Диагностика аномалии Денди-Уокера по МРТ, КТ
Добавил пользователь Алексей Ф. Обновлено: 21.01.2026
Аномалии формирования центральной нервной системы — отнюдь не редкость. Эта патология занимает второе место по частоте после врожденных дефектов развития сердца и сосудов и в большинстве случаев представлена гидроцефалией самого разного происхождения. Помимо тяжелых нарушений, сопровождающих патологическое развития мозга, пороки ЦНС несут высокий риск смертельного исхода, а по данным статистики они лидируют по числу смертей в младенческом возрасте.
Синдром Денди-Уокера — одна из разновидностей нарушения формирования головного мозга еще во время внутриутробного развития. И хотя частота его относительно невелика (всего 1 случай на 25-30 тысяч младенцев), диагностируется порок едва ли не у каждого десятого малыша с врожденной гидроцефалией, которая и служит одним из основных проявлений патологии.
Синдром Денди-Уокера — это порок задней черепной ямки, при этом основные структурные изменения касаются мозжечка, ликвороотводящих путей, четвертого желудочка мозга. Аномалия диагностируется во время беременности посредством ультразвукового осмотра, после чего женщине может быть предложено прерывание беременности по медицинским показаниям.
Конечно, любые отклонения в развитии плода — это всегда большой стресс и переживания для родителей, однако в случае врожденного порока мозга надеяться на чудо не приходится — прогноз серьезный, а смертность высока. Малыши с сочетанными пороками мозга и других органов погибают в раннем возрасте как от мозговой дисфункции, так и от присоединившейся инфекции.
Аномалия Денди-Уокера часто сочетается с другими нарушениями и генетическими заболеваниями, зачастую несовместимыми с жизнью — микроцефалия (недоразвитие полушарий мозга), мозговые грыжи. У младенца может быть диагностирован генетически обусловленный поликистоз почек, недоразвитие зрительных нервов и глазных яблок со слепотой, аномалии сердечно-сосудистой системы.
Все эти неблагоприятные факторы, возможность сочетанной патологии многих органов делают синдром Денди-Уокера серьезнейшей проблемой в случае, если малышу дадут возможность родиться. Лечение патологии, как правило, симптоматическое, направленное на поддержание главных систем жизнеобеспечения и борьбу с инфекционными осложнениями. В редких случаях применяют хирургическую операцию, которая лишь облегчает явления гидроцефалии, но не ликвидирует ее полностью.
Почему возникает синдром Денди-Уокера?
Причины аномалий развития задней черепной ямки до сих пор не выяснены, однако выделен ряд факторов, способствующих подобным врожденным порокам:
- Инфицирование во время беременности цитомегаловирусом, перенесенная краснуха;
- Употребление алкоголя, курение, наркомания во время беременности;
- Экстрагенитальная патология, особенно — сахарный диабет у будущей мамы.
Под действием перечисленных причин или среди полного благополучия может возникнуть спонтанная мутация в генах, предрасполагающая к нарушению развития мозга. Особенно высок риск пороков при действии неблагоприятных факторов во время первого триместра гестации, когда и происходит закладка основных структур центральной нервной системы.
В части случаев синдром Денди-Уокера носит наследственно-обусловленный характер, то есть возникает из-за дефекта генов и может передаваться по наследству как рецессивный признак. Если первая беременность протекала с формированием этой патологии, то риск повторного порока в последующем возрастает до 25%.
Что происходит с мозгом при синдроме Денди-Уокера?
Анатомически классический вариант мальформации задней черепной ямки включает:
- Гидроцефальный синдром разной степени выраженности;
- Кистозную полость в задней части черепа с расширенным четвертым желудочком мозга;
- Отсутствие или недоразвитие червя мозжечка, недоразвитие его полушарий.
Червь мозжечка — это структура, расположенная между его половинами и несущая в себе проводящие нервные волокна. При аномалии Денди-Уокера он может быть представлен небольшой щелью или широким пространством между гемисферами органа. При неполном отсутствии червя щелевидное расширение образуется лишь в нижней его части. На фоне патологии этого отдела наблюдается недостаточное развитие и мозжечковых полушарий.
Именно дефект мозжечкового червя в виде расщелины считается характерным признаком аномалии Денди-Уокера, позволяющим отличать ее от недоразвития на фоне других пороков мозга.
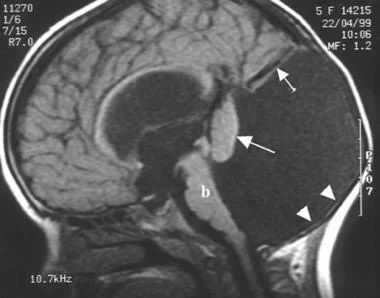
Киста четвертого мозгового желудочка может самопроизвольно вскрыться в 3-ий желудочек или субарахноидальное пространство. В этом случае симптомы окклюзии ликворных путей несколько ослабнут. Выраженность гидроцефального синдрома вариабельна — от небольшого расширения желудочковой системы до высокой степени окклюзионной гидроцефалии с отсутствием возможности для циркуляции ликвора.
