвеси № 5, тромбоконцентрат 27 доз, комбинированная антибактериальная терапия , противогрибковые препараты, ацикловир. В дальнейшем проведено ещё четыре пятидневных курса лечения дакогеном со снижением дозы до 35 мг. После курсов развивалась умеренная тромбоцитопения без геморрагического синдрома, получала переливание тромбоконцетрата. Последний курс лечения дакогеном проведен в марте 2010 года.
На фоне проведенных 5 курсов лечения высокотехнологичным препаратом, восстанавливающим баланс генов, дакогеном, получен клинико-гематологический эффект: исчезла потребность в гемотрансфузиях, нормализовались показатели гемограммы. Диагностирована гематологическая ремиссия МДС. Длительность ответа составляет 14 месяцев. Всё это время больная наблюдается у гематолога в поликлинике, лечения не получает.
аутоТГСК — трансплантация аутологичных гемопоэтических стволовых клеток
АХМЛ — атипичный хронический миелолейкоз
ГКСФ — гранулоцитарный колониестимулирующий фактор
КМ — костный мозг
МДС — миелодиспластический синдром
ОМЛ — острый миелоидный лейкоз
Интенсификация индукционной и постремиссионной терапии — принципиальное условие повышения выживаемости больных онкогематологическими заболеваниями [1]. Назначение мегадоз цитостатиков способствует максимальной редукции патологического клона, а длительная поддерживающая терапия удерживает резидуальные лейкозные клетки на уровне, сопряженном с минимальной вероятностью развития рецидива. При этом значительная кумулятивная доза химиопрепаратов может явиться причиной развития вторичных неоплазий [2—4]. Не исключен и другой сценарий негативного воздействия агрессивной химиотерапии (ХТ), а именно селекция предшествующих (суб-)клонов [5, 6].
Наиболее частыми вариантами вторичных неоплазий являются миелодиспластический синдром (МДС) и острый миелоидный лейкоз (ОМЛ).
В классификации ВОЗ от 2001 г. предложены 2 варианта вторичных ОМЛ и МДС в зависимости от провоцирующего фактора, в качестве которых выступают алкилирующие препараты (циклофосфан, алкеран) и/или лучевая терапия и ингибиторы топоизомеразы (этопозид, антрациклины) [3].
В первом случае вторичные неоплазии выявляются через 4—7 лет после соответствующего лечения, проявляются преимущественно в виде МДС или ОМЛ с диспластическими изменениями, в большинстве случаев характеризуются несбалансированными хромосомными аберрациями, в основном -5/5q- и -7/7q- и отличаются рефрактерностью к ХТ.
Во втором случае вторичные неоплазии представлены преимущественно ОМЛ, отличительными признаками которого являются короткий латентный период (медиана 2—3 года), моноцитарная природа бластных клеток, сбалансированные хромосомные аберрации в виде транслокаций с частым вовлечением 11q23 или 21q22 и эффективность начальной ХТ, идентичной результатам лечения больных ОМЛ de novo с соответствующим кариотипом.
В классификации ВОЗ от 2008 г. не рекомендовано выделять самостоятельные варианты вторичных МДС и ОМЛ, так как больные могут получать любые из перечисленных лекарственных средств [4]. Кроме того, повреждающее действие на гемопоэтические клетки оказывают и другие цитостатики, например, антиметаболиты: 6-меркаптопурин, метотрексат, флударабин, цитарабин [7, 8]. Не исключено развитие вторичных неоплазий вследствие применения гранулоцитарного колониестимулирующего фактора (ГКСФ) [8—10]. Применение ГКСФ для мобилизации гемопоэтических стволовых клеток в периферическую кровь и в посттрансплантационном периоде для укорочения периода агранулоцитоза в совокупности с миелоаблативным режимом кондиционирования рассматривается как возможная причина развития вторичных неоплазий после трансплантации аутологичных гемопоэтических стволовых клеток (аутоТГСК) [11].
Вторичные МДС и ОМЛ возникают у 0,8—6,2% больных хроническими лимфопролиферативными неоплазиями и множественной миеломой при наблюдении в течение 20 лет. Вторичные МДС и ОМЛ составляют 10—20% от общего числа миелоидных неоплазий [6]. Менее чем у 5% больных вторичными неоплазиями могут диагностироваться другие, чем МДС и ОМЛ, варианты заболеваний. Описаны случаи вторичного Ph+ хронического миелолейкоза, смешанных миелоидных неоплазий (МДС/МПН). Причина разнообразия в характере молекулярно-генетических повреждений, определяющих клинико-гематологический фенотип вторичной неоплазии, эффективность ХТ, морфологические особенности при трансформации [7, 11—17].
В литературе приведены единичные случаи вторичного атипичного хронического миелолейкоза (АХМЛ). Так, A. Takeshita и соавт. [18] описали 61-летнего больного острым промиелоцитарным лейкозом, у которого после достижения полной ремиссии развился вторичный АХМЛ, трансформировавшийся в последующем в М0 вариант ОМЛ. S. Wang и соавт. [19] у 10 (8%) из 134 больных АХМЛ и неклассифицируемым МДС/МПН зафиксировали указание в анамнезе на предшествующее назначение ХТ и/или лучевой терапии (варианты заболевания не уточнены).
В связи с этим несомненный практический интерес представляет собственное наблюдение, когда АХМЛ явился результатом естественного развития вторичного МДС.
Больная Л., 67 лет, в ноябре 2012 г. госпитализирована в гематологическую клинику ФГБУ РосНИИГТ ФМБА в связи с бластемией, выявленной на фоне прогрессирующей слабости, кровоточивости десен и геморрагий на коже.
При поступлении состояние тяжелое. Бледность кожи и слизистых оболочек. На коже туловища и конечностей петехии и экхимозы разной степени давности. Периферические лимфатические узлы не увеличены. Пульс ритмичный, 94 уд/мин. Приглушенный I тон. В легких везикулярное дыхание. Печень и селезенка не пальпируются.