Многие специалисты отмечают, что у большинства малышей с синдромом Денди-Уокера при рождении гидроцефалии как таковой нет, а формируется она и прогрессивно нарастает в течение первых нескольких месяцев жизни, поэтому факт отсутствия гидроцефального синдрома сразу после родов при наличии диагностированной внутриутробно патологии не является поводом для пересмотра диагноза и необоснованных надежд, с этим связанных.
Более, чем в половине случаев синдрома Денди-Уокера у детей помимо описанных структурных аномалий обнаруживаются и другие дефекты мозга — недоразвитие или отсутствие мозолистого тела, мозговые кисты, недоразвитие или отсутствие извилин, смещения серого вещества относительно его правильной локализации, что еще больше усугубляет течение и без того тяжелой патологии.
По данным МРТ было выделено несколько разновидностей синдрома Денди-Уокера:
- Классический тип аномалии — задняя черепная ямка расширена, четвертый желудочек кистозно изменен, червь мозжечка частично или полностью недоразвит, полушария его гипоплазированы, а намет находится выше, чем в норме, желудочковая система не сообщается с подпаутинным пространством, часто наблюдаются мозговые кисты и отсутствие мозолистого тела, практически у всех пациентов есть гидроцефалия, возможно сдавление стволовых структур. Порок проявляется клинически уже с рождения и имеет неблагоприятный прогноз.
- Вариант Денди-Уокера — морфологические признаки выражены меньше, чем при классической форме, гипоплазирован нижний отдел червя мозжечка, желудочки сообщаются с кистой и ликворными пространствами, обеспечивая отток ликвора, поэтому гидроцефалия наблюдается редко. Задняя черепная ямка имеет нормальные размеры, стволовые структуры не сдавливаются.
- Киста кармана Блейка — расширение желудочковой системы с гидроцефальным синдромом, киста расположена под или за мозжечком, червь развит относительно хорошо. Четвертый желудочек расширен, но не сообщается с затылочной ликворной цистерной.
- Mega cisterna magna — вариант очагового расширения подпаутинного пространства в задней и нижней частях задней ямки черепа с увеличением объема затылочной цистерны, которая сообщается с четвертым желудочком и субарахноидальным пространством.
Проявления заболевания
Симптоматика синдрома Денди-Уокера разнообразна. Возможны как практически нормальное развитие ребенка после рождения, так и грубые неврологические изменения, влекущие тяжелую инвалидность и даже смерть. По некоторым данным, нормальное развитие интеллекта бывает в половине случаев изолированного порока, возможно даже случайное обнаружение синдрома при обследовании взрослых.
дети с синдромом Денди-Уокера
Внутриутробное течение патологии определяется степенью поражения мозга, нарастанием гидроцефалии, наличием других пороков развития. Прогноз значительно хуже при диагностике синдрома до рождения. При глубоких нарушениях в формировании мозга на первый план среди других проявлений выступает гидроцефальный синдром:
- Увеличение диаметра головы;
- Выбухание родничка.
Увеличение диаметра черепа происходит, главным образом, за счет затылочной области, в которой образуется киста, вызывающая истончение и растяжение костной основы. При выраженной гидроцефалии голова малыша активно растет на протяжении первых двух месяцев, параллельно происходит расхождение швов между костями спереди или в заднем отделе. Кроме того, характерны:
- Повышение нервной возбудимости (рефлексов);
- Глазодвигательные расстройства — нистагм, косоглазие;
- Приступы остановки дыхания;
- Парез лицевого нерва.
Симптомы мозжечковых нарушений у новорожденных выявить невозможно, и даже тяжелый дефект формирования мозжечковых структур далеко не всегда вызывает значимые признаки атаксии (двигательных расстройств), которая регистрируется всего у трети пациентов.
Значительно чаще, нежели двигательные нарушения, возникают расстройства психической деятельности, интеллекта, которые проявляются на фоне общей двигательной «неловкости». У 25% больных с гипоплазированным мозжечком имеются признаки аутизма, в связи с чем специалисты пытаются найти взаимосвязь между изменениями мозжечка и аутизмом у детей.
Дети с гидроцефалией в раннем возрасте беспокойны, плохо спят, характерен монотонный крик, усиление рефлексов, плавающие движения глаз и их закатывание, выраженность сосудов роговицы, заметная подкожная венозная сеть по мере роста размеров головки. Спонтанная двигательная активность новорожденных может быть ослаблена, возможны судороги и тетрапарез из-за гипертонуса мышц.
В более старшем возрасте становится заметным отставание в психическом и интеллектуальном развитии, дети не могут обучаться, быстро устают, плохо усваивают новую информацию, что делает процесс адаптации крайне затруднительным. В тяжелых случаях обучение невозможно совсем, в связи с чем ребенок нуждается в постоянной посторонней помощи, уходе и рассмотрении вопроса об инвалидности.
Моторное развитие заметно замедлено. При тяжелых формах аномалии дети не могут своевременно научиться переворачиваться, ползать, садиться и ходить, не удерживают взгляд на игрушках, быстро устают и часто плачут. Возможны нарушения питания с гипотрофией, общее снижение иммунитета, частые инфекционные заболевания.
Сочетание порока нервной системы с другими аномалиями развития органов предрасполагает к серьезным осложнениям, в числе которых не только мозговая дисфункция, слабоумие, судорожный синдром, но и сердечная недостаточность, склонность к пневмониям при пороках сердца, хроническая почечная недостаточность и уремия при врожденном поликистозе, что усугубляет явления отека мозга и может послужить причиной гибели пациента.
При тяжелой окклюзионной гидроцефалии смерть может наступить в раннем младенчестве от отека головного мозга, фатальных аритмий, остановки дыхания на фоне компрессии стволовых структур, тяжелой пневмонии и других инфекционных осложнений.
У взрослых возможно постепенное нарастание гидроцефалии с краниалгиями, снижением памяти и внимания, раздражительностью, склонностью к депрессии, утренней тошнотой и рвотой на высоте головной боли. В тяжелых случаях бывает судорожный синдром. Возможны проблемы с координацией и выполнением мелких движений, неуверенность при ходьбе, зрительные расстройства.
Диагностика и лечение
Диагностика синдрома Денди-Уокера основывается на результатах ультразвукового осмотра, при этом важно обнаружить аномалию еще во время эмбрионального развития. УЗИ становится информативным после 18 недели гестации, но в некоторых случаях заподозрить патологию можно и раньше — уже на 14-15 неделях эмбрионального развития.
Диагностическими критериями при аномалии задней черепной ямки считаются:
- Наличие крупной кистозной полости, включающей четвертый мозговой желудочек, в задней части черепа;
- Отсутствие или аномальное развитие червя мозжечка;
- Гипоплазия мозжечковых гемисфер, наличие широкой щели между ними;
- Расширение желудочковой системы (гидроцефалия).
Для постановки диагноза синдрома Денди-Уокера необходимы:
- УЗИ (нейросонография);
- МРТ для определения анатомических особенностей четвертого желудочка мозга;
- Консультация офтальмолога;
- Осмотр нейрохирурга;
- УЗИ сердца для исключения врожденных аномалий;
- Консультация генетика и определение кариотипа при возможных генетических мутациях.
Лечение патологии определяется симптоматикой и тяжестью проявлений. Если гидроцефалии нет, а внутричерепное давление в пределах нормы, то оправдано динамическое наблюдение невролога, педиатра или нейрохирурга, каких-либо медикаментов не требуется.
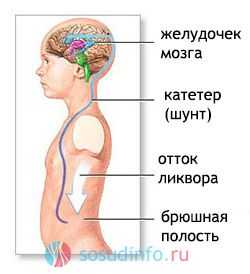
шунтирование для нивелирования гидроцефалии
При нарастании гидроцефалии и внутричерепного давления показаны шунтирующие хирургические операции для отвода ликвора из черепа в грудную или брюшную полость. Медикаментозное лечение включает применение диуретиков (диакарб, маннитол), ноотропных средств (пирацетам, пантогам), антиконвульсантов (депакин).
В случае гипертонуса показаны физиотерапевтические и водные процедуры, массаж, специальные упражнения. Важен тщательный уход и постоянное наблюдение за малышом, создание спокойной обстановки при беспокойном поведении и нарушениях сна.
При тяжелых формах течения патологии с отставанием в интеллектуальном развитии детям показана работа с дефектологами-педагогами, психологом по индивидуальной программе, исключающей избыток информации и умственное перенапряжение.
Прогноз при синдроме Денди-Уокера зависит от ряда причин: времени установления диагноза, наличия других пороков и хромосомных болезней, степени окклюзии ликворных путей. Смертность и заболеваемость после рождения выше в тех случаях, когда аномалия сочетается с другими дефектами и обнаружена до рождения малыша.
Гидроцефалия и внутричерепная гипертензия — ключевые моменты в определении прогноза, которые влияют и на развитие пациента, и на продолжительность и качество его жизни. В случае изолированного поражения мозга без признаков гидроцефалии прогноз благоприятный. Ребенок может развиваться по возрасту, а иногда аномалия и вовсе выявляется у взрослых при обследовании по поводу других причин.
В связи с тем, что причины порока так и не выяснены, проводить специфическую профилактику не представляется возможным. Конечно, нужно соблюдать здоровый образ жизни, особенно, женщинам, планирующим беременность или уже забеременевшим с исключением вредных привычек, неблагоприятных влияний внешней среды. Важно своевременно выявить и пролечить цитомегаловирусную инфекцию, герпес, а в случае краснухи, которой женщина заболела при беременности, врачи предложат аборт по медицинским показаниям из-за высокого риска сочетанных пороков.
Решать вопрос о сохранении беременности в том случае, если синдром возник случайно, у плода абсолютно здоровой женщины, придется будущей маме и ее семье. Решение всегда дается сложно, но следует знать, что аномалия нервной системы и нормальное развитие и рост ребенка — скорее, исключение из правил.