В анализе крови гемоглобин 108 г/л, лейкоциты 206·10 9 /л из них бласты 97%, тромбоциты 34·10 9 /л. В пунктате костного мозга (КМ) миелокариоциты 178·10 9 /л, бласты 94,4%. В 41% бластных клеток положительная реакция на активность миелопероксидазы и в 95% бластных клеток положительная реакция на фосфолипиды, PAS-реакция отрицательная. Иммунофенотип бластных клеток CD34 - , DR - , cyMPO + , CD38 + , CD13 + , CD33 + , CD11c + .
Кариотип 46, ХХ [20]. Выявлена мутация FLT3-TKD. Мутаций FLT3-ITD и в гене NPM1, а также слитых генов (BCR/ABL) p190 и (BCR/ABL) p210 не обнаружено. Активность лактатдегидрогеназы (ЛДГ) в сыворотке крови 5,75 мккат/л (верхняя граница нормы 3,3 мккат/л).
Диагностирован ОМЛ без созревания, соответствующий М1 варианту по классификации FAB. Прогноз по шкале ELN промежуточный [20].
После подписания больной информированного согласия инициирован прием гидроксимочевины в суточной дозе 100 мг/кг и аллопуринола 600 мг/сут, проведены 3 сеанса бластафереза. Назначен курс ХТ, в котором по причине быстрого снижения содержания лейкоцитов доза цитарабина (Ара-Ц) уменьшена на 50%: по 100 мг внутривенно 2 раза в день в течение 5 последовательных дней в комбинации с идарубицином по 12 мг/м 2 внутривенно в течение 3 последовательных дней.
При контрольном обследовании, проведенном через 1 мес после завершения ХТ, содержание бластов в миелограмме 1,6%, мутация FLT3-ITD не обнаружена. Констатирована полная ремиссия.
В последующем проведено 2 курса 7+3 с круглосуточным внутривенным введением Ара-Ц по 100 мг/м 2 /сут и болюсным внутривенным введением митоксантрона по 12 мг/м 2 и 2 консолидирующих курса монотерапии Ара-Ц в суммарной дозе 6 г/м 2 на курс.
В июне 2013 г. на фоне сохраняющейся полной ремиссии осуществлена заготовка аутологичных гемопоэтических стволовых клеток периферической крови. Для праймирования использована комбинация циклофосфана в суммарной дозе 2 г/м 2 с ГКСФ. В сентябре 2013 г. после миелоаблативного режима кондиционирования BuCph выполнена аутоТГСК. Восстановление кроветворения зафиксировано на 18—25-й день. Начата поддерживающая терапия интерфероном-α по 1·10 6 МЕ 3 раза в неделю.
С декабря 2012 г. по июль 2014 г. отмечены следующие лабораторные находки: тромбоцитопения (40—70)·10 9 /л, ассоциированная с интерферонотерапией. Дисплазия единичных клеток нейтрофильного ростка как следствие Х.Т. Нормальный кариотип. Отсутствие мутаций в генах FLT3 и NPM1.
В августе 2014 г., т. е. через 19 мес после констатации полной ремиссии, в контрольной миелограмме бласты составили 14,8%. В анализе крови лейкоцитов 3,7·10 9 /л, тромбоцитов 15·10 9 /л, бластов нет. Кариотип нормальный. Мутаций в генах FLT3 и NPM1 не обнаружено. Диагностирован поздний рецидив ОМЛ. Назначен реиндукционный курс 7+3: Ара-Ц 100 мг/м 2 2 раза в день и идарубицин 12 мг/м 2 /сут.
Через 1 мес после окончания ХТ гемоглобин 96 г/л, лейкоциты 4,3·10 9 /л, тромбоциты 144·10 9 /л, в пунктате КМ миелокариоциты 175·10 9 /л, бласты 0,4%. Констатирована вторая ремиссия ОМЛ. Обращало внимание наличие в миелограмме более 10% клеток нейтрофильного ряда с признаками дисгранулопоэза в виде пельгероидности ядер созревающих форм и скудности специфической зернистости в цитоплазме нейтрофилов, а также умеренных проявлений дизэритропоэза в виде присутствия мегалобластоидных элементов.
Проведены второй курс 7+3 и консолидирующий курс монотерапии Ара-Ц в дозе 8 г на курс. Одновременно отмечены нарастание признаков дизэритропоэза, появление микрогенераций мегакариоцитов с гиполобулярным ядром, тенденция к тромбоцитопении. При очередном цитогенетическом исследовании в 20 проанализированных метафазах обнаружена делеция длинного плеча 5-й хромосомы del (5)(q14q31). Активность ЛДГ в сыворотке крови в пределах нормы. По совокупности результатов исследования в феврале 2015 г., т. е. через 26 мес после верификации ОМЛ, диагностирован вторичный МДС. По критериям, разработанным в MD Anderson Cancer Center, вариант прогноза промежуточный, вероятность безлейкозной выживаемости в течение одного года 84% [21].
Больной назначен децитабин по 20 мг/м 2 внутривенно однократно в сутки в течение 5 последовательных дней. При госпитализации для проведения 3-го курса выявлена трехростковая цитопения, из-за чего введение децитабина отложено. На фоне коррекции показателей крови развился пароксизм мерцательной аритмии, не купированный внутривенным введением кордарона. Констатирован переход мерцательной аритмии в постоянную форму.
С мая 2015 г. больная переведена на терапию малыми дозами Ара-Ц по 20 мг подкожно каждые 12 ч в течение 10—14 дней. Прием леналидомида, назначение которого было обосновано персистирующей del (5q), сопровождался развитием кожной реакции II—III степени, которая возобновилась при попытке повторного применения. В анализах крови сохранялись умеренная анемия и тромбоцитопения, диспластические изменения в нейтрофилах и тромбоцитах. Периодически проводились трансфузии донорских эритроцитов.