В абсолютном большинстве случаев детям и родителям приходится бороться с гидроцефалией, зачастую требуется не одна дорогостоящая и сложная операция, тогда как ее эффективность и прогноз все равно могут оставаться сомнительными.
Видео: примеры детей с синдромом Денди-Уокера
Классическая мальформация Денди-Уокера
Мальформация Денди-Уокера — наиболее частая мальформация задней черепной ямки, характеризующаяся следующей патологической триадой:
1) Гипоплазия червя с его низким расположением.
2) Кистозное расширение четвёртого желудочка, выдающаяся в заднюю черепную ямку.
3) Увеличенная в размерах задняя черепная ямка
Эпидемиология
Генетически обусловленное заболевание встречаемое с частотой 1:30000 и определяется преимущественно у женщин.
Клинические проявления
Постнатальный период:
Медленное моторное развитие, краниомегалия.
В более старшем возрасте:
Гидроцефалия, мозжечковая симптоматика, реже поражение черепных нервов.
Патология
Генетически обусловленное заболевание.
Ассоциация
Не входящие в комплекс мальформации патологические изменения ЦНС встречаются в 70% случаев.
Кортикальная дисплазия
Полимикрогирия
Дисгенезия мозолистого тела
Липома мозолистого тела
Шизэнцефалия
Затылочное энцефалоцеле
Пояснично-крестцовое менингоцеле
Сирингомиелия
Не входящие в комплекс мальформации патологические изменения не-ЦНС встречаются в ~25% случаев.
Волчья пасть
Ангиомы лица
Низко посаженные уши
Полидактилия или синдактилия
Аномалия развития сердца
Радиологические находки
УЗИ
При антенатальном УЗИ типичны следующие находки:
Увеличение цистерны магна (>10 мм).
Полная аплазия червя.
Пространства между полушариями мозжечка выглядит треугольной формы.
МРТ
МРТ является методом выбора при диагностике мальформации Денди-Уокера.
Гипоплазия червя с его низким расположением.
Кистозное расширение четвёртого желудочка, выдающаяся в заднюю черепную ямку. При данном состоянии доли мозжечка подвержены антеро-латеральному смещению.
Увеличенная в размерах задняя черепная ямка
У 75-90% пациентов выявляется обструктивная гидроцефалия с наиболее частой причиной — стеноз на уровне водопровода.
Дифференциальный диагноз
Мега цистерна магна
Эпидермоидная киста
Арахноидальная киста
Гипоплазия червя при аномалии Джуберта
Изолированный четвёртый желудочек
Киста Блейка
Лечение
Хирургическое вмешательство и дренирование избытка ЦСЖ в брюшную полость.
Мега цистерна магна
Мега цистерна магна — это фокальное увеличение субарахноидального пространства в нижних и задних отделах задней черепной ямки и чаще является случайной находкой при визуализации головного мозга.
Эпидемиология
При нейровизуализации в постнатальном периоде мега цистерна магна выявляется примерно у ~1% населения.
Патология достоверно связана
— инфарктом миокарда
— воспалительные процессы/инфекция (в особенности — цитомегаловирус).
— хромосомные аномалии (трисомия 18 — Синдром Эдвардса)
Однако, если при визуализации изменений со стороны желудочков не выявлено, то говорят о хорошем прогнозе.
Клиника
Нет специфических симптомов.
Патология
Считается, что мега цистерна магна — это результат недоразвития (замедленного развития) кармана Блейка.
Радиологические находки
УЗИ
При скрининге в антенатальном периоде выявляется расширение позади мозжечкового пространства, заполненного жидкостью.
обычно >10 мм (некоторые до 12 мм)
Для исключения Денди-Уокера внимательная оценка анатомии червя.
Аномалия Киари ( Синдром Арнольда-Киари )
Аномалия Киари (синдром Арнольда-Киари) — заболевание, при котором структуры головного мозга, расположенные в задней черепной ямке, опущены в каудальном направлении и выходят через большое затылочное отверстие. В зависимости от типа аномалия Киари может проявляться головной болью в затылке, болью в шейном отделе, головокружением, нистагмом, обмороками, дизартрией, мозжечковой атаксией, парезом гортани, снижением слуха и ушным шумом, нарушением зрения, дисфагией, дыхательными апноэ, стридором, расстройствами чувствительности, гипотрофией мышц и тетрапарезом. Аномалия Киари диагностируется путем проведения МРТ головного мозга, шейного и грудного отделов позвоночника. Аномалия Киари, сопровождающаяся стойким болевым синдромом или неврологическим дефицитом, подлежит хирургическому лечению (декомпрессия задней черепной ямки или шунтирующие операции).
МКБ-10
Общие сведения
В области соединения черепа с позвоночным столбом находится большое затылочное отверстие, на уровне которого ствол головного мозга переходит в спинной мозг. Выше этого отверстия локализуется задняя черепная ямка. В ней расположен мост, продолговатый мозг и мозжечок. Аномалия Киари связана с выходом части анатомических структур задней черепной ямки в просвет большого затылочного отверстия. При этом происходит сдавление находящихся в этой области структур продолговатого и спинного мозга, а также нарушение оттока цереброспинальной жидкости из головного мозга, приводящее к гидроцефалии. Вместе с платибазией, ассимиляцией атланта и др. аномалия Киари относится к врожденным порокам развития краниовертебрального перехода.
Аномалия Киари встречается по различным данным у 3-8 человек на 100 тысяч населения. В зависимости от типа аномалия Киари может диагностироваться в первые дни после рождения ребенка или стать неожиданной находкой у взрослого пациента. В 80% случаев аномалия Киари сочетается с сирингомиелией.
Причины
До сих пор аномалия Киари остается заболеванием, об этиологии которого в неврологии нет единого мнения. Ряд авторов считает, что аномалия Киари связана с уменьшенным размером задней черепной ямки, приводящим к тому, что по мере роста расположенных в ней структур они начинают выходить через затылочное отверстие. Другие исследователи предполагают, что аномалия Киари развивается в результате увеличенных размеров головного мозга, который при этом как бы выталкивает содержимое задней черепной ямки через затылочное отверстие.
Спровоцировать переход незначительно выраженной аномалии в выраженную клиническую форму может гидроцефалия, при которой за счет увеличения желудочков увеличивается общий объем мозга. Поскольку аномалия Киари наряду с дисплазией костных структур краниовертебрального перехода сопровождается недоразвитием связочного аппарата этой области, любая черепно-мозговая травма может приводить к усугублению вклинения миндалин мозжечка в затылочное отверстие с манифестацией клинической картины заболевания.
Классификация
Аномалия Киари подразделяется на 4 типа:
- Аномалия Киари I характеризуется опущением миндалин мозжечка ниже большого затылочного отверстия. Обычно она проявляется у подростков или во взрослом возрасте. Зачастую сопровождается гидромиелией — скоплением цереброспинальной жидкости в центральном канале спинного мозга.
- Аномалия Киари II проявляется в первые дни после рождения. Кроме миндалин мозжечка при этой патологии через большое затылочное отверстие выходят также червь мозжечка, продолговатый мозг и IV желудочек. Аномалия Киари II типа намного чаще сочетается с гидромиелией, чем первый тип, и в подавляющем большинстве случаев связана с миеломенингоцеле — врожденной спинномозговой грыжей.
- Аномалия Киари III отличается тем, что опустившиеся через большое затылочное отверстие мозжечок и продолговатый мозг, располагаются в менингоцеле шейно-затылочной области.
- Аномалия Киари IV заключается в гипоплазии (недоразвитии) мозжечка и не сопровождается его смещением в каудальном направлении. Некоторые авторы относят эту аномалию к синдрому Денди-Уокера, при котором гипоплазия мозжечка сочетается с наличием врожденных кист задней черепной ямки и гидроцефалией.
Аномалия Киари II и Киари III часто наблюдается в комбинации с другими дисплазиями нервной системы: гетеротопией коры головного мозга, полимикрогирией, аномалиями мозолистого тела, кистами отверстия Можанди, перегибом сильвиевого водопровода, гипоплазией подкорковых структур, намета и серпа мозжечка.
Симптомы аномалии Киари
Наиболее часто в клинической практике встречается аномалия Киари I типа. Она проявляется ликворногипертензионным, церебеллобульбарным и сирингомиелическим синдромами, а также поражением черепно-мозговых нервов. Обычно аномалия Киари I манифестирует в период полового созревания или уже во взрослом возрасте.
Для ликворногипертензионного синдрома, которым сопровождается аномалия Киари I, характерна головная боль в затылке и шейной области, усиливающаяся во время чихания, кашля, натуживания или напряжения мышц шеи. Может наблюдаться рвота, не зависящая от приема пищи и ее характера. При осмотре пациентов с аномалией Киари выявляется повышенный тонус мышц шеи. Среди мозжечковых нарушений наблюдаются нарушение речи (дизартрия), нистагм, мозжечковая атаксия.
Поражение ствола мозга, расположенных в нем ядер черепно-мозговых нервов и их корешков проявляются снижением остроты зрения, диплопией, расстройством глотания, снижением слуха по типу кохлеарного неврита, системным головокружением с иллюзией вращения окружающих предметов, ушным шумом, синдромом сонных апноэ, повторяющимися кратковременными потерями сознания, ортостатическим коллапсом. Пациенты, у которых имеется аномалия Киари, отмечают усиление головокружения и ушного шума при поворотах головой. Поворот головы у таких больных может спровоцировать обморок. Может отмечаться атрофические изменения половины языка и парез гортани, сопровождающийся осиплостью голоса и затруднением дыхания. Возможен тетрапарез с большим снижением мышечной силы в верхних конечностях, чем в нижних.