В сентябре 2015 г., т. е. через 6 мес после диагностики вторичного МДС, в крови гемоглобин 114 г/л, тромбоциты 244·10 9 /л, лейкоциты 13,2·10 9 /л, из них миелоциты 5%, метамиелоциты 10%, палочкоядерные 13%, сегментоядерные 65%, лимфоциты 5%, моноциты 2%. При морфологическом исследовании периферической крови выявлены анизо- и пойкилоцитоз, полихроматофилия и базофильная пунктуация эритроцитов, со стороны клеток гранулоцитарного ряда — пельгероидность ядер и скудность специфической зернистости в цитоплазме. В пунктате КМ миелокариоциты 140·10 9 /л. В миелограмме клетки нейтрофильного ряда 83,8%, клетки эритроидного ряда 10,6%, мегакариоциты 30 в препарате. При цитогенетическом исследовании обнаружена моносомия 17-й хромосомы. Кариотип 45, ХХ, del (5)(q14q31),-17 [20]. Мутации JAK2V617 °F, MPLW551L и CALR (del, ins) не выявлены. Концентрация ЛДГ в сыворотке крови 6,6 мккат/л. Печень и селезенка не увеличены.
Отдельные показатели периферической крови и КМ представлены в таблице.
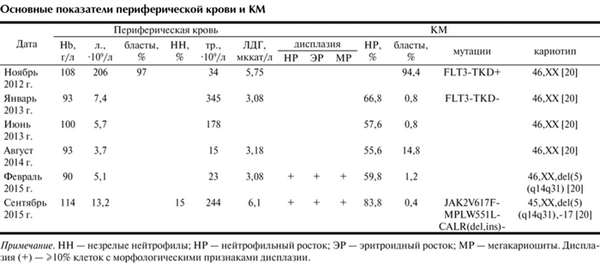
Основные показатели периферической крови и КМ Примечание. НН — незрелые нейтрофилы; НР — нейтрофильный росток; ЭР — эритроидный росток; МР — мегакариоциты. Дисплазия (+) — ≥10% клеток с морфологическими признаками дисплазии.
Обнаруженные изменения сохранялись в последующие месяцы наблюдения, содержание лейкоцитов повышалось до 40·10 9 /л. Персистирующий характер лейкоцитоза >13·10 9 /л, циркуляции незрелых миелоидных клеток в количестве >10%, выраженных признаков дисгранулопоэза в отсутствие моноцитоза, базофилии и слитого гена BCR/ABL служили основанием для диагностики трансформации вторичного МДС в аХМЛ.
В связи с отсутствием избытка костномозговых бластов продолжены циклы терапии малыми дозами Ара-Ц, к которой добавлен прием гидроксимочевины в межкурсовой период. Периодически для коррекции анемии проводятся трансфузии донорских эритроцитов.
Алгоритм лечения больных ОМЛ дает возможность выбора интенсивности лечебного пособия [1]. Для пожилых больных наиболее часто используемой опцией является терапия низкой интенсивности, что обусловлено сопутствующими заболеваниями и неблагоприятными хромосомными аберрациями, ассоциированными с низкой эффективностью индукционных курсов [22, 23]. Напротив, при благоприятных условиях приоритетными признаются стандартные протоколы ХТ, применение которых существенно повышает выживаемость [24].
В свою очередь агрессивная ХТ рассматривается как принципиальное, но не единственное условие возникновения вторичных неоплазий. Дополнительными являются механизмы, обусловливающие генетическую нестабильность, вероятность развития которых увеличивается с возрастом. Это нарушение процессов восстановления ДНК, внутриклеточного метаболизма и транспортировки цитостатиков, укорочение длины теломер, развитие ОМЛ из предшествующего МДС [7, 11, 25].
Предполагается, что прогнозировать развитие вторичных неоплазий можно посредством молекулярно-генетического мониторинга, используя методы детекции минимального объема клонального гемопоэза до клинико-гематологической манифестации. К ним относятся стандартное кариотипирование, FISH и изучение профиля экспрессии генов [26]. В этом случае появляется возможность кардинально изменить лечебную тактику: выполнить трансплантацию аллогенных гемопоэтических стволовых клеток (аллоТГСК) вместо запланированной аутоТГСК, исключить цитостатики с повреждающим кроветворные клетки и клетки гемопоэтического микроокружения действием.
Описанный случай еще раз подтверждает клинико-гематологическую вариабельность вторичных миелоидных неоплазий, которые могут быть представлены не только МДС и ОМЛ, но и АХМЛ.
Необходимо отметить, что в отличие от упоминаний в литературе, когда АХМЛ был первым проявлением вторичной неоплазии [18, 19], в собственном наблюдении АХМЛ — результат дальнейшего развития вторичного МДС. Обнаружение клональной эволюции с появлением дополнительной хромосомной аберрации в клетках с ранее детектированной делецией длинного плеча 5-й хромосомы дает основание предполагать выраженную генетическую нестабильность с возникновением дополнительных молекулярно-генетических повреждений, чем селекцию нового клона на фоне продолжающейся терапии.
Международной рабочей группой по изучению смешанных миелоидных неоплазий сделано заключение об отсутствии молекулярных аберраций специфичных для больных АХМЛ. Тем не менее не исключено, что развитие АХМЛ может быть сопряжено с мутацией генов SETBP1 или ASXL1 [27, 28]. Установлено, что в отличие от больных ювенильным миеломоноцитарным лейкозом и хроническим миеломоноцитарным лейкозом мутация гена SETBP1 является более частой находкой у больных АХМЛ: в 3, 6—15 и 25% случаях соответственно [27]. Напротив, мутации генов CBL, CSF3R, SRSF2 и ТЕТ2 происходят редко [29].
Для прогнозирования течения АХМЛ разными авторами предложены практически одни и те же показатели [19, 30, 31]. Снижение общей выживаемости ассоциировано с возрастом ≥65 лет, количеством гемоглобина менее 100 г/л, лейкоцитозом ≥50·10 9 /л и высоким процентным содержанием незрелых клеточных элементов в периферической крови. Негативными предикторами безлейкозной выживаемости являются зависимость от трансфузий донорских эритроцитов, спленомегалия, избыточное содержание костномозговых бластов. Возможно ли применять данные критерии для оценки прогноза вторичного АХМЛ, неизвестно, тем более что среди выделенных параметров отсутствуют данные цитогенетического исследования.