В случаях, когда аномалия Киари I сочетается с сирингомиелией, наблюдается сирингомиелический синдром: нарушения чувствительности по диссоциированному типу, онемения, мышечные гипотрофии, тазовые нарушения, нейроартропатии, исчезновение брюшных рефлексов. При этом некоторые авторы указывают на несоответствие размера и местонахождения сирингомиелической кисты распространенности расстройств чувствительности, степени выраженности парезов и мышечной гипотрофии.
Аномалия Киари II и Киари III имеют сходные клинические проявления, которые становятся заметны с первых минут жизни ребенка. Аномалия Киари II сопровождается шумным дыханием (врожденный стридор), периодами кратковременной остановки дыхания, двусторонним нейропатическим парезом гортани, нарушением глотания с забросом жидкой пищи в нос. У новорожденных аномалия Киари II проявляется также нистагмом, повышением мышечного тонуса в верхних конечностях, цианозом кожных покровов, возникающим во время кормления. Двигательные расстройства могут быть выражены в различной степени и прогрессировать вплоть до тетраплегии. Аномалия Киари III имеет более тяжелое течение и зачастую является не совместимым с жизнью нарушением развития плода.
Диагностика
Неврологический осмотр и стандартный перечень неврологических обследований (ЭЭГ, Эхо-ЭГ, РЭГ) не дают специфических данных, позволяющих установить диагноз «аномалия Киари». Как правило, они выявляют лишь признаки значительного повышения внутричерепного давления, т. е. гидроцефалию. Рентгенография черепа выявляет только костные аномалии, которыми может сопровождаться аномалия Киари. Поэтому до внедрения в неврологическую практику томографических методов исследования диагностика этого заболевания представляла для невролога большие затруднения. Теперь врачи имеют возможность поставить таким пациентам точный диагноз.
Следует отметить, что МСКТ и КТ головного мозга при хорошей визуализации костных структур краниовертебрального перехода не позволяют достаточно точно судить о мягкотканных образованиях задней черепной ямки. Поэтому единственным достоверным методом диагностики аномалии Киари на сегодняшний день является магнитно-резонансная томография. Ее проведение требует обездвиженности пациента, поэтому у маленьких детей она проводится в состоянии медикаментозного сна. Кроме МРТ головного мозга для выявления менингоцеле и сирингомиелических кист необходимо также проведение МРТ позвоночника, особенно его шейного и грудного отделов. При этом проведение МРТ исследований должно быть направлено не только на диагностику аномалии Киари, но и на поиск других аномалий развития нервной системы, которые часто с ней сочетаются.
Лечение аномалии Киари
Бессимптомно протекающая аномалия Киари не нуждается в лечении. В случаях, когда аномалия Киари проявляется лишь наличием болей в шее и затылочной области, проводят консервативную терапию, включающую анальгетические, противовоспалительные и миорелаксирующие препараты. Если аномалия Киари сопровождается неврологическими нарушениями (парезы, расстройства чувствительности и мышечного тонуса, нарушения со стороны черепно-мозговых нервов и пр.) или не поддающимся консервативной терапии болевым синдромом, то показано ее хирургическое лечение.
Наиболее часто в лечении аномалии Киари применяется краниовертебральная декомпрессия. Операция включает расширение затылочного отверстия за счет удаления части затылочной кости; ликвидацию сдавления ствола и спинного мозга за счет резекции миндалин мозжечка и задних половин двух первых шейных позвонков; нормализацию циркуляции цереброспинальной жидкости путем подшивания в твердую мозговую оболочку заплаты из искусственных материалов или аллотрансплантата. В некоторых случаях аномалия Киари лечится при помощи шунтирующих операций, направленных на дренирование цереброспинальной жидкости из расширенного центрального канала спинного мозга. Цереброспинальная жидкость может отводиться в грудную или брюшную полость (люмбоперитонеальное дренирование).
Прогноз
Важное прогностическое значение имеет тип, к которому относится аномалия Киари. В некоторых случаях аномалия Киари I может на протяжении всей жизни пациента сохранять бессимптомное течение. Аномалия Киари III в большинстве случаев приводит к летальному исходу. При появлении неврологических симптомов аномалии Киари I, а также при аномалии Киари II большое значение имеет своевременное проведение хирургического лечения, поскольку возникший неврологический дефицит плохо восстанавливается даже после успешно проведенной операции. По различным данным эффективность хирургической краниовертебральной декомпрессии составляет 50-85%.
Сосудистые мальформации головного мозга — их виды, симптомы и удаление

В переводе с английского языка, слово мальформация означает неправильное развитие чего-либо. Медики под этим термином подразумевают разные патологические отклонения, которые влекут за собой сбои в функциональности или изменения в строении каких-либо органов.
По большому счету, мальформации появляются при рождении или в ходе развития человека. Но не исключено, что они могут возникнуть в результате травм или как последствие серьезных болезней.
Сосудистые мальформации головного мозга
Существует несколько видов мальформаций, и одна из них относится к патологии сосудов головного мозга. Этот вид заболевания характеризуется изменениями артерий, вен и их коммуникации. Также, как и в остальных случаях, патологические изменения проявляют себя как хроническое расстройство и появляются в результате некоторых болезней.
Основной особенностью мальформаций сосудов головного мозга является то, что они напрямую связаны с центральной нервной системой, и появляются либо в головном, либо в спинном мозге.
Внешне болезнь выглядит, как узлы или переплетения сферической формы и разных размеров, которые затрудняют движение крови по сосудам.
Согласно медицинской статистике, такая аномалия в хронической форме проявляется у 15-19 новорожденных на 1000 человек.
О причинах в теории
Точных причин появления патологии у младенцев пока не выявлено, но существует одна теория-предположение.
Окончательное формирование всей структуры сосудов головного мозга плода происходит к 20 неделе его развития в утробе. Значит, сосудистая сеть в это время находится в нестабильной форме и склонна к деформации, особенно под воздействием негативных факторов.
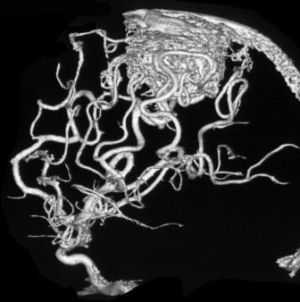
Сосудистая мальформация на МРТ
К примеру, причиной этого может служить серьезная болезнь матери:
- инфекционное заболевание;
- родовой травматизм беременной матери;
- серповидноклеточная анемия.
А также в список провоцирующих факторов можно включить медикаменты или наркотические средства, которые мама принимала во время вынашивания ребенка.
Точных причин, которые напрямую влияют на развитие болезни на сегодняшний день не обнаружено.
Общая симптоматика
Зачастую пациенты живут с сосудистой мальформацией долгие годы и не знают о наличии этого заболевания. Заболевание имеет свойство не проявляться долгое время. Именно в этот период, затаившаяся болезнь прогрессирует, увеличивая свои размеры. И в случае «созревания», происходит давление на ГМ (головной мозг), что приводит к проявлению симптомов.
Общие симптомы сосудистой мальформации:
- апатическое состояние и депрессивность;
- вялость и быстрое истощение;
- сбои координации человека;
- нарушение моторики;
- повышенное давление внутричерепной коробки с пульсацией в голове;
- зрительные нарушения;
- приступы судороги;
- шумы в голове;
- слуховые или обонятельные галлюцинации;
- головные боли и головокружения.
Если недуг появился при рождении, то он начинает себя проявлять в возрасте 12-14 лет, когда ребенок растет физически. В любых других случаях, он может дать о себе знать даже в среднем возрасте (47-65 лет). Существует несколько видов мальформаций, от которых напрямую зависит симптоматика.
Артериовенозная мальформация
Это патология, которая затрагивает соединение вен и артерий. Суть нарушения заключается в том, что при соединении вен и артерий отсутствуют капилляры. Она напрямую связана с центральной нервной системой, но ее проявление возможно даже в отдаленных областях.
К примеру, часто бывают случаи, когда нарушение может проявить себя при пороке сердца, между луковицей аорты и легочным стволом.
Причинами АВМ могут служить:
- повреждения сосудов головы в результате черепно-мозговых травм;
- хронические структурные нарушения сосудистой системы;
- отложения жирных элементов на стенках сосудов.
АВМ считается самой серьезной разновидностью мальформация сосудистой системы и имеет симптомы, по которым ее можно определить:
- тошнота и частая рвота;
- потеря сознания;
- кровоизлияние внутри ГМ;
- резкие головные боли;
- парциальные и генерализованные судороги;
- частичная потеря зрения и слуха;
- обонятельные галлюцинации;
- сбои речевых функций.
Полностью устранить болезнь такого вида можно только лишь на начальных стадиях ее развития. После диагностирования, зная все тонкости имеющейся патологии, лечащий врач должен назначить метод лечения. В большинстве случаев делают проводят три вида операций:
- оперативное вмешательство;
- радиохирургия;
- эмболизация.
Болезнь Денди-Уокера
Аномалия Денди-Уокера характеризуется гипертрофией мозжечка или ликворной опухолью задней черепной ямки. Аномалия вызывается в основном генетически, но бывают случаи, когда она проявляется из-за вирусных инфекций, воздействия алкоголя или диабета беременной женщины.
Развитие синдрома может проявляться следующими симптомами:
- тошнота;
- агрессия;
- появление судорог;
- расстройство зрения;
- сбои координации движения;
- нарушение согласованности движений мышц;
- колебания движений глаз (нистагм).
Вылечить болезнь можно только с помощью хирургической операции. Она заключается в искусственном вентрикулоперитонеальном шунтировании для уменьшения давления ликвора в мозгу.
Но при этом есть риск, что некоторые появившиеся дефекты могут остаться на всю жизнь.
Мальформация Арнольда Киари
Суть аномалии лежит в том, что миндалины мозжечка ГМ опускаются в отверстие затылочной области и при этом сдавливает продолговатый мозг.
До 2005 года считалось, что эта мальформация является врожденной. Но после соответственных исследований было установлено, что синдром Киари возникает в результате напряжения концевой нити, что приводит к натяжению спинного мозга.