Лечение больных с вторичными неоплазиями предполагает 3 опции: поддерживающая терапия, ХТ и аллоТГСК. S. Lim и соавт. [32] приводят результаты аллоТГСК у 10 больных МДС/МПН, включая 2 больных АХМЛ, с медианой возраста 43 года (28—53 лет). При медиане наблюдения в 47,5 мес 5-летняя общая, безрецидивная и «бессобытийная» выживаемость составила 42,2, 51,9 и 46,7% соответственно. Так как основной причиной неэффективности лечения был рецидив, развившийся преимущественно после режима кондиционирования со сниженной интенсивностью, авторы делают заключение о целесообразности применения миелоаблативных режимов. При невозможности выполнения аллоТГСК эффективными могут оказаться гипометилирующие препараты, иммуномодуляторы, ингибиторы JAK½ [27, 33, 34]. Так, К. Dao и соавт. [34] описали 75-летнего больного с диагнозом АХМЛ и мутацией CSF3R-T618I, у которого прием руксолитиниба с постепенным увеличением суточной дозы с 20 до 40 мг сопровождался улучшением соматического статуса, уменьшением размеров селезенки, увеличением количества гемоглобина и тромбоцитов.
Заключение
Представленный случай свидетельствует о вариабельности клинического течения вторичных миелоидных неоплазий. В частности, продемонстрирована возможность возникновения АХМЛ как этапа естественного развития вторичного МДС. Дальнейшее накопление результатов молекулярно-генетического мониторинга и данных изучения функционального состояния клеток гемопоэтической ниши позволит расшифровать механизмы формирования клинико-гематологического фенотипа вторичной неоплазии.
РА — рефрактерная анемия
РАКС — рефрактерная анемия с кольцевыми сидеробластами
РЦМД — рефрактерная цитопения с мультилинейной дисплазией
Анализ молекулярно-генетических повреждений с целью прогнозирования течения миелодиспластического синдрома (МДС) обусловлен их непосредственным вовлечением в патогенез заболевания [1—4]. Согласно Международной прогностической шкале (International Prognostic Scoring System — IPSS) анализ кариотипа, количество бластов в костном мозге (КМ) и тяжесть цитопении позволяют сформировать 3 группы больных, различающихся по выживаемости и риску трансформации МДС в острый миелоидный лейкоз (ОМЛ) [5].
В рамках IPSS выделены 3 прогностических варианта кариотипа, которые являются самостоятельными маркерами риска. В группу благоприятного прогноза отнесены нормальный кариотип, делеция длинного плеча 5-й и 20-й хромосом, потеря хромосомы Y. Группа неблагоприятного прогноза объединяет аберрации 7-й хромосомы и комплексный кариотип, для верификации которого необходимо наличие 3 независимых аномалий и более. Все другие цитогенетические повреждения включены в группу промежуточного прогноза.
Основной акцент в IPSS сделан на морфологических показателях и, прежде всего, на количестве бластов. Максимальные оценки 1,5 и 2 балла присвоены 11—20 и 21—30% бластов соответственно. В то же время неблагоприятные варианты кариотипа оценены на 1 балл. Вместе с тем негативное влияние, оказываемое неблагоприятными цитогенетическими поломками на выживаемость больных с МДС, может быть сопоставимо с избытком бластов [6, 7]. Более того, дальнейшее снижение общей выживаемости с вероятностью прогрессии заболевания ассоциировано с так называемым моносомным кариотипом [8, 9]. Неслучайно в шкале, разработанной H. Kantarjian и соавт. [10], представлен только один цитогенетический маркер — неблагоприятный кариотип, которому наряду с глубокой тромбоцитопенией присвоено наибольшее количество баллов.
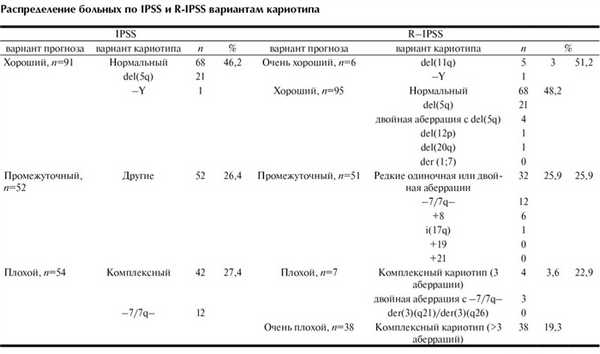
Накопленные клинические данные способствуют расширению спектра хромосомных аберраций с установленным прогностическим потенциалом [11—13]. Так, испанской рабочей цитогенетической группой выявлено неблагоприятное течение заболевания у пациентов с аномалиями 3q21q26 (медиана выживаемости 2,6 года и 100% вероятность трансформации в ОМЛ в течение 5 лет) и минимальный риск прогрессии у больных с del(17p) [11]. Германо-австрийской группой по изучению МДС определен прогноз многих редко встречаемых хромосомных аномалий, например, +1/1q+, t(1q), t(7q), del(9q), моносомия и трисомия 21-й хромосомы и др. [12, 13]. Объединение данных этих групп с результатами исследований IMRAW и ICWG послужило базой для разработки новой прогностической шкалы кариотипа (revised IPSS, R-IPSS), содержащей 5 самостоятельных вариантов кариотипа вместо 3 в IPSS (см. таблицу) [14]. Медиана продолжительности жизни больных с вариантами R-IPSS кариотипа составила 60,8, 48,5, 24, 14 и 5,7 мес (р<0,0001). Предполагается, что данная шкала составит основу для модификации IPSS [15].
Помимо прогностического значения, кариотип является важным диагностическим инструментом. Обнаружение ряда несбалансированных (-7q/7q-, -5/5q-, i(17q), t(17p), -13/13q-) и сбалансированных (inv(3)(q21q26.2), t(11;16)(q23;p13.3), t(3;21)(q26.2;q22.1)) аберраций делает диагноз МДС более убедительным [16]. Кроме того, кариотип может быть ассоциирован с эффективностью отдельных препаратов, например леналидомида, у больных с делецией длинного плеча 5-й хромосомы [17, 18].
Целью данной работы была оценка у больных с МДС распространенности хромосомных аберраций, представленных в R-IPSS.
Материалы и методы
Для решения поставленной цели проанализированы результаты цитогенетических исследований клеток КМ у больных с МДС, выполненных в период с 1995 по октябрь 2011 г.
Критериями включения были диагноз первичного МДС; заключение об исследовании морфологических препаратов КМ, позволяющее верифицировать вариант заболевания по классификации ВОЗ [16].
Критериями исключения были количество бластов в КМ ≥20%, транслокация t(8;21) и инверсия inv(16).
Для изучения кариотипа применен стандартный метод GTG. При обнаружении ≥3 независимых хромосомных аберраций идентифицировали комплексный кариотип. Случаи с 2 аутосомными моносомиями и более или 1 аутосомной моносомией в комбинации с ≥1 структурным повреждением расценивали как моносомный кариотип [19].
Термин «двойная» аберрация использовали для обозначения 2 независимых хромосомных аномалий, а термин «редкие аберрации» — для обозначения группы цитогенетических повреждений, которые в R-IPSS специально не оговорены и включены в состав промежуточного кариотипа как «любая другая одиночная или двойная аберрация» (см. таблицу).
При анализе сопряженности кариотипа с возрастом больные были распределены в 3 группы: моложе 50 лет, 51 год—60 лет и 61 год и старше.
Статистическую обработку данных проводили с использованием программ Excel и Statisticа. Различие между отдельными показателями принимали достоверным при р
Результаты
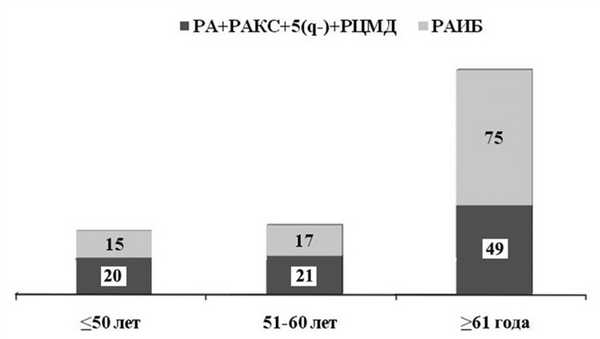
В соответствии с установленными критериями из базы данных отобраны результаты обследования 197 больных с de novo МДС с медианой возраста 64 года (от 14 до 86 лет). Большинство больных были старше 50 лет (рис. 1). Рисунок 1. Распределение больных в возрастных группах в зависимости от количества бластов в КМ. Здесь и на рис. 4: РА — рефрактерная анемия; РАКС — рефрактерная анемия с кольцевыми сидеробластами; РЦМД — рефрактерная цитопения с мультилинейной дисплазией; РАИБ — рефрактерная анемия с избытком бластов.
У 90 (45,7%) больных верифицированы варианты менее чем с 5% бластов в КМ: РА (n=28), РАКС (n=7), делеция длинного плеча 5-й хромосомы (n=9) и РЦМД (n=46). В морфологических препаратах КМ остальных 107 (54,3%) больных количество бластов было 5% и более.
Соотношение морфологических вариантов у больных моложе 60 лет было приблизительно одинаковым. После 61 года число больных с бластами ≥5% существенно увеличивалось (р=0,034; см. рис. 1). Выявлена корреляция возраста больных с МДС с бластозом в КМ (r=0,159; р=0,024).
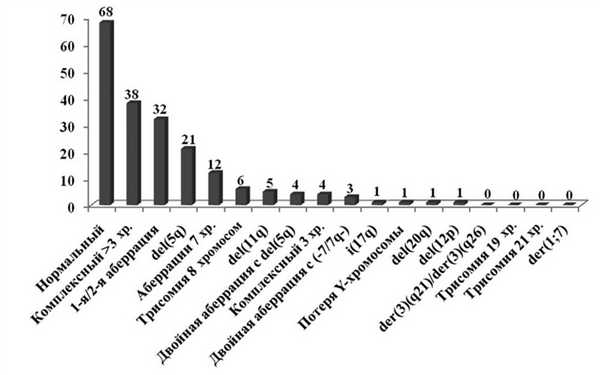
Повреждения кариотипа выявлены у 129 (65,5%) больных. Число случаев с отдельными хромосомными аберрациями представлено на рис. 2. Рисунок 2. Частота отдельных аберраций. Наиболее частыми были нормальный (34,5%) и комплексный кариотип с >3 аберрациями (19,3%), редкие аберрации (16,2%), делеция длинного плеча 5-й хромосомы (10,7%) и повреждения 7-й хромосомы (6,1%).
Выявлено 29 одиночных и двойных аберраций промежуточного варианта R-IPSS. За исключением таких аберраций, как моносомии 5-й хромосомы, делеции длинного плеча 10-й и 13-й хромосом, каждая из которых определена у 2 больных, остальные повреждения (ХХ;YY, -Х, i(Х)(р10), del(Y)(q11.2), +9, +10, +11, +15, del(8)(q24), del(9)(q13), del(16)(q22), add(3)(q29), add(12)(р13), inv(9)(p23;q13), t(1;2)(q23;q37), t(1;3)(q10;q10), t(3;5)(q12;q11.2), t(3;17)(q27;q25)) были одиночными находками.
У 2 больных с 2 аберрациями и у 22 с множественными хромосомными поломками верифицирован моносомный кариотип (12,2%).
Для изучения ассоциации кариотипа с возрастом и бластозом выбраны варианты, обнаруженные не менее чем у 10% больных: нормальный и комплексный кариотип с >3 аберрациями, редкие аберрации, моносомный кариотип и del(5q). По результатам исследования морфологических препаратов КМ больные распределены в 2 группы: с
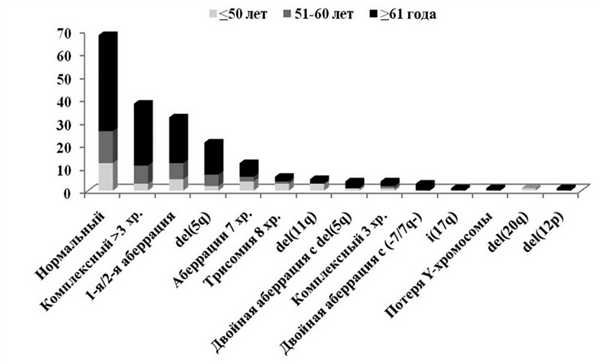
Указанные варианты кариотипа были более частой находкой у больных старше 60 лет (рис. 3). Рисунок 3. Возраст и кариотип. Тем не менее не обнаружено корреляций кариотипа с возрастом.