А также существует версия, что мозжечок смещается к верхней части канала позвоночника, тем самым нарушая стабильную циркуляцию ликвора и блокируя сигналы, поступающие к другим органам.
Этому заболеванию свойственны симптомы поражения мозжечка и продолговатого отдела ГМ:
- головная боль с проявлением простудных симптомов;
- затылочная боль, спускающаяся к плечевому поясу;
- потеря равновесия;
- снижение активности моторики рук;
- онемение верхних конечностей.
Как и при любой подобной болезни, лечение будет производиться только после обследования пациента. Это зависит от степени ее проявления.
Врачами может быть назначено консервативное лечение лекарственными препаратами, которое будет направлено на устранение первичных симптомов. Но, при прогрессии болезни проблема решается оперативным вмешательством.
Мальформация Арнольда Киари II степени характеризуется большими размерами задней части черепа, по сравнению с передней. За счет увеличенных габаритов также и увеличено большое затылочное отверстие, в которое впадает нижняя часть мозжечка. Это приводит к их встречному сдавливанию со спинным мозгом.
Определить такую мальформацию можно с помощью МРТ, КТ (область затылка, позвонков шеи) и проявления симптомов:
- посторонние шумы в ушах;
- головокружение и истощенность;
- повышенное давление внутри головы;
- периодические нистагмы.
Хирургическая операция направлена на удаление мешающей части кости в задней области черепа. Появляется больше места для мозга, вследствие чего снижается давление. Такая операция считается серьезной, но не слишком трудной. Она длится около 2 часов.
Общий подход к лечению
Как уже упомянуто выше, основной базой лечения этого заболевания является три вида операций: микрохирургические, радиохирургические и эндоваскулярные.
Целесообразность применения того или иного вида вмешательства определяется несколькими факторами:
- место нахождения мальформации;
- габариты патологии и внешняя форма;
- наличие кровоизлияний ГМ в истории болезней;
- тип патологии;
- лабораторные анализы пациента.
Подробней о каждом методе:
- Микрохирургическая операция - заключается в полном иссечении патологии, но является одним из опаснейших способов из-за возможных внутренних кровотечений. Если заболевание касается артерий и вен, то проводится трепанация черепа. Параллельно с сосудистым узлом также удаляется внутричерепная гематома. Такие операции проводятся под наркозом высокого уровня.
- Радиохирургия - такая операция возможна только, если АВМ в диаметре не более 30 мм. Принято выполнять для удаления при вновь выявленных и остаточных изменениях. Терапевтические лучи подаются постепенно, с удержанием. В результате попадания оптимального количества лучей, сосуды «заращиваются» и движение крови по ним останавливается. Процесс лечения может проводиться по частям, растянувшись 1-3 года.
- Эмболизация - проводится перед хирургической операцией, для профилактики кровоизлияний. Через тазобедренный прокол внедряется тонкий катетер, направляется к сосудам, которые снабжают ГМ кровью и подводится к самой патологии. Через трубку подается эмболизирующий материал и блокирует пораженные места сосудов и останавливает подачу кровотока.
В некоторых ситуациях в курс лечения включается и консервативный метод с использованием аптечных препаратов.
Чем опасны мальформации?
Нужно отметить, что сосудистая мальформация чревата осложнениями. Среди них можно выделить три, которые представляют опасность для жизни больного:
- Кислородное голодание ГМ. Из-за него происходит отмирание клеток головного мозга (полное или частичное). Сперва это приводит к инфаркту мозга, который проявляется потерей памяти, зрения, координации движения. При отсутствии лечения, человек впадает в кому или сопор.
- Истончение сосудов. Вследствие этого может произойти разрыв сосуда с кровоизлиянием в результате. Кровь попадает в полость черепа, что характеризуется нарушением кровообращения мозга. Происходит геморрагический инсульт.
- Паралич. При сдавливании спинного мозга есть риск развития паралича.
Профилактика осложнений
Профилактические меры здесь направлены на предупреждение развития осложнений. Это можно сделать так:

- не подвергаться психоэмоциональным воздействиям, которые провоцируют стрессы;
- отказаться от физических нагрузок, связанных с поднятием тяжестей;
- контролировать артериальное давление и не давать ему повышаться, для снижения АД разрешено использовать аптечные препараты для гипертоников;
- употреблять пищу с большим содержанием натрия;
- отказаться от вредных привычек и употребления алкогольных напитков.
Для более подробных рекомендаций следует обратиться к вашему лечащему врачу.
Читайте также:
- Дифференциация поражений тройничного нерва. Похожие на тройничную невралгию синдромы
- Блокада синаптического пути торакальных больных. Показатели адекватности анестезии
- КТ, МРТ при плоскоклеточном раке задней стенки ротоглотки
- Снятие оттиска с верхней беззубой челюсти по методу ЦИТО. Этапы снятия оттиска.
- Хронический алкоголизм и кишечные инфекции. Абстинентный синдром в виде кишечной патологии