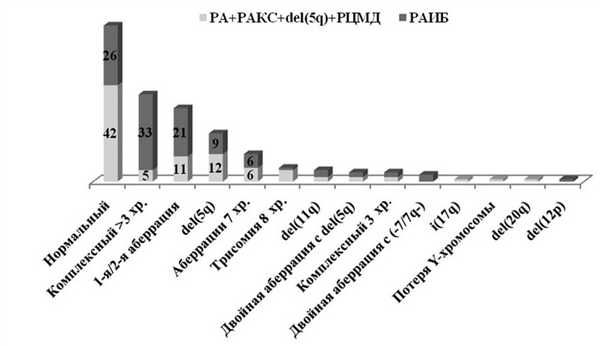
При анализе отдельных морфологических вариантов выявлено, что нормальный кариотип чаще встречался у больных без избытка бластов в КМ. Напротив, в пунктате КМ большинства больных с >3 аберрациями, редкими аберрациями и моносомным кариотипом количество бластов было повышенным. У больных с del(5q) соотношение морфологических вариантов было практически одинаковым (рис. 4). Рисунок 4. Варианты МДС и кариотип.
Из прогностических IPSS-вариантов кариотипа самой многочисленной была группа благоприятного прогноза (46,2%). Причина — наибольшее число случаев с нормальным кариотипом и del(5q) среди обследованных больных. В группах с промежуточным и неблагоприятным кариотипом число больных было приблизительно в 2 раза меньше, но не различалось между собой: 26,4 и 27,4% соответственно (см. таблицу).
По критериям R-IPSS в группы с очень благоприятным и неблагоприятным кариотипом вошли единичные больные: 3 и 3,6% соответственно. Группа благоприятного кариотипа по-прежнему оставалась самой большой (48,2%). Больных с промежуточным и очень неблагоприятным кариотипом было меньше: 25,9 и 19,3% соответственно (см. таблицу).
После объединения вариантов R-IPSS с очень благоприятным и благоприятным кариотипом, а также неблагоприятным и очень неблагоприятным кариотипом достоверных различий по распределению больных по 3 прогностическим вариантам кариотипа 2 шкал не выявлено (р=0,613; см.таблицу).
МДС — гетерогенная группа клональных заболеваний, различающихся по характеру естественного течения и риску трансформации в ОМЛ [20]. Вариабельность МДС отражена в классификациях FAB и ВОЗ [16, 21]. Более совершенным прогностическим инструментом служат специальные шкалы, обязательным элементом которых является кариотип [5, 10, 22].
Наибольшее распространение получила IPSS [5]. Несмотря на многообразие цитогенетических повреждений, встречающихся у больных МДС, в IPSS приводится прогностический потенциал нормального и комплексного кариотипа и 4 аберраций. Все остальные повреждения, включая одиночные делеции, моно- и трисомии, транслокации и разные комбинации, объединены в группу промежуточного кариотипа.
Очевидным представляется преимущество R-IPSS, в которой представлены 10 ранее не упоминаемых аберраций и сформированы 5 самостоятельных вариантов кариотипа [14]. Эффективность стратификации больных с de novo МДС по цитогенетическим критериям R-IPSS подтверждена результатами ряда исследований [23].
Наряду с этим обращает внимание перемещение аберраций 7-й хромосомы в группу промежуточного кариотипа. Сомнительным также представляется нахождение в пределах одного варианта одновременно случаев с del(5q) и del(5q) с дополнительной аберрацией. Данные литературы не позволяют однозначно трактовать влияние дополнительной аберрации на риск трансформации в ОМЛ и выживаемость больных с del(5q). Однако указания на возможное снижение выживаемости обусловливают необходимость дальнейшего накопления клинического материала [24—27]. Не менее важным является также то, что больные, чьи результаты цитогенетического исследования использованы для создания R-IPSS, получали только поддерживающую терапию. Это снижает вероятность прогнозирования выживаемости больных при проведении специальной терапии [28].
По результатам собственного анализа установлено, что чаще других у больных с de novo МДС встречались нормальный и комплексный кариотипы с повреждением 4 хромосом и более, редкие одиночные и двойные аберрации, делеция длинного плеча 5-й хромосомы и повреждения 7-й хромосомы (необходимо подчеркнуть, что речь идет о случаях с аномалиями 5-й и 7-й хромосом как единственном повреждении, выявленным стандартным цитогенетическим методом).
Эти данные, а также преимущественное обнаружение множественных хромосомных поломок у больных с избыточным количеством бластов в КМ согласуются с данными других исследователей [11—13, 29].
Несмотря на недостатки новой прогностической шкалы, следует признать, что, используя критерии R-IPSS, удалось стратифицировать 84% больных, в то время как по шкале IPSS — только 74% больных (см. таблицу).
Наиболее многочисленной была группа благоприятного R-IPSS-кариотипа. Меньше всего было больных с очень благоприятным кариотипом. Это совпадает с результатами других исследований [23, 28] и обусловлено наибольшим числом больных с нормальным кариотипом и del(5q). Численность состава других вариантов кариотипа варьирует в разных исследованиях, что зависит от целей отбора больных, например, для лечения гипометилирующими препаратами в исследовании GFМ (Groupe Francophone Des Myelodysplasies) [28].
Предложенные 5 вариантов кариотипа в совокупности с возрастом, количеством бластов в КМ и тяжестью цитопении со снижением разграничительного уровня абсолютного числа нейтрофилов с 1,5·109 до 0,8·109/л позволяют существенно улучшить качество стратификации больных с МДС на прогностические группы [15]. Вместе с тем одновременно с расширением спектра выявляемых цитогенетических поломок с установленным прогностическим потенциалом существует вероятность, что многие из них не будут выявлены в соответствии с критериями R-IPSS, т.е. в виде единственных повреждений кариотипа. Так, 3 из 17 хромосомных аберраций, представленных в R-IPSS, обнаружены менее чем у 1% больных (-Y, del(12p), del(20q)) и 4 аберрации не обнаружены ни в одном случае (der(1;7), +19, +21, der(3)(q21)/der(3)(q26)). Нельзя исключить значение числа больных в исследовании и его одноцентровый характер. Однако увеличение распространенности отдельных аберраций до 2% [14, 30], вероятно, не меняет ситуацию в целом.
Следовательно, учитывая многообразие хромосомных поломок и редкость отдельных повреждений, необходимо задать вопрос о том, насколько оправданным является выделение новых прогностических вариантов кариотипа. Так, в состав группы «редкие аберрации» вошли 29 цитогенетических аномалий (-5 (у 2 больных), del(10)(q23) (у 2 больных); del(13)(q22)/del(13)(q14) (у 2 больных); остальные абберации выявлены только по 1 больному: ХХ;YY; -Х; i(Х)(р10); del(Y)(q11.2); +9; +10; +11; +15; del(8)(q24); del(9)(q13); del(16)(q22); add(3)(q29); add(12)(р13); inv(9)(p23;q13); t(1;2)(q23;q37); t(1;3)(q10;q10); t(3;5)(q12;q11.2); t(3;17)(q27;q25); t(9;20)(р24;q12); t(11;21)(q24;q22); t(16;17)(р13;q21); -Y, ins(11)(q13); -5,del(6)(q23); del(3)(q22),del(11)(q23); del(12)(р12),add(16)(q); +2mar). Малочисленность 2 новых R-IPSS-вариантов кариотипа послужила причиной того, что распределение больных в группы благоприятного, промежуточного и неблагоприятного кариотипов практически не различалось при применении 2 прогностических шкал (см. таблицу). Кроме того, R-IPSS не оговаривает такой крайне неблагоприятный кариотип, как моносомный [19], который был идентифицирован у 12% больных и включал случаи, не соответствующие критериям очень неблагоприятного кариотипа.
Таким образом, результаты цитогенетического обследования 197 больных демонстрируют несомненное преимущество R-IPSS в стратификации больных с de novo МДС по сравнению с IPSS. Вместе с тем выявлен целый ряд недостатков новой прогностической шкалы кариотипа из-за многообразия хромосомных аберраций у больных с МДС. Оправданным представляется включение в состав разрабатываемых шкал дополнительных показателей, позволяющих прогнозировать естественное течение МДС и вероятность ответа на терапию. К ним следует отнести концентрацию ферритина, лактатдегидрогеназы и β2-микроглобулина в сыворотке крови, фиброз КМ, потребность больных в трансфузиях эритроцитной массы [22, 31—34]. Перспективным является изучение мутационного статуса генов [1]. Выявлены мутации, ассоциированные со снижением выживаемости и благоприятным течением МДС [1—4, 35, 36].
Заключение
Дальнейшее уточнение прогностического потенциала отдельных мутаций и комбинации повреждений нескольких генов, изучение сопряженности мутаций с возрастом, морфологическими и цитогенетическими вариантами позволит качественно изменить стратификацию больных с МДС на группы риска.
Миелодиспластический синдром
Миелодиспластический синдром - группа гематологических заболеваний, при которых наблюдаются цитопения, диспластические изменения костного мозга и высокий риск возникновения острого лейкоза. Характерные симптомы отсутствуют, выявляются признаки анемии, нейтропении и тромбоцитопении. Диагноз устанавливается с учетом данных лабораторных анализов: полного анализа периферической крови, гистологического и цитологического исследования биоптата и аспирата костного мозга и т. д. Дифференциальный диагноз может представлять значительные затруднения. Лечение - переливание компонентов крови, химиотерапия, иммуносупрессивная терапия, пересадка костного мозга.
Общие сведения
Миелодиспластический синдром - группа заболеваний и состояний с нарушениями миелоидного кроветворения и высоким риском развития острого лейкоза. Вероятность развития увеличивается с возрастом, в 80% случаев данный синдром диагностируется у людей старше 60 лет. Мужчины страдают несколько чаще женщин. У детей миелодиспластический синдром практически не встречается. В последние десятилетия гематологи отмечают увеличение заболеваемости среди лиц трудоспособного возраста. Предполагается, что причиной «омоложения» болезни могло стать существенное ухудшение экологической обстановки.
До недавнего времени лечение миелодиспластического синдрома было только симптоматическим. Сегодня специалисты разрабатывают новые методы терапии, однако эффективное лечение этой группы болезней все еще остается одной из самых сложных проблем современной гематологии. Пока прогноз при миелодиспластическом синдроме, в основном, зависит от особенностей течения болезни, наличия или отсутствия осложнений. Лечение осуществляют специалисты в сфере онкологии и гематологии.
Причины и классификация миелодиспластического синдрома
С учетом причин развития различают два типа миелодиспластического синдрома: первичный (идиопатический) и вторичный. Идиопатический вариант выявляется в 80-90% случаев, диагностируется преимущественно у пациентов старше 60 лет. Причины возникновения установить не удается. В числе факторов риска первичного миелодиспластического синдрома - курение, повышенный уровень радиации при выполнении профессиональных обязанностей или проживании в неблагоприятной экологической зоне, частый контакт с бензином, пестицидами и органическими растворителям, некоторые наследственные и врожденные заболевания (нейрофиброматоз, анемия Фанкони, синдром Дауна).
Вторичный вариант миелодиспластического синдрома наблюдается в 10-20% случаев, может возникать в любом возрасте. Причиной развития становится химиотерапия или радиотерапия по поводу какого-то онкологического заболевания. В число лекарственных средств с доказанной способностью вызывать миелодиспластический синдром включают циклофосфан, подофиллотоксины, антрациклины (доксорубицин) и ингибиторы топоизомеразы (иринотекан, топотекан). Вторичный вариант отличается более высокой резистентностью к лечению, более высоким риском развития острого лейкоза и более неблагоприятным прогнозом.
В современной редакции классификации ВОЗ различают следующие типы миелодиспластического синдрома:
- Рефрактерная анемия. Сохраняется более полугода. В анализе крови бласты отсутствуют либо единичные. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная анемия с кольцевыми сидеробластами. Сохраняется более полугода. В анализе крови бласты отсутствуют. В костном мозге дисплазия эритроидного ростка.
- Рефрактерная цитопения с многолинейной дисплазией. В анализе крови тельца Ауэра отсутствуют, бласты отсутствуют либо единичные, выявляются панцитопения и увеличение количества моноцитов. В костном мозге диспластические изменения менее 10% клеток в 1 миелоидной клеточной линии, бластов менее 5%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-1. В анализе крови тельца Ауэра отсутствуют, бластов более 5%, цитопения и увеличение количества моноцитов. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 5-9%, телец Ауэра нет.
- Рефрактерная анемия с избытком бластов-2. В анализе крови увеличение количества моноцитов, цитопения, бластов 5-19%, могут выявляться тельца Ауэра. В костном мозге дисплазия одной либо нескольких клеточных линий, бластов 10-19%, обнаруживаются тельца Ауэра.
- Неклассифицируемый миелодиспластический синдром. В анализе крови цитопения, бласты отсутствуют либо единичные, тельца Ауэра отсутствуют. В костном мозге дисплазия одного мегакариоцитарного либо гранулоцитарного ростка, бластов более 5%, тельца Ауэра отсутствуют.
- Миелодиспластический синдром, ассоциированный с изолированной делецией 5q. В анализе крови анемия, бластов более 5%, возможен тромбоцитоз. В костном мозге более 5% бластов, тельца Ауэра отсутствуют, изолированная делеция 5q.
Симптомы миелодиспластического синдрома
Клиническая симптоматика определяется степенью нарушений миелопоэза. При мягко протекающих расстройствах возможно длительное бессимптомное или стертое течение. Из-за слабой выраженности клинических проявлений некоторые больные не обращаются к врачам, и миелодиспластический синдром обнаруживается во время проведения очередного медицинского осмотра. При преобладании анемии наблюдаются слабость, одышка, плохая переносимость физических нагрузок, бледность кожных покровов, головокружения и обморочные состояния.
При миелодиспластическом синдроме с тромбоцитопенией возникает повышенная кровоточивость, отмечаются десневые и носовые кровотечения, на коже появляются петехии. Возможны подкожные кровоизлияния и меноррагии. Миелодиспластический синдром с выраженными нейтропенией и агранулоцитозом проявляется частыми простудами, стоматитом, синуситом или стрептодермией. В тяжелых случаях возможно развитие пневмонии или сепсиса. Инфекционные заболевания нередко вызываются грибками, вирусами или условно-патогенными микробами. У каждого пятого пациента с миелодиспластическим синдромом выявляется увеличение лимфоузлов, селезенки и печени.
Диагностика миелодиспластического синдрома
Диагноз выставляется с учетом данных лабораторных исследований: анализа периферической крови, биопсии костного мозга с последующим цитологическим исследованием, цитохимических и цитогенетических тестов. В анализе периферической крови больных миелодиспластическим синдромом обычно обнаруживается панцитопения, реже выявляется дву- или одноростковая цитопения. У 90% пациентов наблюдается нормоцитарная либо макроцитарная анемия, у 60% - нейтропения и лейкопения. У большинства больных миелодиспластическим синдромом отмечается тромбоцитопения.
При исследовании костного мозга количество клеток обычно нормальное либо повышенное. Уже на ранних стадиях обнаруживаются признаки дизэритропоэза. Количество бластов зависит от формы миелодиспластического синдрома, может быть нормальным либо увеличенным. В последующем наблюдаются дисгранулоцитопоэз и дисмегакариоцитопоэз. У некоторых больных признаки дисплазии костного мозга выражены очень слабо. В процессе цитогенетического исследования у ¾ больных выявляются хромосомные нарушения. Дифференциальный диагноз миелодиспластического синдрома проводят с В12-дефицитной анемией, фолиево-дефицитной анемией, апластической анемией, острым миелолейкозом и другими острыми лейкозами.
Лечение и прогноз при миелодиспластическом синдроме
Тактика лечения определяется выраженностью клинической симптоматики и лабораторных изменений. При отсутствии явных признаков анемии, геморрагического синдрома и инфекционных осложнений осуществляется наблюдение. При миелодиспластическом синдроме с выраженной анемией, тромбоцитопенией и нейтропенией, а также при высоком риске возникновения острого лейкоза назначают сопроводительную терапию, химиотерапию и иммуносупрессивную терапию. При необходимости осуществляют пересадку костного мозга.
Сопроводительная терапия является самым распространенным методом лечения миелодиспластического синдрома. Предусматривает внутривенные инфузии компонентов крови. При длительном применении может провоцировать повышение уровня железа, влекущее за собой нарушения деятельности жизненно важных органов, поэтому переливания гемокомпонентов производят при одновременном приеме хелаторов (лекарственных средств, связывающих железо и способствующих его выведению).
Иммуносупрессоры эффективны при лечении миелодиспластического синдрома с отсутствием хромосомных аномалий, наличием гена HLA-DR15 и гипоклеточном костном мозге. Химиотерапию применяют при невозможности трансплантации костного мозга. Высокие дозы препаратов используют при трансформации миелодиспластического синдрома в острый лейкоз, а также при рефрактерных анемиях с избытком бластов при нормоклеточном и гиперклеточном костном мозге, низкие - при невозможности пересадки костного мозга. Наряду с перечисленными средствами пациентам назначают гипометилирующие средства (азацитидин). Наиболее надежным способом достижения полноценной длительной ремиссии является трансплантация костного мозга.
Прогноз зависит от типа миелодиспластического синдрома, количества хромосомных аномалий, необходимости в регулярных переливаниях компонентов крови, выраженности клинических проявлений и наличия осложнений. Различают 5 групп риска. Средняя выживаемость больных миелодиспластическим синдромом, входящих в группу с самым низким уровнем риска, составляет более 11 лет; с самым высоким - около 8 месяцев. Вероятность отторжения костного мозга после трансплантации - около 10%.
Читайте также: